
Abstract
Background: Cerebellar low-grade astrocytomas (CLGAs) of childhood are benign tumours and are usually curable by surgical resection alone or combined with adjuvant radiotherapy. Objective: To undertake a retrospective study of our children with CLGA to determine the optimum schedule for surveillance imaging following initial surgery. In this report we describe the phenomenon of spontaneous regression of residual tumour and discuss its prognostic significance regarding future imaging. Materials and methods: A retrospective review was conducted of children treated for histologically proven CLGA at Great Ormond Street Hospital from 1988 to 1998. Results: Of 83 children with CLGA identified, 13 (15.7%) had incomplete resections. Two children with large residual tumours associated with persistent symptoms underwent additional treatment. Eleven children were followed by surveillance imaging alone for a mean of 6.83 years (range 2–13.25 years). Spontaneous tumour regression was seen in 5 (45.5%) of the 11 children. There were no differences in age, gender, symptomatology, histological grade or Ki-67 fractions between those with spontaneous tumour regression and those with progression. There was a non-significant trend that larger volume residual tumours progressed. Conclusions: Residual tumour followed by surveillance imaging may either regress or progress. For children with residual disease we recommend surveillance imaging every 6 months for the first 2 years, every year for years 3, 4 and 5, then every second year if residual tumour is still present 5 years after initial surgery. This would detect not only progressive or recurrent disease, but also spontaneous regression which can occur later than disease progression.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cerebellar low-grade astrocytoma (CLGA) is the most common childhood brain tumour and accounts for approximately 40% of paediatric posterior fossa tumours. It is the second most common posterior fossa tumour after medulloblastoma. The majority of CLGAs are classified by the World Health Organisation as grade I pilocytic or grade II fibrillary astrocytomas. They have been considered benign tumours since their original description by Cushing in 1931 [1], and they are associated with good outcomes for both survival and function. In the modern era, studies have shown long-term survival in more than 90% of patients [2–4]. Complete surgical resection at first operation, low histological grade, location of tumour outside the brainstem and shorter intraoperative time have all been associated with improved survival [2, 3, 5–7].
We have previously demonstrated that complete resection of medulloblastoma and ependymoma correlates with improved outcome [8, 9], but the evidence for a better prognosis with complete resection for CLGAs is not established [4, 10–13]. This is due to the paradoxical observation that long-term survival has been observed in a higher proportion of children than might be expected if a complete surgical resection (as judged by either the surgical or the postoperative radiological assessments) was the sole guarantor of cure. Indeed incomplete resection rates of between 11.3% and 35% have been reported [2, 3, 6]. We suggest that it is the phenomenon of postoperative regression occurring without further adjuvant treatment which accounts for this apparent “mismatch”.
Individual cases of spontaneous regression of incompletely resected CLGAs have been observed in some published series [2, 6, 10, 13, 14], but the biological behaviour of the residual tumour remains unclear and the optimum treatment for postoperative residual tumour has yet to be established. The aim of this study was to investigate in more detail the phenomenon of spontaneous regression of residual tumour and to determine its significance particularly with regard to prognosis and the future (surveillance) imaging of this group.
Materials and methods
Patient selection
Clinical details of all children with histologically proven CLGA, diagnosed and treated at Great Ormond Street Hospital between January 1988 and September 1998 (in order to allow a minimum follow-up period of 5 years) were obtained from the Department of Neurosurgery operative and oncology databases. The age at diagnosis, gender, histology, date of first resection and extent of tumour clearance, and tumour progression/regression were taken from the department of radiology information systems, neurosurgery database and patients’ clinical records. The available imaging and radiology reports of all patients were reviewed by a paediatric neuroradiologist (D.S.) and those children with residual postoperative tumour were identified. The neuroimaging of those children with incomplete resection were re-reviewed by two paediatric neuroradiologists (R.G. and D.S.).
Histopathological criteria
The histopathology for the patients with residual tumour after the initial surgical resection was reviewed by a specialist paediatric neuropathologist (B.H.). Tumours were classified as pilocytic or fibrillary astrocytomas. Pilocytic tumours were classified according to the World Health Organisation classification of pilocytic astrocytoma occurring in children and young adults and histologically characterized by a biphasic pattern of variable proportions of compacted bipolar cells with Rosenthal fibres and multipolar cells with microcysts and granular bodies. Fibrillary or diffuse tumours had a uniform and densely compacted cellular organization with mild-to-moderate degrees of nuclear hyperchromatism and atypia [15]. The percentages of nuclei stained for Ki-67 monoclonal antibody levels were measured for all incompletely resected tumours.
Imaging
Patients were managed according to standard oncology protocols. The preoperative MRI (Magnetom, Vision and Symphony, Siemens, Erlangen, Germany) protocol for the brain included axial T2-weighted (T2-W), coronal precontrast T1-weighted (T1-W), three-plane post-contrast T1-W fast spin-echo images and coronal FLAIR images. Although our current oncology protocol includes an immediate postoperative MRI performed within 48 h, all children with residual tumour in this series were scanned before this era, and we therefore chose the scan performed at 3–6 months postoperatively for our assessment of the presence or absence of tumour, comparing it with the preoperative scan in order to avoid misinterpretation of postsurgical enhancement as residual tumour. Surveillance imaging commenced at 3–6 months postoperatively and was repeated 3- to 6-monthly for the first year, and then yearly or 2-yearly thereafter for at least 5 years.
Approximate of tumour volumes were calculated using the formula 4/3πr3 on the assumption of approximately spherical geometry, where r is the mean of the sum of the AP, craniocaudal and right-to-left diameters of the lesion, measured to within 0.1 cm by reference to the scale on the printed film. Interval change in tumour volume was also rated visually on a separate occasion by a neuroradiologist (R.G.).
Definition of terms
Complete tumour excision was defined as the absence of residual tumour on postoperative MRI regardless of the surgeon’s assessment of extent of resection. Radiologically, an incomplete resection was defined as the presence of a residual contrast-enhancing mass or nodule on the earliest postoperative imaging and which correlated with tumour seen on the original scan. Linear enhancement at the resection margins, which may be seen immediately postoperatively on a scan performed under the same general anaesthetic and may persist for several years following surgery, was not considered to represent residual disease. Tumour progression was defined as an increase in tumour volume observed on serial scans and spontaneous regression was defined as a reduction in tumour volume in the absence of further surgery or adjuvant radiotherapy. Surveillance imaging was defined as scans carried out on a routine basis for the detection of recurrent or residual disease in the absence of new symptoms or signs.
Results
In total, 86 children were treated for CLGA between January 1988 and September 1998. Of these, three returned to their home countries following surgery, leaving a cohort of 83 patients who had long-term follow-up at our hospital. All 83 children survived a mean follow-up period of 6 years 10 months. Their mean age was 5.5 years (range 16 months to 15 years). None of the children within the cohort had neurofibromatosis 1 (NF1) and none had evidence of craniospinal metastasis at presentation. Further clinical and radiological details and surveillance imaging strategies for the overall cohort are discussed elsewhere [12].
Of the 83 children, 13 (15.5%) were judged to have had an incomplete resection because of the presence of MRI-visible residual disease on the scan performed 3 months or so following their surgery, and 12 were also deemed to have had an incomplete resection according to the neurosurgeon’s observations. Two children had such large residual tumour volumes associated with persistent symptoms that they received further early radiotherapy. The remaining 11 children with known residual disease were managed initially with a policy of surveillance imaging for a mean of 6 years 10 months (range 2–13 years 3 months).
Spontaneous tumour regression was observed in 5 (45.5%) of these 11 children at 7, 16, 29, 45 and 50 months postoperatively (Fig. 1) with a mean of 32 months. Further surveillance imaging was then stopped for two children (one at 14 months and the other at 5 years) and continued for a maximum period of 5 years 11 months for the remaining three. In one of the remaining six children, the postoperative residual tumour was observed to be stable at 14 months after the original resection. In the other five children, tumour progression was observed on subsequent surveillance scans at 7, 9, 12, 13 and 20 months, respectively, at which point they were treated with either further surgery (n=1), adjuvant radiotherapy (n=1) or a combination of both (n=3). None of the tumours which spontaneously regressed has shown evidence of subsequent progression, either clinical or radiological, over a period of up to 12 years since their initial surgery.

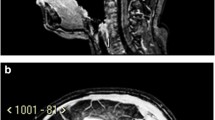
A child aged 16 months at the time of diagnosis. Residual CLGA in the right cerebellar hemisphere (volume approximately 2 cm3) on (a) coronal and (b) sagittal post-contrast T1-W images obtained 12 weeks after surgery. The residual disease was smaller on a scan at 50 months and had completely regressed on the next scan (c) performed at 87 months
There were no differences in age, numbers of children under 3 years or gender between the two groups (Table 1). Most patients were asymptomatic with the exception of one child with small volume residual disease (which later regressed spontaneously) who developed persistent postoperative headaches. The majority of tumours within the overall cohort (80 of 83) and most of the incompletely resected tumours under surveillance (10 of 11) were grade I pilocytic astrocytomas. One of the incomplete resections which later progressed was a diffuse fibrillary tumour. The Ki-67 fraction was <2% in all cases and did not predict residual tumour progression. There was an association of subsequent progression of residual disease with a larger postoperative tumour volume compared to tumours that were stable or regressed (Table 2), although with the small numbers involved statistical comparisons were not possible.
Discussion
The most important finding in this study was that following an incomplete resection of residual CLGA in childhood, 5 of 11 residual tumours kept under surveillance underwent spontaneous regression. This observation is consistent with two previous studies [2, 6] and supports the value of surveillance imaging in this group of children as opposed to exposing them to the risks of further treatment, whether surgical or adjuvant radiotherapy.
Due-Tonnessen et al. [2] looked at a cohort of 82 patients with CLGA (very similar to our own) and found spontaneous regression of residual postoperative tumour in 9 of 16 patients (56%). Although most of the residual tumours were documented by the operating surgeon, this is comparable with our findings of a spontaneous regression rate of 45.5% for residual tumours assessed by MRI. It is not clear whether some of their “true recurrent” tumours (5 of 79 patients with “total resections”) may in fact have represented regrowth of residual tumour as little reference is made to the timing of early postoperative imaging. Despite the high incidence of spontaneous regression, the authors suggest that a second operation should be performed unless there is a high risk of surgical morbidity/mortality.
Smoots et al. [6] described spontaneous tumour regression in 4 (24%) of 17 patients with residual disease at a mean of 21 months (range 3–49 months) confirmed and followed-up on surveillance CT or MRI imaging. In 9 (53%) of the 17 patients with residual tumour on postoperative imaging the residual tumour continued to progress. They found that residual tumours with a smaller postoperative volume had a reduced risk of progression and a non-significant trend for smaller lesions to regress spontaneously was noted. We concur with their findings, but the small numbers involved do not allow valid statistical comparisons.
The main difficulty of this work and work by others [2, 6] is that we relied on imaging alone, and not histology, to determine the presence of postoperative tumour residuum. However, we believe that we were looking at spontaneous regression of residual tumour and not resolution of postoperative enhancement for a number of reasons. Firstly, the “tissue” subsequently progressed in just under 50% of the children implying that the postoperative imaging had certainly demonstrated residual tumour in these cases. Secondly, we made a careful assessment of the residual “tissue” using preoperative imaging for comparison. We believe that we were dealing with postoperative MRI-visible residual tumour and not postoperative enhancement when there was focal nodular or mass-like enhancement at the site of the original tumour. Surgically induced postoperative enhancement is usually thin and linear, typically occurring 3–5 days after surgery [16] and may occur earlier [17] or even intraoperatively [18] (as we have also observed under the same general anaesthetic) due to disruption of the blood–brain barrier and later to neovascularization. Nodular enhancement, however, has been shown to mimic tumour and also typically develops 3–5 days after surgery [19] (hence the rationale for completing all immediate postoperative imaging within 48–72 h as recommended by the UKCCSG) [20], although this may also be seen as a transient intraoperative phenomenon particularly in the presence of repeated electrocoagulation [18]. We used the postoperative scan at 3 months to determine the extent of residual tumour because by then any surgically induced nodular enhancement can be expected to have resolved [21], and we made careful comparison with the preoperative scan in order to confirm there was tumour previously at that site. Thirdly, our incomplete resection rate of 15.5% of childhood CLGA is at the low end of the published data of incomplete resection rates which range from 11.3% to 35% [2, 3, 6], which suggests that we were not over-diagnosing postoperative residual tumour. In our larger cohort of 84 patients there was a 96% concordance (95% confidence interval 89–99%) of the reporting of the presence or absence of postoperative tumour when compared with the surgeon’s operative report [12].
We did not find statistically significant independent variables (symptomatology, age, gender, histological grade or the Ki-67 fraction) as predictors of spontaneous regression. Although for the small numbers involved valid statistical comparisons could not be made, tumour progression was observed more commonly for those with larger volumes of residual disease and did not occur in children with residual tumour volumes below 10 cm3. It has been suggested that the Ki-67 monoclonal antibody fraction, a labelling index for neuronal proliferation, might predict the biological behaviour of postoperative residual tumours with a higher index correlating with tumours that later progressed. Studies of adult patients with all grades of astrocytic tumour found Ki-67 to be of some prognostic significance independent of tumour grade [22–25], while one large study of 96 patients with supratentorial fibrillary astrocytomas did not find Ki-67 helpful in predicting survival or response to radiotherapy [26]. In children with low-grade optic pathway gliomas, the proliferative labelling index did not predict progression-free survival and overall survival [27] while in one study of 39 pilocytic astrocytomas there was a non-significant trend that a negative proliferative index was associated with fewer progressions of residual tumour [28]. As expected, the majority of the residual tumours in our study were pilocytic astrocytomas and differences in Ki-67 did not determine subsequent tumour behaviour. The numbers of children with fibrillary astrocytomas in our cohort were too small to analyse.
Spontaneous involution of other types of low-grade astrocytoma is well-recognized in children with NF1, both for optic pathway gliomas and at other intracerebral sites [29–33]. There are descriptions of individual cases of low-grade astrocytoma regression in children and young adults without NF1 [30, 33, 34]. For optic pathway gliomas this phenomenon was first described by Hudson in 1912, an observation which later generated discussion as to whether they represented tumours or were in fact hamartomatous lesions. Neither the mechanisms of spontaneous tumour regression nor of spontaneous involution of residual tumour following surgery are known. Although programmed cell death or apoptosis may be the common denominator, possible mechanisms include immunological, genetic and biological factors. However, in some settings and for some tumours, for example neuroblastomas and squamous cell carcinomas, apoptosis is not a prerequisite.
Terminal differentiation, in association with inflammation or apoptosis, whereby terminally differentiated cells are no longer capable of cell division and are therefore mortal, is another potential mechanism for spontaneous regression. Immunological mechanisms including cell-mediated and humoral responses to tumour-associated antigens can trigger apoptosis resulting in spontaneous regression. The observation that autoimmune manifestations may occur concurrently with spontaneous tumour regression provides additional evidence of an immune-mediated phenomenon. Failure of angiogenesis resulting in vascular compromise, and genomic instability, for example telomerase inhibition, are other possible causes and all offer potential targets for treatment [35, 36]. The possible effects of surgery on the residual low-grade astrocytoma are also speculative and explanations ranging from impaired vascular supply to reduced concentrations of autocrine growth factors, ultimately reducing the proliferative potential of residual tumour have been proposed [6, 13].
Treatment and imaging recommendations for children with residual tumour following their initial surgery
Spontaneous tumour regression includes cases in which the disease is not necessarily cured but remains dormant and may recur, as well as those in which regression is complete (Table 3). Although none of the residual tumours that spontaneously regressed in our cohort had subsequently recurred at the time of this report, it remains uncertain whether the tumour had entirely disappeared and as the long-term outcome of these patients is not yet known it would be prudent to keep them under surveillance.
A “watch and wait” policy (surveillance imaging) which would detect those that either regress spontaneously or progress is in our experience to be recommended for those children with residual disease who do not have symptoms due to that residual disease (for whom further surgery with or without adjuvant therapy should be considered). Spontaneous regression was observed at the earliest at 7 months and at the latest at 4 years 2 months, while progression of residual tumour occurred within 2 years of initial surgery in all five children and was detected at the earliest at 7 months. In addition to the early postoperative scan performed at 24–48 h after surgery, we recommend surveillance imaging at 6-monthly intervals for the first year. Six-monthly intervals for the subsequent 2 years would detect those with late tumour progression and yearly for a further 2 years would reliably detect those children in whom late spontaneous regression occurs. We believe that if residual tumour is still seen on an MRI performed at 5 years following initial surgery then continuing surveillance imaging at 2-yearly intervals after that would be prudent, bearing in mind the paucity of information on the long-term prognosis of this group of patients. This regimen is in contrast to our recommendation for scanning at 6 months then 1, 2, 3.5 and 5 years for children following complete excision [12].
In conclusion, residual tumour after initial surgical resection for which a “watch and wait” policy (surveillance imaging) is followed may either regress or progress. We recommend surveillance imaging at 6-monthly intervals for the first 2 years for all children with known residual disease. Scanning should then be continued at 3, 4 and 5 years after initial surgery, then every second year if residual tumour is still present at 5 years after surgery. This regimen would detect not only those children with progressive or recurrent disease, but also those with spontaneous regression, a phenomenon which can occur later than disease progression.
References
Cushing H (1931) Experiences with the cerebellar astrocytoma. A critical review of seventy-six cases. Surg Gynecol Obstet 52:129–204
Due-Tonnessen BJ, Helseth E, Scheie D, et al (2002) Long term outcome after resection of benign cerebellar astrocytomas in children and young adults (0–19 years): report of 110 consecutive cases. Pediatr Neurosurg 37:71–80
Pencalet P, Maixner W, Sainte-Rose C, et al (1999) Benign cerebellar astrocytomas in children. J Neurosurg 90:265–273
Palma L, Russo A, Celli P (1984) Prognosis of the so-called “diffuse” cerebellar astrocytomas. Neurosurgery 15:315–317
Desai KI, Nadkami TD, Muzumdar DP, et al (2001) Prognostic factors for cerebellar astrocytomas in children: a study of 102 cases. Pediatr Neurosurg 35:311–317
Smoots D, Russell Geyer J, Lieberman DM, et al (1998) Predicting disease progression in childhood cerebellar astrocytoma. Childs Nerv Syst 14:636–648
Sgouros S, Fineron PW, Hockley AD (1995) Cerebellar astrocytoma of childhood; long term follow up. Childs Nerv Syst 11:89–96
Saunders DE, Hayward RD, Phipps KP, et al (2003) Surveillance neuroimaging of intracranial medulloblastoma in children: how effective, how often, and for how long? J Neurosurg 99:280–286
Good CD, Wade AM, Hayward RD, et al (2001) Surveillance neuroimaging of childhood intracranial ependymoma: how effective, how often, and for how long? J Neurosurg 94:27–32
Dirven CM, Mooj JJ, Molenaar WM (1997) Cerebellar pilocytic astrocytomas: a treatment protocol based upon analysis of 73 cases and a review of the literature. Childs Nerv Syst 13:17–23
Fernandez C, Figarwalla-Branger D, Girard N, et al (2003) Pilocytic astrocytomas in children: prognostic factors—a retrospective study of 80 cases. Neurosurgery 53:544–553
Saunders DE, Phipps KP, Wade AM, et al (2005) Surveillance imaging strategies following surgery and/or radiotherapy for childhood cerebellar low-grade astrocytomas. J Neurosurg 102:172–178
Schneider JH, Raffel C, McComb JG (1992) Benign cerebellar astrocytomas of childhood. Neurosurgery 30:58–62
Davis CH, Jogelkar VM (1981) Cerebellar astrocytomas in children and young adults. J Neurol Neurosurg Psychiatry 44:820–828
Kleihues P, Cavenee WK (2000) Astrocytic tumours. World Health Organization classification of tumours. Pathology and genetics of tumours of the nervous system. IARC Press, Lyon
Cairncross JG, Pexman W, Rathbone MP (1985) Post surgical contrast enhancement mimicking residual brain tumour. Can J Neurol Sci 12:75
Nicoletti GF, Barone F, Passanassi M, et al (1994) Linear contrast enhancement at the operative site on early post operative CT after removal of brain tumors. J Neurosurg Sci 38:131–135
Knauth M, Aras N, Wirtz CR, et al (1999) Surgically induced intracranial contrast enhancement: potential source of diagnostic error in intra-operative MR imaging. AJNR 20:1547–1553
Rollins NK, Nisen P, Shapiro KN (1998) The use of early post operative MR in detecting residual juvenile cerebellar pilocytic astrocytoma. AJNR 19:151–156
Griffiths PD (1999) A protocol for imaging paediatric brain tumours. United Kingdom Children’s Cancer Study Group (UKCCSG) and Societe Francaise D’Oncologie Pediatrique (SFOP) panelists. Clin Oncol 11:290–294
Forsting M, Albert FK, Kunze S, et al (1993) Extirpation of glioblastomas: MR and CT follow-up of residual tumor and regrowth patterns. AJNR 14:77–87
McKeever PE, Strawderman MS, Yamini B, et al (1998) MIB-1 proliferation index predicts survival among patients with grade II astrocytoma. J Neuropathol Exp Neurol 57:931–936
Shinoda J, Sakai N, Nakatani K, et al (1998) Prognostic factors in supratentorial WHO grade II astrocytomas in adults. Br J Neurosurg 12:318–324
Wakimoto H, Aoyagi M, Nakayama T, et al (1996) Prognostic significance of Ki-67 labelling indices obtained using MIB-1 monoclonal antibody in patients with supratentorial astrocytomas. Cancer 77:373–380
Wessels PH, Hopman AH, Kubat B, et al (2003) Proliferation and aneusomy predict survival of young patients with astrocytoma grade II. Br J Cancer 89:128–134
Hilton DA, Love S, Barber R, et al (1998) Accumulation of p53 and Ki-67 expression do not predict survival in patients with fibrillary astrocytomas or the response of these tumors to radiotherapy. Neurosurgery 42:724–729
Czech T, Slavc I, Aicholzer M, et al (1999) Proliferative activity as measured by MIB-1 labeling index and long-term outcome of visual pathway astrocytomas in children. J Neurooncol 42:143–150
Dirven CM, Koudstaal J, Mooj JJ, et al (1998) The proliferative potential of the pilocytic astrocytoma: the relation between MIB-1 labelling and clinical and neuroradiological follow-up. J Neurooncol 37:9–16
Gottschalk S, Tavakolian R, Buske A, et al (1999) Spontaneous remission of chiasmatic/hypothalamic masses in neurofibromatosis type 1: report of two cases. Neuroradiology 41:199–201
Parsa CF, Hoyt CS, Lesser RL, et al (2001) Spontaneous regression of optic gliomas: thirteen cases documented by serial neuroimaging. Arch Ophthalmol 119:516–529
Schmandt SM, Packer RJ, Vezina LG, et al (2000) Spontaneous regression of low-grade astrocytomas in childhood. Pediatr Neurosurg 32:132–136
Venes JL, Latack J, Kandt RS (1984) Postoperative regression of opticochiasmatic astrocytoma: a case for expectant therapy. Neurosurgery 15:421–423
Borit A, Richardson EP Jr (1982) The biological and clinical behaviour of pilocytic astrocytomas of the optic pathways. Brain 105(Pt 1):161–187
Hoffmann HJ (1994) Management and outcome of low-grade astrocytomas of the midline in children: a retrospective review. Neurosurgery 35:343–344
Folkman J (1995) Seminars in medicine of the Beth Israel Hospital, Boston: clinical applications of research on angiogenesis. N Engl J Med 333:1757–1763
Elston DM (2004) Mechanisms of regression. Clin Med Res 2:85–88
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gunny, R.S., Hayward, R.D., Phipps, K.P. et al. Spontaneous regression of residual low-grade cerebellar pilocytic astrocytomas in children. Pediatr Radiol 35, 1086–1091 (2005). https://doi.org/10.1007/s00247-005-1546-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00247-005-1546-z