
Abstract
Purpose
Pilocytic astrocytoma (PA) is a World Health Organization grade I neoplasm that generally follows a benign course. However, in some patients, PA exhibits an aggressive clinical course. Here, we examined the clinical course of pediatric and adult PAs with progression at a single institution.
Methods
Between 1995 and 2013, 39 patients with PA were treated. Nineteen were pediatric patients (mean age, 12 years; range, 1–17 years) with a male-to-female patient ratio of 10:9, while 20 were adults (mean age, 36.4 years; range, 19–65 years) with a male-to-female ratio of 9:11. We analyzed and compared tumor location, extent of tumor resection, adjuvant treatment, and clinical course in all patients.
Results
In the 19 pediatric patients, tumors were located in the cerebellar vermis, cerebellar hemisphere, optic pathways plus hypothalamus, hypothalamus, brainstem, and the temporal lobe in 6 (31.6 %), 5 (26.3 %), 3 (15.8 %), 2 (10.5 %), and 2 (10.5 %) patients and 1 (5.3 %) patient, respectively. The mass was totally, subtotally, or partially resected in 11 (57.9 %), 2 (10.5 %), and 4 (21.1 %) patients, respectively; biopsies were performed in 2 (10.5 %) patients. Immediate postoperative adjuvant treatment was carried out in 6 patients. Tumor progression was detected in 3 patients at 3.0, 4.6, and 5.2 years after treatment, respectively, without significant symptoms. In the 20 adult patients, tumors were located in the cerebellar hemisphere, cerebellar vermis, hypothalamus, brainstem, cerebral hemisphere, and lateral ventricle in 5 (25 %), 4 (20 %), 3 (15 %), 3 (15 %), 3 (15 %), and 2 (10 %) patients, respectively. The mass was totally, subtotally, or partially resected in 11 (55 %) and 6 (30 %) patients and 1 (5 %) patient, respectively; biopsies were performed in 2 patients. Immediate adjuvant treatment was carried out in 2 patients. Progression was detected in 3 patients at 0.3, 0.9, and 2.5 years after treatment, respectively, with progressive neurologic symptoms. There was one case of disease-related mortality during follow-up among the adult patients.
Conclusion
Most of the PA cases evaluated in this study were benign. However, tumor progression in adult PAs followed a more aggressive clinical course than those in pediatric PAs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pilocytic astrocytoma (PA) is a World Health Organization grade I neoplasm with an expected benign course and a 10-year survival rate of >95 % [1, 2]. PAs are more common in pediatric patients than in adult patients and account for up to 25 % of all pediatric brain tumors. Tumors are typically diagnosed between 5 and 14 years of age and are primarily located in the deep cerebral midline structures, brainstem, and cerebellum. PAs are generally slow-growing, well-circumscribed tumors that are curable by complete surgical resection and are typically thought to behave in an indolent fashion [2].
The majority of available data have been derived from pediatric patients, with few studies focusing on adult PAs [3–7]. There is some controversy regarding the clinical behavior of adult PAs. Whereas some studies have indicated that adult PAs follow an indolent benign course after initial surgery, others have reported more aggressive clinical behavior in adult PAs compared to pediatric PAs [3–9] and have suggested that adult PAs have the potential for rapid tumor recurrence, progression, and malignant transformation.
We recently observed three adult patients with PA who showed relatively rapid progression and clinical deterioration at our institution. Therefore, we performed this retrospective analysis of all pediatric and adult patients with PA who underwent surgery for their disease at our institution over a 15-year period. We primarily focused on the clinical course of pediatric and adult PAs with progression.
Materials and methods
We retrospectively analyzed 39 patients with PA who were treated between 1995 and 2013. PA was histologically diagnosed as grade I according to the World Health Organization classification [1]. Out of all patients, one adult was associated with type 1 neurofibromatosis. The institutional review board approved this study (CNUHH-2015-088). Nineteen of the 39 (48.7 %) patients were pediatric patients, with a mean age of 12 years (range, 1–17 years). The male-to-female patient ratio was 10:9 (with male and female patients representing 52.6 and 47.4 % of the pediatric patients, respectively). The mean follow-up duration was 96.7 months (range, 8–213 months); tumor progression occurred in 3 (15.8 %) of the 19 patients. Twenty (51.3 %) of the 39 patients were adults, with a mean age of 36.4 years (range, 19–65 years). The male-to-female adult patient ratio was 9:11 (with male and female patients representing 45.0 and 55 % of the adult patients, respectively). The mean follow-up duration was 69.4 months (range, 8–197 months); there were 3 (15 %) patients with tumor progression.
The collected data, which included pre-operative neurologic symptoms, tumor location, extent of tumor resection, and adjuvant treatment, were obtained from individual medical records and imaging studies during follow-up. We focused on the clinical courses of cases of PA with progression in pediatric and adult patients.
As an evaluation of initial treatment, we analyzed the extent of tumor resection and immediate adjuvant treatment. Gross total tumor resection, subtotal tumor resection, and partial tumor resection were defined as 100 %, less than 100 % but equal to or greater than 50 %, and less than 50 % macroscopic removal of the tumor mass, respectively. Determination of the extent of tumor resection was based primarily on the judgment of the neurosurgeons and was validated by review of magnetic resonance imaging (MRI) scans within 3 months after treatment. The radiologic findings were subsequently assessed every 6 months or yearly by performing gadolinium-enhanced MRI. After operation and/or postoperative adjuvant treatment, the development of new lesions was regarded as recurrence and the increase in size over that of the original residual volume as progression.
Results
Clinical characteristics of pediatric and adult PAs
The clinical characteristics are summarized in Table 1.
Among the 19 pediatric patients, headache was the most common symptom, reported in 7 (36.8 %) patients, followed by nausea/vomiting and gait disturbance, reported in 3 (15.8 %) patients each; visual field defects were observed in 2 (10.5 %) patients, and strabismus, dizziness, seizure, and posterior neck pain were observed in 1 (5.3 %) patient each. Tumors were located in the cerebellar vermis, cerebellar hemisphere, and optic pathways plus hypothalamus in 6 (31.6 %), 5 (26.3 %), and 3 (15.8 %) patients, respectively; in the hypothalamus or brainstem in 2 (10.5 %) patients each; and the temporal lobe in 1 (5.3 %) patient. The mass was totally, subtotally, or partially resected in 11 (57.9 %), 2 (10.5 %), and 4 (21.1 %) patients, respectively; biopsies were performed in 2 (10.5 %) patients. Immediate postoperative adjuvant treatment was administered to 6 (31.6 %) patients. Of 3 patients with optic pathways plus hypothalamic PA, 1 patient each was treated with chemotherapy with vinblastine, vincristine, 5-fluorouracil (5-FU), cyclophosphamide, and VP-16; chemotherapy with vincristine and carboplatin; and conventional radiotherapy with 45 Gy. Of 2 patients with hypothalamic PA, 1 patient was treated with gamma knife radiosurgery (10 Gy, 40 % isodose) and the other was treated with chemotherapy (vincristine, etoposide, 5-FU, and cyclophosphamide). One patient with brainstem PA received radiotherapy with 57.6 Gy. During the follow-up period, progression was detected in 3 patients. Out of them, 2 patients with optic pathways plus hypothalamic PAs showed progression after 3 and 4.6 years, respectively, but were stable symptomatically. One patient underwent radiologic follow-up without treatment, and 1 patient received radiotherapy with 50.4 Gy for progressive tumor. One patient with brainstem PA showed progression after 5.4 years and was treated with chemotherapy for enlargement of the mass in the absence of symptoms. There was no disease-related mortality in the pediatric patients during follow-up.
Among the 20 adult patients, headache was the most common symptom, reported in 7 (35.0 %) patients, followed by dizziness and gait disturbance, reported in 4 (20.0 %) and 2 (10.0 %) patients, respectively. Decreased mental status and motor weakness were reported in 2 (10.0 %) patients each, and nausea/vomiting, visual field defects, and hemibody sensory changes were reported in 1 (5.0 %) patient each. Tumors were located in the cerebellar hemisphere, cerebellar vermis, hypothalamus, brainstem, cerebral hemisphere, and the lateral ventricle in 5 (25 %), 4 (20 %), 3 (15 %), 3 (15 %), 3 (15 %), and 2 (10 %) patients, respectively. Tumors in cerebral hemispheres were located in the frontal, parietal, or temporal lobes in 1 patient each. The mass was totally, subtotally, or partially resected in 11 (55 %) and 6 (30 %) patients and 1 (5.0 %) patient, respectively; biopsies were performed in 2 (10 %) patients. Immediate postoperative adjuvant treatment was administered to 2 (10.0 %) patients with hypothalamic PA, conventional radiotherapy with 55.8 Gy was administered to 1 patient, and chemotherapy with vincristine and carboplatin was administered to 1 patient. Progression was detected in 3 patients with progressive symptoms: in 1 patient with a hypothalamic tumor at 0.3 years, in 1 patient with a cerebellar and left cerebellar peduncle tumor at 0.9 years, and in 1 patient with a brainstem tumor at 2.5 years following treatment. There was one instance of disease-related mortality among the patients with adult hypothalamic PA.
Three cases of pediatric PA with progression
Case I
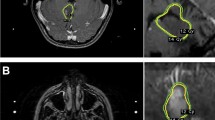
A 12-year-old boy complained of strabismus for 1 year. Brain MRI showed a heterogeneously enhancing mass within the optic pathways plus hypothalamic area, and PA was pathologically diagnosed via craniotomy and biopsy (Fig. 1a). Pathologically, there was sparse cellularity and a large number of Rosenthal fibers in a myxoid background (Fig. 3a). Chemotherapy with vincristine and carboplatin was administered for 19 months, resulting in a partial response (Fig. 1b). However, 4.6 years later, he complained of a mild headache and dizziness, and the follow-up brain MRI showed an enlarged cystic mass (Fig. 1c). Radiologic follow-up was performed without any treatment, and the size of the cystic mass gradually decreased over 2 years (Fig. 1d).

Three cases of pediatric PA with progression. Case I, optic pathways plus hypothalamic PA: Brain MRI showed a heterogeneously enhancing mass within the optic pathways plus hypothalamic area on T1 post-contrast sequences (a). There was a partial response after chemotherapy (b). After 4.6 years, follow-up brain MRI showed an enlarged cystic mass (c). The cystic mass gradually decreased in size over 2 years without treatment (d). Case II, optic pathways plus hypothalamic PA: Brain MRI showed a heterogeneously enhancing mass within the optic pathways plus hypothalamic area on T1 post-contrast sequences (e). Three years later, follow-up brain MRI showed a nodular enhanced lesion after chemotherapy (f). At 1.1 years after radiotherapy, an enlarged cystic lesion without visual deterioration was observed (g). The cystic lesion slowly enlarged over 1.3 years without changes in visual symptoms (h). Case III, brainstem PA: Brain MRI showed a mass diffusely involving the medulla on T1 post-contrast sequences (i). There was a partial response after radiotherapy (j). After 5.2 years, follow-up brain MRI showed a slight enlargement of the enhanced lesion; the patient did not have any symptoms (k). After chemotherapy, radiologic follow-up showed a decrease in the size of the mass; the patient was symptom-free for 3 years after chemotherapy (l)
Case II
A 7-year-old boy complained of decreased visual acuity for several years. Brain MRI showed a heterogeneously enhancing mass within the optic pathways plus hypothalamic area (Fig. 1e). The tumor was partially resected, and PA was pathologically diagnosed. Chemotherapy with vinblastine, vincristine, 5-FU, cyclophosphamide, and VP-16 was administered for 27 months, leading to a partial response. After 3 years, follow-up brain MRI showed a nodular enhancing lesion (Fig. 1f). Radiotherapy with 50.4 Gy was performed for this lesion. During radiologic follow-up 1.1 years after radiotherapy, an enlarged cystic lesion without visual deterioration was observed (Fig. 1g). The cystic lesion slowly enlarged over 1.3 years without changes in visual symptoms (Fig. 1h). Microscopic cyst fenestration was carried out for this lesion.
Case III
A 12-year-old boy complained of posterior neck pain for 6 months. Brain MRI showed a mass diffusely involving the medulla (Fig. 1i). PA was pathologically diagnosed via biopsy. Conventional radiotherapy with 57.6 Gy was performed, leading to a partial response (Fig. 1j). However, 5.2 years later, follow-up brain MRI showed a slight enlargement of the enhancing lesion without any symptoms (Fig. 1k). Chemotherapy with vincristine, VP-16, 5-FU, and cyclophosphamide was administered. Radiologic follow-up revealed a decrease in the mass size, and the patient was symptom-free for 3 years after chemotherapy (Fig. 1l).
Three cases of adult PA with progression
Case I
A 25-year-old man with type I neurofibromatosis complained of headache and dizziness for 2 months. Brain MRI showed a hypothalamic lesion associated with heterogeneous enhancement of the ventricular wall (Fig. 2a). An endoscopic biopsy was performed. Pathologically, there was a sparsely cellular tumor whose cells had piloid cytoplasm (Fig. 3b). Chemotherapy with vincristine and carboplatin was administered for 4 months. Follow-up brain MRI showed enlargement of the mass associated with ventricular enhancement, and neurologic symptoms were aggravated with diplopia and gait disturbance (Fig. 2b). A repeat biopsy was performed, yielding the same pathologic diagnosis of PA. Conventional radiotherapy with 45 Gy was then performed. Four months after the radiotherapy, follow-up MRI showed further progression of the lesion in the hypothalamus and spread to all four ventricles (Fig. 2c). Chemotherapy with procarbazine, CCNU, and vincristine was administered for 1 year and resulted in a partial response. However, the mass continued to progress and the patient died due to progression of the brain lesion at 3.4 years after the initial diagnosis (Fig. 2d).

Three cases of adult PA with progression. Case I, hypothalamic PA: Brain MRI showed a hypothalamic lesion associated with heterogeneous enhancement of the ventricular wall on T1 post-contrast sequences (a). Follow-up brain MRI showed enlargement of the mass, with ventricular enhancement, and neurologic symptoms were aggravated after chemotherapy for 4 months (b). Four months after radiotherapy, follow-up MRI showed a further progressive lesion in the hypothalamus which spread to all four ventricles (c). The mass progressed for 1 year, even after chemotherapy (d). Case II, brainstem PA: Brain MRI showed a mass diffusely involving the medulla and the fourth ventricle with heterogeneous enhancement on T1 post-contrast sequences (e). The tumor was subtotally resected (f). After 2.5 years, follow-up MRI showed an enlarged mass; clinically, the patient had developed a gait disturbance (g). Case III, cerebellar PA: Brain MRI showed a heterogeneously enhancing mass in the cerebellum and left middle cerebellar peduncle on T1 post-contrast sequences (h). One year later, after partial resection, brain MRI showed tumor enlargement (i). Radiologic follow-up revealed a mass enlarging with progressive symptoms over 3 years without treatment (j). The mass continued to progress 0.5 years after radiotherapy (k)

Pathologic findings of PAs. a Pediatric case I showed the sparse cellularity and a large number of Rosenthal fibers in the myxoid background (hematoxylin and eosin staining, original magnification ×200). b Adult case I showed the sparsely cellular tumor whose cells had piloid cytoplasm (hematoxylin and eosin staining, original magnification ×200). c Adult case III showed the sparse cellularity and contained Rosenthal fibers (hematoxylin and eosin staining, original magnification ×200)
Case II
A 65-year-old man complained of headache for 3 months. Brain MRI showed a mass diffusely involving the medulla and the fourth ventricle with heterogeneous enhancement (Fig. 2e). The tumor was subtotally resected (Fig. 2f). After 2.5 years, the patient experienced gait disturbance. A follow-up MRI showed an enlarged mass (Fig. 2g), and surgery was repeated. The patient died after surgery due to pulmonary edema and sepsis.
Case III
A 65-year-old woman complained of headache and gait disturbance for 3 months. Brain MRI showed a heterogeneously enhancing mass in the cerebellum and left middle cerebellar peduncle (Fig. 2h). The mass was partially resected. Pathologically, the tumor was sparsely cellular and contained Rosenthal fibers (Fig. 3c). One year later, the patient complained of dizziness and body imbalance, and brain MRI showed tumor enlargement (Fig. 2i). She refused treatment, and radiologic follow-up showed that the mass continued to enlarge with progressive symptoms over 3 years (Fig. 2j). Conventional radiotherapy with 54 Gy was then performed. However, after 0.5 years, the mass continued to progress (Fig. 2k).
Discussion
The characteristics of pediatric and adult PAs differ. Most pediatric PAs occur in the cerebellum and present as well-circumscribed lesions with indolent behavior [2, 11]. Radiation and chemotherapy have been used to successfully control even progressive and recurrent pediatric PAs [10]. In contrast, adult PAs are rare and, thus, little is known regarding their clinical behavior. In adult patients, supratentorial lobar tumors are more frequent and are commonly located in the temporal or parietal regions [2, 3, 7]. Reported differences in the clinical behaviors of pediatric and adult PAs are inconsistent. Some reports have suggested that adult PAs follow a relatively benign course, similar to pediatric PAs [3, 8, 9]. Adult patients with supratentorial PAs are reported to have a favorable prognosis, with regard to survival and neurologic function, and it has been suggested that radiotherapy is not required for these patients following gross or subtotal resection [3]. However, adult PAs are often reported to not be benign. Recurrence rates are high, and tumor-related deaths are frequent [4, 7]. Adult PAs have been reported to show a recurrence rate of 30 % [4], with relatively rapid recurrence and malignant transformation noted in these patients. Another study reported recurrence and malignant transformation rates of 30 and 50 %, respectively, in patients who required secondary resection [7]. Therefore, studies support that some PAs do not follow an indolent clinical course in adult patients. In this study, adult patients had the same frequency of progression as the pediatric patients, but those tumors which progressed did so more rapidly and aggressively than the examples in children. Disease-related death was observed only in the adult patient group during follow-up.
Several immunohistochemical and molecular genetic markers have been studied as potential prognostic indicators in PA, and the expression of MIB-1, myelin basic protein, and matrilin-2 protein correlate with aggressive behavior [12–14]. In our 3 cases of adult PAs with progression, the initial Ki-67 labeling index was less than 0.5 %, a level comparable to that of pediatric PAs with progression.
Complete surgical resection is likely curative for the majority of patients with PA and is therefore the primary objective of the neurosurgeon whenever possible. Patients who undergo biopsy or subtotal tumor resection experience recurrence or progression, making the extent of resection a major prognostic factor in the clinical course of the disease. Therefore, an aggressive surgical resection should be attempted whenever possible. A 75 % progression-free survival rate at 5 years and overall survival rates of 95.6 and 82 % at 10 and 20 years, respectively, have been reported after gross total resection [2]. The clinical course after subtotal resection can be less predictable, but most patients show long-term stable disease. In adult PA, gross total resection has also been shown to be associated with improved survival [6, 7]. However, some reports have suggested that the extent of tumor resection was not related to improved survival [3, 5]. Given the relatively frequent recurrence and high progression rates, initial adjuvant radiation may be recommended as postsurgical treatment for adult patients with PA. One study showed that although adjuvant radiotherapy for adult patients with PA significantly prolonged progression-free survival, compared with observation alone, it did not affect overall survival [5]. On the contrary, radiation therapy may also predispose the patient to the malignant transformation of PA. In one review study, juvenile PA did not recur spontaneously, but malignant transformation only emerged after treatment with radiation [15]. Additionally, another study showed that malignant transformation was limited to adult patients treated with radiation [4]. Therefore, we should consider both possibilities of rapid recurrence or progression, and malignant transformation after radiotherapy. A comprehensive postoperative follow-up is essential for adult patients with PA, and adjuvant radiotherapy should be recommended cautiously after recurrence or progression.
Conclusion
Most PAs are benign. However, comprehensive postoperative follow-up is essential for adult patients with PA, as some exhibit aggressive clinical behavior.
References
Scheithauer BW, Hawkins C, Tihan T, VandenBerg SR, Burger PC (2007) Pilocytic astrocyoma. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) WHO classification of tumours of the central nervous system. IARC, Lyon, pp. 14–21
Burkhard C, Di Patre PL, Schuler D, Schuler G, Yasargil MG, Yonekawa Y, Lutolf UM, Kleihues P, Ohgaki H (2003) A population-based study of the incidence and survival rates in patients with pilocytic astrocytoma. J Neurosurg 98:1170–1174
Brown PD, Buckner JC, O'Fallon JR, Iturria NL, Brown CA, O'Neill BP, Scheithauer BW, Dinapoli RP, Arusell RM, Abrams RA, Curran WJ, Shaw EG, North Central Cancer Treatment Group, Mayo Clinic (2004) Adult patients with supratentorial pilocytic astrocytomas: a prospective multicenter clinical trial. Int J Radiat Oncol Biol Phys 58: 1153–1160
Ellis JA, Waziri A, Balmaceda C, Canoll P, Bruce JN, Sisti MB (2009) Rapid recurrence and malignant transformation of pilocytic astrocytoma in adult patients. J Neuro-Oncol 95:377–382
Ishkanian A, Laperriere NJ, Xu W, Millar BA, Payne D, Mason W, Sahgal A (2011) Upfront observation versus radiation for adult pilocytic astrocytoma. Cancer 117:4070–4079
Johnson DR, Brown PD, Galanis E, Hammack JE (2012) Pilocytic astrocytoma survival in adults: analysis of the Surveillance, Epidemiology, and End Results Program of the National Cancer Institute. J Neuro-Oncol 108:187–193
Stuer C, Vilz B, Majores M, Becker A, Schramm J, Simon M (2007) Frequent recurrence and progression in pilocytic astrocytoma in adults. Cancer 110:2799–2808
Afra D, Muller W, Slowik F, Firsching R (1986) Supratentorial lobar pilocytic astrocytomas: report of 45 operated cases, including 9 recurrences. Acta Neurochir 81:90–93
Bell D, Chitnavis BP, Al-Sarraj S, Connor S, Sharr MM, Gullan RW (2004) Pilocytic astrocytoma of the adult—clinical features, radiological features and management. Br J Neurosurg 18:613–616
Gnekow AK, Kortmann RD, Pietsch T, Emser A (2004) Low grade chiasmatic-hypothalamic glioma-carboplatin and vincristin chemotherapy effectively defers radiotherapy within a comprehensive treatment strategy—report from the multicenter treatment study for children and adolescents with a low grade glioma—HIT-LGG 1996—of the Society of Pediatric Oncology and Hematology (GPOH). Klin Padiatr 216:331–342
Dirven CM, Mooij JJ, Molenaar WM (1997) Cerebellar pilocytic astrocytoma: a treatment protocol based upon analysis of 73 cases and a review of the literature. Childs Nerv Syst 13:17–23
Bowers DC, Gargan L, Kapur P, Reisch JS, Mulne AF, Shapiro KN, Elterman RD, Winick NJ, Margraf LR (2003) Study of the MIB-1 labeling index as a predictor of tumor progression in pilocytic astrocytomas in children and adolescents. J Clin Oncol 21:2968–2973
Wong KK, Chang YM, Tsang YT, Perlaky L, Su J, Adesina A, Armstrong DL, Bhattacharjee M, Dauser R, Blaney SM, Chintagumpala M, Lau CC (2005) Expression analysis of juvenile pilocytic astrocytomas by oligonucleotide microarray reveals two potential subgroups. Cancer Res 65:76–84
Sharma MK, Watson MA, Lyman M, Perry A, Aldape KD, Deak F, Gutmann DH (2006) Matrilin-2 expression distinguishes clinically relevant subsets of pilocytic astrocytoma. Neurology 66:127–130
Parsa CF, Givrad S (2008) Juvenile pilocytic astrocytomas do not undergo spontaneous malignant transformation: grounds for designation as hamartomas. Br J Ophthalmol 92:40–46
Acknowledgments
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT and Future Planning (2014R1A1A1004469).
Conflict of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ryu, HH., Jung, TY., Lee, GJ. et al. Differences in the clinical courses of pediatric and adult pilocytic astrocytomas with progression: a single-institution study. Childs Nerv Syst 31, 2063–2069 (2015). https://doi.org/10.1007/s00381-015-2887-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-015-2887-z