
Abstract
5S rRNA is the smallest nucleic acid component of the large ribosomal subunit, contributing to ribosomal assembly, stability, and function. Despite being a model for the study of RNA structure and RNA–protein interactions, the evolution of this universally conserved molecule remains unclear. Here, we explore the history of the three-domain structure of 5S rRNA using phylogenetic trees that are reconstructed directly from molecular structure. A total of 46 structural characters describing the geometry of 666 5S rRNAs were used to derive intrinsically rooted trees of molecules and molecular substructures. Trees of molecules revealed the tripartite nature of life. In these trees, superkingdom Archaea formed a paraphyletic basal group, while Bacteria and Eukarya were monophyletic and derived. Trees of molecular substructures supported an origin of the molecule in a segment that is homologous to helix I (α domain), its initial enhancement with helix III (β domain), and the early formation of the three-domain structure typical of modern 5S rRNA in Archaea. The delayed formation of the branched structure in Bacteria and Eukarya lends further support to the archaeal rooting of the tree of life. Remarkably, the evolution of molecular interactions between 5S rRNA and associated ribosomal proteins inferred from a census of domain structure in hundreds of genomes established a tight relationship between the age of 5S rRNA helices and the age of ribosomal proteins. Results suggest 5S rRNA originated relatively quickly but quite late in evolution, at a time when primordial metabolic enzymes and translation machinery were already in place. The molecule therefore represents a late evolutionary addition to the ribosomal ensemble that occurred prior to the early diversification of Archaea.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
5S ribosomal RNA (rRNA) is an integral component of the large subunit of the ribosome. It harbors fundamentally important functions during protein synthesis. Results of cross-linking studies suggest that 5S rRNA may serve as a signal transducer between the peptidyl transferase center and domain II of the large rRNA subunit that is responsible for translocation (Bogdanov et al. 1995; Dokudovskaya et al. 1996), and between regions of 23S rRNA responsible for principal ribosomal functions (Kouvela et al. 2007). 5S rRNA may also be a determinant of stability for the large subunit (Holmberg and Nygard 2000). Evolutionarily, SINE3, a class of short interspersed elements (SINEs), are derived from 5S rRNA (Kapitonov and Jurka 2003). However, detailed functions of 5S rRNA are still lacking (Bogdanov et al. 1995; Barciszewska et al. 2000, 2001; Szymanski et al. 2003) and the origins and evolutionary history of the molecule have not been explored.
5S rRNA is the smallest RNA component of the ribosome (~120-nucleotides long) and associates not only with the large rRNA subunit but also with several ribosomal proteins. Studies of the 5S rRNA molecule began in the 1980s, when Fanning and Traut (1981) attempted to purify cross-linked 5S-protein complexes. 5S rRNA interacts in the ribosome with various ribosomal proteins to form a stable complex, three (L15, L18, and L25) in Bacteria (Christiansen and Garrett 1986), one or two in Archaea (Smith et al. 1978; McDougall and Wittmann-Liebold 1994), and one in Eukarya (Deshmukh et al. 1993; Wool 1986). 5S rRNA has been used as a model molecule for studies on RNA structure, RNA–RNA, and RNA–protein interactions, and as a phylogenetic marker (Hunt et al. 1984; Hori et al. 1985; Hori and Osawa 1987; Küntzel et al. 1981, 1983; Nearhos and Fuerst 1987; Villanueva et al. 1985). The molecule appears to act as a seventh domain in the large ribosomal subunit, conferring stability to the entire 3-dimensional (3D) structure (the structure of 23S rRNA contains six domains). In fact, genetic deletions in 5S rRNA decrease substantially cell viability, especially when compared to the 16S and 23S rRNA subunits (Ammons et al. 1999). This stability is most notable in interactions with domains II and V of 23S rRNA, which are involved in translocation and peptide bond formation, respectively. Experiments performed using 5S rRNA mutants indicate that the molecule might also be involved in signal transmission during the translation process (Sergiev et al. 2000).
Due to its universally conserved structure, 5S rRNA molecules can be substituted by molecules in other species, restoring in every case the biological activity of the ribosome (Erdmann et al. 1986; Teixido et al. 1989). Because the nucleotide sequences of 5S rRNA are highly conserved throughout nature, phylogenetic analysis alone provided an initial model for its secondary structure (Fox and Woese 1975). This model was later on refined (Luehrsen and Fox 1981; but see Hannock and Wagner 1982). Structurally, 5S rRNA can always be folded into a common secondary structure. This structure contains five helices (I–V) (labeled S1–S5 in this study), two hairpin loops (C, D), two internal loops (B, E), and a multiloop (hinge) region (A) connecting helices I, II, and V. This 3-branched general structure has been confirmed by a number of structural studies and comparative sequence analyses. The three branches are occasionally addressed collectively as the α, β, and γ domains (Joachimiak et al. 1990). Limited tertiary interactions exist that are centered on loop A and the domain containing helices II and III. Furthermore, the crystal structure of the large subunit from Haloarcula marismortui (Ban et al. 2000) allowed verification of the secondary structure of 5S rRNA inferred from phylogenetic analysis and structural studies in solution. Most of the base pairs predicted by comparative sequence analysis were detected in the crystal structure. Furthermore, several 3D structural models of 5S rRNA have been proposed (reviewed in Barciszewska et al. 2000), but all differ in many aspects from the model derived from the H. marismortui 50S subunit (Ban et al. 2000). Although programing algorithms for 5S rRNA secondary structure predictions have been improved, the predicted structures are not always satisfactory (Azad et al. 1998; Mathews et al. 1999; Ding and Lawrence 1999). However, the generic 3-domain structure of 5S rRNA has been consistently recovered and confirmed. Finally, Gabashvili et al. (2003) revealed the structural dynamics of 5S rRNA with alternative conformations complementary or additional to those observed by crystallography (Yusupov et al. 2001; Brodersen et al. 2002; Ramakrishnan 2002; Yonath 2002; and references therein) and other experimental methods (Lodmell and Dahlberg 1997; Frank and Agrawal 2000).
In the present study, we apply an award-winning phylogenetic method that reconstructs evolutionary history directly from molecular structure to study the evolution of 5S rRNA (Caetano-Anollés 2002a). This novel cladistic approach produces intrinsically rooted trees that ‘‘embed structure and function directly into phylogenetic analysis’’ (Pollock 2003). The method has been applied widely to study the structural evolution of two crucial molecules, rRNA (Caetano-Anollés 2002a, b) and tRNA (Sun and Caetano-Anollés 2008a, b, c), has been improved during studies of other functional RNA molecules (Caetano-Anollés 2005; Sun et al. 2007), and has been extended to the study of molecular repertoires of protein domains at both the fold and the fold superfamily levels (Caetano-Anollés and Caetano-Anollés 2003; recently reviewed in Caetano-Anollés et al. 2009). Here we dissect for the first time the structure of 5S rRNA, reconstructing intrinsically rooted phylogenetic trees of molecules and substructures (Fig. 1). These trees not only reveal the evolutionary history of the molecule, but also identify ancestral functional and structural components that were crucial for its workings during early life.

General methodological approach of phylogenetic analysis. The structure of 5S rRNA molecules with its 3 domains (α, β, and γ) and its five helical segments (I–V) can be decomposed into substructures, such as coaxial stem tracts and unpaired regions that can be studied using features (characters) that describe molecular geometry (e.g., length of stems or unpaired regions). These ‘shape’ characters are coded and assigned ‘character states’ according to an evolutionary model that polarizes character transformation towards an increase in molecular order (character argumentation). Coded characters (s) are arranged in data matrices, which can be transposed and subjected to cladistic analyses to generate rooted phylogenies of either molecules or substructures. Phylogenetic trees of molecules describe how the structure of entire molecules diversifies. Trees of substructures describe how substructures in molecules have evolved and can be used to generate evolutionary heat maps of secondary structure that color secondary structures with molecular ancestries derived directly from the trees. Tracing of ancestry information on 3D structural models provides information on the age of inter- and intra-molecular contacts that exist in molecular complexes, such as the ribosome. Helical stems and loops of the secondary structure of 5S rRNA molecules are portrayed by bars and circles, respectively
Materials and Methods
Data
The entire set of 1,371 5S rRNA sequences was retrieved from the 5S rRNA Database (http://rose.man.poznan.pl/5SData/; September 2005 edition; Szymanski et al. 2002). We used the program RNAfold in the Vienna RNA package (Hofacker 2003) to fold the RNA molecules and predict the minimum free energy (mfe) structure among alternative structural topologies. Like many other currently available RNA-folding programs, RNAfold cannot fold each individual molecule in the dataset into the 3-domain mfe structure typical of 5S rRNA, even if many 3-armed structural topologies are found at higher (unstable) free energy levels. We therefore selected for further study approximately one half of the available sequences (666), which folded into 3-armed mfe structures and were compatible with 5S rRNA phylogeny and known 3D crystallographic models. These sequences represent a comprehensive sampling of molecules in the three superkingdoms of life (89 Archaea, 168 Bacteria, and 409 Eukarya).
Phylogenetic Characters, Character Coding, and Taxa Selection
Forty-six structural characters were scored (Table 1). Character homology was determined by the relative position of substructures in the secondary structures and coded character states were based on the length (number of bases or base pairs) and number of these substructures. Character states were defined in alphanumerical format with numbers from 0 to 9 and letters from A to E. Missing substructures were given the minimum state (0). Partitioned data matrices were constructed based on taxonomy (Archaea, Bacteria, or Eukarya) or types of characters (stabilizing characters, i.e., stems, or de-stabilizing characters, including bulges, hairpins, and other single-stranded regions). The data matrix of coded characters is provided in Table S1 as Supplementary Online Material.
Character Argumentation
Structural features were treated as linearly ordered multistate characters that were polarized by invoking an evolutionary tendency toward molecular order. The validity of character argumentation and the use of maximum parsimony (MP) has been discussed in detail elsewhere (Caetano-Anollés 2001; 2002a, b; 2005; Sun and Caetano-Anollés 2008a, b, c; Sun et al. 2009). Operationally, polarization was determined by fixing the direction of character state change using a transformation sequence that distinguishes ancestral states as those thermodynamically more stable. Maximum character states were defined as the ancestral states for stems and G · U base pairs (i.e., structures stabilizing the 5S rRNAs). Minimum states (0) were treated as the ancestral states for bulges, hairpin loops, and other unpaired regions (i.e., structures de-stabilizing the 5S rRNAs).
Phylogenetic Analysis
All data matrices were analyzed using equally weighted MP as the optimality criterion in PAUP* (Swofford 2003). Note that a more realistic weighting scheme should consider for example the evolutionary rates of change in structural features. However, this requires the measurement of evolutionary parameters along individual branches of the tree and the development of an appropriate quantitative model. In the absence of this information, it is most parsimonious and preferable to give equal weight to the relative contribution of each character. The use of MP (the preference of solutions that require the least amount of change) is particularly appropriate and can outperform maximum likelihood (ML) approaches in certain circumstances (Steel and Penny 2000). MP is precisely ML when character changes occur with equal probability but rates vary freely between characters in each branch. This model is useful when there is limited knowledge about underlying mechanisms linking characters to each other (Steel and Penny 2000). Furthermore, the use of large multi-step character state spaces decreases the likelihood of revisiting a same character state on the underlying tree, making MP statistically consistent. Depending on the number of taxa in each matrix, MP tree reconstructions were sought using either exhaustive, branch-and-bound, or heuristic search strategies. When the heuristic search strategy was used, 1,000 heuristic searches were initiated using random addition starting taxa, with tree bisection reconnection (TBR) branch swapping and the MulTrees option selected. One shortest tree was saved from each search. Hypothetical ancestors were included in the searches for the most parsimonious trees using the ancstates command. A ‘‘total evidence’’ approach (Kluge 1989; Kluge and Wolf 1993), also called ‘‘simultaneous analysis’’ by Nixon and Carpenter (1996), was applied in phylogenetic analyses to combine both sequence and structure data of the complete and partitioned matrices. Sequences were aligned using Clustal X (Jeanmougin et al. 1998) and manually adjusted as necessary. The goal of this analysis was to provide stronger support for the phylogenetic groupings recovered from analyses of structural data. Bootstrap support (BS) values (Felsenstein 1985) were calculated from 105 replicate analyses using “fast” stepwise addition of taxa in PAUP*. The g 1 statistic of skewed tree length distribution calculated from 104 random parsimony trees was used to assess the amount of nonrandom structure in the data (Hillis and Huelsenbeck 1992).
Evolutionary relationships derived from trees of substructures were traced in generic 2D and 3D models of 5S rRNA secondary structure that we here call evolutionary heat maps of ancestry. Because reconstructed trees were intrinsically rooted, we established the relative age (ancestry) of each substructure by measuring a distance in nodes from the hypothetical ancestral substructure on a relative 0–1 scale (node distance, nd). To do this, we used a perl script that counts the number of nodes from the base of the tree to its leaves and divides this number by the maximum number of nodes that is possible in a lineage of the tree (Caetano-Anollés 2002b). Ancestry values were divided in classes, giving them individual hues in a color scale that was then used to color substructures in a generic 3-domain secondary structure model of 5S rRNAs or 3D crystallographic models.
Phylogenomic Analysis of Protein Architecture
A census of the genomic sequence of 584 organisms, including 46 Archaea, 397 Bacteria, and 141 Eukarya, assigned protein structural domains corresponding to 1,453-fold superfamilies to protein sequences using advanced linear hidden Markov models of structural recognition in superfamily and a probability cutoff E of 10−4. Fold superfamilies were defined according to the structural classification of proteins (version 1.69; Murzin et al. 1995). The census was used to build data matrices of genomic abundance of domains, which were coded as linearly ordered multistate phylogenetic characters. Data matrices were used to build universal trees of protein architectures with established methodology (Caetano-Anollés and Caetano-Anollés 2003). The reconstruction of these large trees is computationally hard and their visualization challenging. We used a combined parsimony ratchet (PR) and iterative search approach to facilitate tree reconstruction (Wang and Caetano-Anollés 2009). A recent review summarizes the general approach and the progression of census data and tree reconstruction in recent years (Caetano-Anollés et al. 2009). The ages of individual domains were given as nd values and were derived directly from the tree of architectures.
Results
Phylogenetic Trees of 5S rRNA Molecules
Phylogenetic analysis of combined structure and sequence data of 666 5S rRNA molecules resulted in 10,000 preset MP trees, each of 11,481 steps. The strict consensus of these trees of molecules showed that superkingdoms Bacteria and Eukarya were both monophyletic and sister to each other, while Archaea was paraphyletic and basal in the tree (Fig. 2; Fig. S1). We re-run the analysis with structure characters treated as linearly ordered but non-polarized (excluding the hypothetical ancestor in the search). The resulting unrooted trees recovered the monophyly of each of the three superkingdoms of life. The topology of many branches was congruent with trees derived from structure or sequence separately (see below). BS values were generally low (<50%) in deep branches of the tree, but many branches closer to the leaves were supported by high bootstrap values. This is an expected result given the size of these trees.

A global phylogenetic tree of 5S rRNA molecules reconstructed from sequence and structure. MP analysis of data from 666 5S rRNA molecules found in superkingdoms Archaea, Bacteria, and Eukarya resulted in 10,000 preset trees, each of 11,481 steps. Consistency index (CI) = 0.074 and 0.072, with and without uninformative characters, respectively; Retention index (RI) = 0.772; Rescaled consistency index (RC) = 0.057; g 1 = −0.131. Terminal leaves are not labeled since they would not be legible (see Fig. S1 for a tree with labeled taxa). Nodes labeled with closed circles have BS values >50%
Phylogenetic reconstructions of trees of molecules derived from either sequence or structure showed distinct phylogenetic signal in these datasets (Fig. S2). Phylogenetic analysis of sequence data resulted in 10,000 preset unrooted MP trees each of 4,909 steps. The strict consensus of these trees revealed the three superkingdoms. BS values were generally low (<50%), but many branches that were close to the leaves were well supported. Phylogenetic analysis of structural characters resulted in 10,000 preset MP trees each of 4,905 steps. The strict consensus of these trees did not show the three superkingdoms being monophyletic. Instead, a paraphyletic group containing 14 archaeal taxa, including Thermococcus (2), Pyrococcus (5), Sulfurococcus (1), Sulfolobus (5), and Desulfurococcus (1), was found at basal positions. The other archaeal taxa were found in a largely unresolved clade. Again, BS values were generally low (<50%) in basal branches, while values were higher closer to the leaves of the tree.
Data matrices of sequence, structure, or combined sequence and structure data were partitioned according to superkingdoms (89 Archaea, 168 Bacteria, or 409 Eukarya). Strict consensus trees showed phylogenetic relationships of taxa were largely maintained in each superkingdom. Statistics of these trees are described in Table S2 and trees can be retrieved from the MANET database (http://manet.illinois.edu). Two partitioned data matrices based on stabilizing (stems and G · U pairs) or de-stabilizing characters (single strands, hairpins, bulges, and multiloops) were also generated but resulted in incongruent phylogenies, indicating that these two types of structures contain different histories and phylogenetic signals. Overall, trees derived from de-stabilizing characters were more resolved than those derived from stabilizing characters. However, the incongruent nodes were all weakly supported (BS < 50%) and the relationships of many groups close to the leaves of the tree were generally congruent. Statistics of these trees are described in Table S3 and trees can be retrieved from MANET. Finally, neighbor-joining (NJ) trees were also generally congruent with those derived from MP analyses; so were trees derived from the data matrices partitioned according to superkingdom.
Phylogenetic Trees of 5S rRNA Substructures
Phylogenetic trees of substructures were reconstructed from geometrical characters describing the complete 5S rRNA dataset (Fig. 3). The tree of stem substructures revealed S1 was the most basal helical segment, followed in order by S3, S2, and S5 and S4. Because RNA structures are defined by a frustrated conformational interplay of stems and loops, this tree of helical stems defines the fundamental scaffold of structural evolution of the entire molecule. Consequently, structural diversification of related substructures had to occur once individual supporting secondary structures had developed. Analyses of G · U pairs placed GU4 at the base of the tree, followed in order by GU1, GU5, and GU2 and GU3. This pattern of G · U pairs was also revealed by phylogenetic analyses of datasets partitioned according to superkingdom (Fig. S3). Analyses of bulges and unpaired regions complemented information derived from other substructures. Remarkably, the 5′ free end was the most ancient unpaired substructure, while the 3′ free end was derived. Phylogenetic analyses of stem substructures derived from partitioned datasets of Bacteria and Eukarya 5S rRNAs, respectively, revealed the same topology as that derived from the complete dataset (Fig. 4). However, the tree of stem substructures derived from the partitioned matrix of 89 Archaea 5S rRNAs showed that stem S5 predated S2. Statistics of partitioned analysis is given in Table S3, and the complete set of trees of substructures is shown in Fig. S3.

Phylogenetic trees of molecular substructures reconstructed from characters describing the geometry of structure in 5S rRNAs. Trees of substructures describe the evolution of stems (S) (7,355 steps; CI = 0.869; RI = 0.570; RC = 0.495; g 1 = −0.861), bulges (B) (4,455 steps; CI = 0.791; RI = 0.536; RC = 0.424; g 1 = −0.418), 5′ and 3′ bulge sections of the molecules (B) (3,183 steps; CI = 0.692; RI = 0.713; RC = 0.494; g 1 = −1.419), loops and free ends (4,626 steps; CI = 0.635; RI = 0.685; RC = 0.435; g 1 = −0.522), and G · U pairs (GU) (3,158 steps; CI = 0.837; RI = 0.630; RC = 0.528; g 1 = −0.915). One minimal-length tree was retained in each case using exhaustive searches derived from equally weighted MP analyses. Bootstrap values >50% are shown for individual nodes. Evolutionary heat maps of secondary structure describe inferences of structural evolution derived directly from the trees. The relative scale describes the number of nodes from the hypothetical ancestor at the base of the tree

Phylogenetic tree of molecular substructures reconstructed from characters describing the geometry of structure in 5S rRNA molecules partitioned according to superkingdom. Trees of substructures describe the evolution of stems in all 666 molecules belonging to the three superkingdoms (7,355 steps; CI = 0.869; RI = 0.570; RC = 0.495; g 1 = −0.861), 89 molecules from Archaea (1,047 steps; CI = 0.837; RI = 0.503; RC = 0.421; g 1 = −0.518), 168 molecules from Bacteria (2,120 steps; CI = 0.874; RI = 0.671; RC = 0.586; g 1 = −1.015), and 409 molecules from Eukarya (4,184 steps; CI = 0.875; RI = 0.520; RC = 0.455; g 1 = −0.875). Bootstrap values >50% are shown for individual nodes
The Age of Ribosomal Proteins Associated with 5S rRNA
In order to study the evolution of the ribosomal protein complement that associates with 5S rRNA, we established the age of individual proteins by tracing their ancestries in a global phylogeny of protein architectures that was reconstructed from a genomic census of protein domain structures in 584 completely sequenced organisms (Caetano-Anollés et al. 2009). This tree describes the history of 1,453 domains defined at fold superfamily level (Fig. S4). The age of fundamental 5S rRNA-linked domains ranged from nd = 0.236 for the translation proteins SH3-like domain (b.34.5) typical of ribosomal protein L21e to nd = 0.328 for the ribosomal protein L5 domain (d.77.1) typical of ribosomal protein L10e. All domain architectures of ribosomal proteins originated during the architectural diversification epoch of the protein world (Wang et al. 2007; Caetano-Anollés et al. 2009). The evolution of 5S rRNA-associated proteins was finally traced on 2D or 3D representations of the 5S rRNA ensemble (Fig. 5). This helped to identify how the history of the 5S rRNA molecule related to the discovery of function and its interactions with protein molecules as the shape of the molecule and its structural domains changed in evolution.

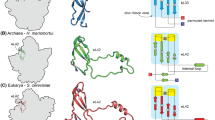
Evolutionary heat maps of 5S rRNA. A Consensus 2D heat map summarizing phylogenetic relationships described in Fig. 3 and contacts of 5S rRNA structural components with ribosomal proteins. The age of proteins derived from a phylogenomic analysis of domain structure (Fig. S4) is given in node distance (nd), with increasing values representing the progression of evolutionary time. A plot describing the age of interacting nucleic acid substructures and protein molecules is given as an inset. B Consensus 3D heat map traced on a 3D model of the Holoarcula marismortui 5S rRNA molecule (PDB entry 1FFK; Ban et al. 2000). Ancestries (nd) derived from trees of substructures were painted directly on the structural model using an ancestry color scale (bar) in ribbons (Carson 1997). Substructures are labeled in the 3D model. C The same model of the 5S rRNA molecule now shows associated ribosomal proteins colored according to their evolutionary age
Discussion
An Archaeal Rooting of the Universal Tree of Life
It is now generally accepted that the world of cellular organisms is tripartite and consists of superkingdoms Archaea, Bacteria, and Eukarya. This view, heralded by the school of Carl Woese in Urbana (Woese et al. 1990), is fundamentally derived from the study of the small subunit of rRNA, an ancient ribosomal molecule that is central to translation. Recent advances in genomic biology have also revealed this tripartite scheme. Phylogenetic analysis of the content and order of genes and the structure of gene products (nucleic acid and protein molecules) uncovered the existence of only three cellular superkingdoms (Doolittle 2005; Caetano-Anollés et al. 2009). However, the root of the universal tree remains controversial and so is the nature of the universal ancestor of all life that this root defines (Woese 1998; Penny and Poole 1999; Glansdorff et al. 2008; Forterre 2009).
Although 5S rRNA sequences have been used to study phylogenetic relationships between organisms at various levels of taxonomical classification, its utility at superkingdom level has been curtailed by the limited phylogenetic signal that is present in the short nucleic acid sequence of these molecules. Furthermore, phylogenetic trees reconstructed from 5S rRNA sequence can only be rooted by inclusion of outgroup taxa, i.e., external hypotheses of relationship, when these can be found. In contrast, analysis of structure has generally deep phylogenetic signal and produces intrinsically rooted trees that can be used to root the universal tree of life (Caetano-Anollés 2002a; Sun and Caetano-Anollés 2008b). In this study, we applied the total evidence approach to combine sequence and structural data in 5S rRNA molecules and infer a universal tree. Remarkably, this tree is rooted paraphyletically in Archaea, and shows that both Bacteria and Eukarya are monophyletic and derived (Fig. 2). Interestingly, a paraphyletic archaeal root of the tree of life has also been suggested by studies of tRNA paralogs (aloacceptors) and other evidence (Xue et al. 2003, 2005; Di Giulio 2007; Wong et al. 2007), tRNA and ribonuclease P (RNase P) structure (Sun and Caetano-Anollés 2008b; F.-J. Sun and G. Caetano-Anollés, unpublished), and phylogenomic studies of protein domains (Wang et al. 2007) and protein domain organization at fold and fold superfamily levels (Wang and Caetano-Anollés 2006, 2009). While the canonical view is that the root of the tree of life lies between the Bacteria and the Archaea, with eukaryotes represented as a long-branched sister group to the Archaea (Brown and Doolittle 1995; Gribaldo and Cammarano 1998; Zhaxybayeva et al. 2005), our results provide additional support to already compelling arguments in favor of the early appearance of Archaea in a diversified world. These arguments are based on an analysis of entire protein repertoires and ancient RNA molecules.
Why is the rooting in Archaea paraphyletic? At first glance, paraphyly could result from loss of phylogenetic signal in the secondary structure of 5S rRNA, or from primordial homoplasy-enhancing processes operating during evolutionary stages prior to the differentiation of the three superkingdoms. However, a more plausible explanation, given that global analyses of protein domains and several non-coding RNA molecules congruently support archaeal paraphyly, is that early diversification of an eukaryal-like communal ancestor involved spatial colonization of unchartered environments unique to the individual primordial lineages. This divergence-by-isolation scenario is particularly plausible close to deep vents in an ancient auxinic ocean, where diverse and more demanding environments were up for grabs. Molecules that were discovered during these early times (e.g., ancient protein domains and tRNA, RNase P, and rRNA molecules) witnessed these processes and recorded their history. This probably occurred before primordial lineages widely ventured into oceans and other environs, processes of primordial lineage homogenization (horizontal transfer, recruitment, etc.) erased unique signals in these ancient molecules, and new molecules and protein architectures established themselves on the evolving primordial world. We note that we have identified three epochs in evolution (Wang et al. 2007; Sun and Caetano-Anollés 2008b), (i) an early architectural diversification epoch in which ancient molecules (including 5S rRNA) emerged and diversified, (ii) a superkingdom specification epoch in which these molecules sorted in emerging archaeal and eukaryal-like lineages, and (iii) an organismal diversification epoch in which increasing numbers of lineage-specific variants of already existing molecules and new molecules and architectures appeared in an increasingly diversified tripartite world. We contend these epochs have left indelible signatures in the make up of ancient molecules such as 5S rRNA. As we will show below, trees of substructures recover a historical timeline that is buried in the structure of the RNA molecule and provides clues on early organismal diversification.
Origin and Evolution of the 5S rRNA Molecule
Phylogenetic trees of substructures revealed clear patterns of evolutionary diversification in the structure of 5S rRNA molecules (Fig. 3). These patterns were summarized in consensus 2D and 3D evolutionary heat maps (Fig. 5A, B) and allowed elaboration of a model for the origin and evolution of 5S rRNA (Fig. 6). This model considers that the modern 3-domain 5S rRNA structure evolved by gradual addition to the growing molecule of structural components (homologous to present day helical and unpaired regions), either by insertion of single or multiple nucleotides or by partial or total duplications. Several salient features are noteworthy:

A model of 5S rRNA evolution. The model is derived directly from phylogenetic trees of substructures and heat maps and shows formation of substructures homologous to present day helical regions in 5S rRNA during the course of evolution. Note distinct evolutionary routes occurring in ancestors of Archaea and in ancestors of Bacteria and Eukarya, which match the topology of phylogenetic trees of molecules
-
1.
The tree of stem substructures showed that helix I (S1) was the most ancient helical segment of 5S rRNA and that it was evolutionarily linked to a 5′-terminal free end. The evolutionary importance of these primordial hairpin structures was originally proposed for tRNA (Bloch et al. 1985; Di Giulio 1992; Dick and Schamel 1995; Eigen and Winkler-Oswatitsch 1981; Hopfield 1978; Tanaka and Kikuchi 2001; Widmann et al. 2005; Woese 1969) and was later emphasized by the genomic tag hypothesis (Weiner and Maizels 1987; Maizels and Weiner 1994). Its significance is also highlighted by recent molecular evolution studies of tRNA (Sun and Caetano-Anollés 2008a) and SINE RNA (Sun et al. 2007), and two undergoing studies that focus on the entire ribosome and RNase P complexes (A. Harish, F.-J. Sun, G. Caetano-Anollés, unpublished). These studies demonstrate that all of these molecules may be modern derivatives of a primitive hairpin structure that probably harbored a multitude of non-specific structural and catalytic functions. Since these primordial structures currently associate with very ancient protein domains, present for example in aminoacyl tRNA synthases, ribosomal proteins, and RNase P proteins (Caetano-Anollés et al. 2009; A. Harish, F.-J. Sun, and G. Caetano-Anollés, unpublished; see analysis of proteins associated with S1 below), these associations could have been operating very early in an ancient ribonucleoprotein world. Alternatively, these hairpins could have acted alone, with proteins interactions appearing later in evolution perhaps to enhance the specificity of the original function.
-
2.
Diversification of unpaired regions (e.g., bulges and loops) somehow followed the growth of stems in the evolving molecule, with the 5′-terminal free end being the most ancient and the 3′-terminal free end being more derived. Remarkably, these same patterns were observed in the evolution of tRNA (Sun and Caetano-Anollés 2008a). Its 5′-terminal end was the most ancient unpaired region, while its 3′-terminal sequence (including the CCA terminus) was added after the entire cloverleaf structure was formed. This observation is important as it matches statistical analyses of tRNA sequences (Tanaka and Kikuchi 2001). In the case of tRNA, it also suggests an evolutionary timing for the establishment of tRNA interactions with CCA-adding enzymes. The fact that tRNA and 5S rRNA share this same evolutionary pattern is more than a coincidence and merits future investigation.
-
3.
Phylogenetic trees suggest the use of weak G · U base pairs in stem regions of the 5S rRNA molecule occurred only after the 3-domain structure was fully realized in evolution (Fig. 3). Consequently, non-canonical base-pairing interactions represent structural features that were introduced late in evolution, probably to help stabilize helical structures. A similar pattern was also observed in the analysis of tRNA molecules (Sun and Caetano-Anollés 2008a). Interestingly, the most ancient G · U substructures in rRNA were associated with S4 and S1 (Fig. 3), helical structures that are unique because they have tandem G · U motifs that stack guanosines (e.g., Gautheret et al. 1995) or stabilize water interactions and mediate nucleotide interactions necessary for helix stability (Betzel et al. 1994).
-
4.
Addition of stem substructures to the evolving molecule was different for Archaea than for Eukarya and Bacteria when analyzing data matrices partitioned according to superkingdom (Fig. 4). Stem S1 was followed by S3 and S5 (in that order) in trees derived from archaeal substructures, while S1 was followed by S3 and S2 (in that order) in trees reconstructed from bacterial or eukaryal molecules. This suggests that primordial 5S rRNA segments homologous to helices I and III extended their helical structure by stacking an additional helical segment (helix II) in the lineage leading to ancestors of Bacteria and Eukarya or added a segment homologous to helix V to produce a branched structure in ancestors of Archaea (Fig. 6). The early generation of a 3-domain structure in the archaeal lineage at the onset of organismal diversification is remarkable and has important implications. When combined with the basal placement of Archaea in the tree of 5S rRNA molecules (Fig. 2), it suggests an early split of the archaeal lineage, which is compatible with a comprehensive analysis of sequence and structure of the tRNA molecule that supports the ancestrality of Archaea (Sun and Caetano-Anollés 2008b), and whole-genome analysis of complements of protein domains and domain combinations that suggest an early split of the archaeal lineage from a architecture-rich communal world (Wang et al. 2007; Wang and Caetano-Anollés 2009). This primordial split is linked to reductive evolutionary tendencies in the make up of archaeal (and then bacterial) genomes that were protracted and ultimately led to the three superkingdoms of life (Wang et al. 2007).
It is particularly noteworthy that the evolutionary history of the tRNA cloverleaf structure also exhibits two distinct evolutionary routes, one delimiting Archaea and the other superkingdoms Bacteria and Eukarya (Sun and Caetano-Anollés 2008a). A similar pattern was also obtained in an ongoing analysis of RNase P RNA (F.-J. Sun and G. Caetano-Anollés, unpublished). In phylogenetic analysis, congruence provides the strongest support that is possible to an evolutionary hypothesis, especially when congruent phylogenetic reconstructions are derived from different kinds of molecular evidence. The fact that now three distinct and ancient RNA molecules produce congruent evolutionary patterns suggests strongly an early rooting of the universal tree of life in Archaea.
Evolution of 5S rRNA Interactions with Ribosomal Proteins and Other Molecules
Protein–RNA interactions are fundamental for the assembly and function of the ribosomal ensemble. 5S rRNA is the only known rRNA species that binds ribosomal proteins before it is incorporated into the ribosome both in prokaryotes and eukaryotes (Szymanski et al. 2003; Smirnov et al. 2008). Central interactions include contacts to eukaryotic ribosomal protein L18, and proteins L5, L18, and L25 in bacteria. The molecule also interacts with non-ribosomal proteins such as the transcription initiator TFIIIA, HSP70, and p43 (Szymanski et al. 2003). Figure 5 describes fundamental RNA–protein interactions, with some interactions traced in a 3D model of structure.
In order to determine when protein–RNA contacts were established in evolution, we timed the appearance of the 3D structure of 5S rRNA-associated ribosomal protein molecules in a tree of protein architectures (Fig. S4) derived from phylogenomic analysis of domain structure at fold superfamily level of structural classification (Caetano-Anollés et al. 2009). A timeline of domain discovery was obtained directly from the tree of domain structure and the age of each domain was given as the number of nodes from the base of the tree in a relative 0–1 scale (node distance, nd), with 0 representing the first domain architecture that originated in the protein world. These timelines are useful. They have been used recently to establish how functions were discovered in evolution of proteins (Caetano-Anollés et al. 2009) or how domain combinations establish in the protein world (Wang and Caetano-Anollés 2009).
Interestingly, the most ancient 5S rRNA-associated protein domain, the translations protein SH3-like domain (b.34.5) present in ribosomal protein L21e of the archaeal molecule (Fig. 5C), appeared quite early in the evolution of proteins (nd = 0.236), but rather late during the ‘architectural diversification’ epoch defined by Wang et al. (2007). This domain associates with helix I (stem S1), the most ancient segment of 5S rRNA molecule. The second most ancient 5S rRNA-associated protein domain was the translational machinery components domain (c.55.4) of ribosomal protein L18 (nd = 0.287). Remarkably, this domain associates with helix III (S3), the second most ancient RNA substructure. Domains associated with more derived helices in the 5S rRNA molecule (d.59.1, d.41.4, and d.77.1) and present in ribosomal proteins L30, L10e, and L5, were all more derived (nd = 0.301–0.328), but closely related in age. This tight relationship between the age of 5S rRNA helices derived from analysis of RNA structure (Fig. 3) and the age of ribosomal proteins obtained from a census of domains in proteomes (Fig. S4) is highly significant (see inset of Fig. 5A). First, it establishes that the 5S rRNA molecule originated quite late in evolution, at a time (nd ~ 0.2) when metabolic enzymes (Caetano-Anollés et al. 2007) and translation machinery (Caetano-Anollés et al. 2009; A. Harish and G. Caetano-Anollés, unpublished) were already in place in the protein world. Second, it shows that the development of the 5S rRNA molecule occurred within a relative short time frame (0.1 nd). Third, it supports the gradual growth of 5S rRNA by addition of helical structural components to the molecule and the model of structural evolution we have proposed (Fig. 6).
Other 5S rRNA-associated domains linked to proteins known to be important for ribosomal function were either more ancient (e.g., p43; b.40.4; nd = 0.019), similar in age to main fundamental ribosomal proteins (e.g., HSP70; b.130.1; nd = 0.347), or more derived, appearing during the ‘organismal diversification’ epoch (e.g., TFIIIA; g.37.1; nd = 0.986) (Fig. S4). For example, the contemporary heat-shock HSP70 binds transiently to 5S rRNA and promotes correct folding of the polypeptide chain (Okada et al. 2000). TFIIIA is involved in initiation of 5S rRNA transcription and forms a 7S RNP complex with the molecule that is exported from the nucleus to the cytoplasm in eukaryotes (Szymanski et al. 2003). The complex acts as a storage particle for 5S rRNA until it is required for ribosomal assembly. Interestingly, protein markers for the nuclear envelope involve proteins (e.g., constituents of the nuclear pore complex) that appeared very late in evolution (nd = 0.82–1.00) (Caetano-Anollés et al. 2009), suggesting they are contemporary to TFIIIA. In amphibian oocytes, 5S rRNA is also stored in larger 42S RNP particles called “thesaurisomes”. Thesaurin b (p43) is an ancient nine-zinc-finger protein component of this complex that shares with TFIIIA RNA-binding activity. Finger-swapping experiments have shown zinc fingers can be exchanged between these proteins without affecting RNA binding (Hamilton et al. 2001). When coupled with our evolutionary genomic analyses, these results suggest recruitment of ancient and use of new domain architectures has enhanced the functional role of the 5S rRNA complex in evolution.
Although most of free energy and specificity of 5S RNA binding to the large ribosomal subunit depend on extensive interactions with proteins, few RNA–RNA interactions do occur and involve the backbones of helical domain γ (stems S4 and S5) (Ban et al. 2000). Our study shows these substructures are derived in the molecule, suggesting 5S rRNA was a late evolutionary addition to the ribosomal ensemble. This is especially so because many ribosomal proteins associated with the small and large subunits of the ribosome are more ancient than the ones here described (Fig. 5), supporting the contention that the 5S rRNA component is indeed derived.
Conclusions
The cladistic method used in this study embeds structure directly in phylogenetic analysis and generates intrinsically rooted phylogenies without the need of outgroups. We have exemplified the potential of this novel phylogenetic approach by focusing on several fundamental molecules that are functionally linked to protein synthesis (reviewed in Sun et al. 2009). The evolutionary analyses of these molecules provide novel insights into important questions surrounding the emergence of cellular life and the origins and evolution of the protein biosynthetic machinery. Here we unveil patterns of origin and diversification in the molecular history of 5S RNA, a molecule that forms a small complex that is at the center of ribosomal assembly and function. Because trees of life generated from these non-coding RNA molecules establish evolution’s arrow, it becomes possible to identify the location of the root on the tree of life. We here show that a common topology emerges from phylogenetic analysis of 5S rRNA that is congruent with topologies generated from other modern RNA molecules and phylogenomic analysis of proteomes. This topology indicates Archaea is the most ancient lineage on Earth. This result is important because the root of the tree of life has been debated over decades, with controversy largely stemming from the various rooting approaches that have been used and the alternative evolutionary scenarios that had been derived (Forterre 2009). We anticipate future studies of molecular structure will focus on all kinds of RNAs, clarifying further questions surrounding origins of modern biochemistry and diversified life. Phylogenetic analyses of molecular structure will also impact the study of function and structure of RNA in interaction with protein molecules, as these are placed within an evolutionary context. Together with evidence derived from molecular, genetic, and biochemical studies, evolutionary insights will enhance our understanding of biological functions and how these are linked to mechanisms embodied in molecular repertoires.
References
Ammons D, Rampersad J, Fox GE (1999) 5S rRNA gene deletions cause an unexpectedly high fitness loss in Escherichia coli. Nucleic Acids Res 27:637–642
Azad AA, Failla P, Hanna PJ (1998) Inhibition of ribosomal subunit association and protein synthesis by oligonucleotides corresponding to defined regions of 18S rRNA and 5S rRNA. Biochem Biophys Res Commun 248:51–56
Ban N, Nissen P, Hansen J, Moore PB, Steitz TA (2000) The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science 289:905–920
Barciszewska MZ, Szymanski M, Erdmann VA, Barciszewski J (2000) 5S ribosomal RNA. Biomacromolecules 1:297–302
Barciszewska MZ, Szymanski M, Erdmann VA, Barciszewski J (2001) Structure and functions of 5S rRNA. Acta Biochim Pol 48:191–198
Betzel C, Lorenz S, Furste JP, Bald R, Zhang M, Schneider TR, Wilson KS, Erdmann VA (1994) Crystal structure of domain A of Thermus flavus 5S rRNA and the contribution of water molecules to its structure. FEBS Lett 351:159–164
Bloch DP, McArthur B, Mirrop S (1985) tRNA–rRNA sequence homologies: evidence for an ancient modular format shared by tRNAs and rRNAs. Biosystems 17:209–225
Bogdanov AA, Dontsova OA, Dokudovskaya SS, Lavrik IN (1995) Structure and function of 5S rRNA in the ribosome. Biochem Cell Biol 73:869–876
Brodersen DE, Clemons WM Jr, Carter AP, Wimberly BT, Ramakrishnan V (2002) Crystal structure of the 30S ribosomal subunit from Thermus thermophilus: structure of the proteins and their interactions with 16S RNA. J Mol Biol 316:725–768
Brown J, Doolittle W (1995) Root of the universal tree of life based on ancient aminoacyl-tRNA synthetase gene duplications. Proc Natl Acad Sci USA 92:2441–2445
Caetano-Anollés G (2001) Novel strategies to study the role of mutation and nucleic acid structure in evolution. Plant Cell Tissue Org Cult 67:115–132
Caetano-Anollés G (2002a) Evolved RNA secondary structure and the rooting of the universal tree of life. J Mol Evol 54:333–345
Caetano-Anollés G (2002b) Tracing the evolution of RNA structure in ribosomes. Nucleic Acids Res 30:2575–2587
Caetano-Anollés G (2005) Grass evolution inferred from chromosomal rearrangements and geometrical and statistical features in RNA structure. J Mol Evol 60:635–652
Caetano-Anollés G, Caetano-Anollés D (2003) An evolutionarily structured universe of protein architecture. Genome Res 13:1563–1571
Caetano-Anollés G, Kim HS, Mittenthal JE (2007) The origin of modern metabolic networks inferred from phylogenomic analysis of protein architecture. Proc Natl Acad Sci USA 104:9358–9363
Caetano-Anollés G, Wang M, Caetano-Anollés D, Mittenthal JE (2009) The origin, evolution and structure of the protein world. Biochem J 417:621–637
Carson M (1997) Ribbons. Methods Enzymol 277:493–505
Christiansen J, Garrett RA (1986) How do protein L18 and 5S RNA interact? In: Hardesty B, Kramer G (eds) Structure functions and genetics of ribosomes. Springer, New York, pp 253–269
Deshmukh M, Tsay Y-F, Paulovich A, Woolford JL Jr (1993) Yeast ribosomal protein L1 required for the stability of newly synthesized 5S rRNA and the assembly of 60S ribosomal subunits. Mol Cell Biol 13:2835–2845
Di Giulio M (1992) On the origin of the transfer RNA molecule. J Theor Biol 159:199–214
Di Giulio M (2007) The tree of life might be rooted in the branch leading to Nanoarchaeota. Gene 401:108–113
Dick TP, Schamel WWA (1995) Molecular evolution of transfer RNA from two precursor hairpins: implications for the origin of protein synthesis. J Mol Evol 41:1–9
Ding Y, Lawrence CE (1999) A Bayesian statistical algorithm for secondary structure prediction. Comput Chem 23:387–400
Dokudovskaya S, Dontsova O, Shpanchenko O, Bogdanov A, Brimacombe R (1996) Loop IV of 5 S ribosomal RNA has contacts both to domain II and to domain V of the 23 S RNA. RNA 2:146–152
Doolittle RF (2005) Evolutionary aspects of whole-genome biology. Curr Opin Struct Biol 15:248–253
Eigen M, Winkler-Oswatitsch R (1981) Transfer-RNA, an early gene? Naturwissenschaften 68:282–292
Erdmann VA, Pieler T, Wolters J, Digweed M, Vogel D, Hartmann R (1986) Comparative structural and functional studies on small ribosomal RNAs. In: Hardesty B, Kramer G (eds) Structure function and genetics of ribosomes. Spring, New York, pp 164–183
Fanning TG, Traut RR (1981) Topography of the E. coli 5S RNA-protein complex as determined by crosslinking with dimethyl suberimidate and dimethyl-3, 3′-dithiobispropionimidate. Nucleic Acids Res 9:993–1004
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Forterre P (2009) The universal tree of life and the Last Universal Cellular Ancestor (LUCA): revolution and counter-revolutions. In: Caetano-Anollés G (ed), Evolutionary genomics and systems biology. Wiley, Hoboken (in press)
Fox GE, Woese CR (1975) 5S RNA secondary structure. Nature 256:505–507
Frank J, Agrawal RK (2000) A ratchet-like inter-subunit reorganization of the ribosome during translocation. Nature 406:318–322
Gabashvili IS, Whirl-Carrillo M, Bada M, Banatao DR, Altman RB (2003) Ribosomal dynamics inferred from variations in experimental measurements. RNA 9:1301–1307
Gautheret RR, Konings D, Gutell R (1995) Pairing motifs in ribosomal RNA. RNA 1:807–814
Glansdorff N, Xu Y, Labedan B (2008) The Last Universal Common Ancestor: emergence, constitution and genetic legacy of an elusive forerunner. Biol Direct 3:29
Gribaldo S, Cammarano P (1998) The root of the universal tree of life inferred from anciently duplicated genes encoding components of the protein-targeting machinery. J Mol Evol 47:508–516
Hamilton TB, Turner J, Barilla K, Romaniuk PJ (2001) Contribution of individual amino acids to the nucleic acid binding activities of Xenopus zinc finger proteins TFIIIA and p43. Biochemistry 40:6093–6101
Hannock J, Wagner R (1982) A structural model of 5S RNA from E. coli based on intramolecular crosslinking evidence. Nucleic Acids Res 10:1257–1269
Hillis DM, Huelsenbeck JP (1992) Signal, noise, and reliability in molecular phylogenetic analyses. J Hered 83:189–195
Hofacker IL (2003) Vienna RNA secondary structure server. Nucleic Acids Res 31:3429–3431
Holmberg L, Nygard O (2000) Release of ribosome-bound 5 S rRNA upon cleavage of the phosphodiester bond between nucleotides A54 and A55 in 5 S rRNA. Biol Chem 381:1041–1046
Hopfield JJ (1978) Origin of the genetic code: a testable hypothesis based on tRNA structure, sequence, and kinetic proofreading. Proc Natl Acad Sci USA 75:4334–4338
Hori H, Osawa S (1987) Origin and evolution of organisms as deduced from 5S ribosomal RNA sequences. Mol Biol Evol 4:445–472
Hori H, Lim B-L, Osawa S (1985) Evolution of green plants as deduced from 5S rRNA sequences. Proc Natl Acad Sci USA 82:820–823
Hunt LT, George DG, Yeh L-S, Dayhoff MO (1984) Evolution of prokaryote and eukaryote lines inferred from sequence evidence. Orig Life 14:657–664
Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ (1998) Multiple sequence alignment with Clustal X. Trends Biochem Sci 23:403–405
Joachimiak A, Nalaskowska N, Barciszewska M, Barciszewski J, Mashkova T (1990) Higher plant 5S rRNAs share common secondary and tertiary structure. A new three domains model. Int J Macromol 12:321–327
Kapitonov VV, Jurka J (2003) A novel class of SINE elements derived from 5S rRNA. Mol Biol Evol 20:694–702
Kluge AG (1989) A concern for evidence and a phylogenetic hypothesis of relationships among Epicrates (Boidae, Serpentes). Syst Zool 38:7–25
Kluge AG, Wolf AJ (1993) Cladistics: what’s in a word? Cladistics 9:183–199
Kouvela E, Gerbanas GV, Xaplanteri MA, Petropoulos AD, Dinos GP, Kalpaxis DL (2007) Changes in the conformation of 5S rRNA cause alternations in principal functions of the ribosomal nanomachine. Nucleic Acids Res 35:5108–5119
Küntzel H, Heidrich M, Piechulla B (1981) Phylogenetic tree derived from bacterial, cytosol and organelle 5S rRNA sequences. Nucleic Acids Res 9:1451–1461
Küntzel H, Piechulla B, Hahn U (1983) Consensus structure and evolution of 5S rRNA. Nucleic Acids Res 11:893–900
Lodmell JS, Dahlberg AE (1997) A conformational switch in Escherichia coli 16S ribosomal RNA during decoding of messenger RNA. Science 277:1262–1267
Luehrsen KR, Fox GE (1981) Secondary structure of eukaryotic cytoplasmic 5S ribosomal RNA. Proc Natl Acad Sci USA 78:2150–2154
Maizels N, Weiner AM (1994) Phylogeny from function: evidence from the molecular fossil record that tRNA originated in replication, not translation. Proc Natl Acad Sci USA 91:6729–6734
Mathews DH, Sabina J, Zuker M, Turner DH (1999) Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol 288:911–940
McDougall J, Wittmann-Liebold B (1994) Comparative analysis of the protein components from 5S rRNA—protein complexes of halophilic archaebacteria. Eur J Biochem 221:779–785
Murzin AG, Brenner SE, Hubbard T, Chothia C (1995) SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol 247:536–540
Nearhos SP, Fuerst JA (1987) Reanalysis of 5S rRNA sequence data for the Vibrionaceae with the clustan program suite. Curr Microbiol 15:329–335
Nixon KC, Carpenter JM (1996) On simultaneous analysis. Cladistics 12:221–241
Okada S, Okada T, Aimi T, Morinaga T, Itoh T (2000) HSP70 and ribosomal protein L2: novel 5S rRNA binding proteins in Escherichia coli. FEBS Lett 485:153–156
Penny D, Poole A (1999) The nature of the last universal common ancestor. Curr Opin Genet Dev 9:672–677
Pollock D (2003) The Zuckerkandl Prize: structure and evolution. J Mol Evol 56:375–376
Ramakrishnan V (2002) Ribosome structure and the mechanism of translation. Cell 108:557–572
Sergiev PV, Bogdanov AA, Dahlberg AE, Dontsova O (2000) Mutations at position A960 of E. coli 23S ribosomal RNA influence the structure of 5S ribosomal RNA and the peptidyltransferase region of 23 S ribosomal RNA. J Mol Biol 299:379–389
Smirnov AV, Entelis NS, Krasheninnikov IA, Martin R, Tarassov IA (2008) Specific features of 5S rRNA structure–Its interactions with macromolecules and possible functions. Biochemistry 73:1418–1437
Smith N, Matheson AT, Yaguchi M, Willick GE, Nazar RN (1978) The 5S rRNA—protein complex from an extreme halophile Halobacterium cutirubrum: purification and characterization. Eur J Biochem 89:501–509
Steel M, Penny D (2000) Parsimony, likelihood, and the role of models in molecular phylogenetics. Mol Biol Evol 17:839–850
Sun F-J, Caetano-Anollés G (2008a) The origin and evolution of tRNA inferred from phylogenetic analysis of structure. J Mol Evol 66:21–35
Sun F-J, Caetano-Anollés G (2008b) Evolutionary patterns in the sequence and structure of transfer RNA: early origins of Archaea and viruses. PLoS Comput Biol 4:e1000018
Sun F-J, Caetano-Anollés G (2008c) Evolutionary patterns in the sequence and structure of transfer RNA: a window into early translation and the genetic code. PLoS ONE 3:e2799
Sun F-J, Fleurdépine S, Bousquet-Antonelli C, Caetano-Anollés G, Deragon J-M (2007) Common evolutionary trends for SINE RNA structures. Trends Genet 23:26–33
Sun F-J, Harish A, Caetano-Anollés G (2009) Phylogenetic utility of RNA structure: evolution’s arrow and emergence of early biochemistry and diversified life. In: Caetano-Anollés G (ed), Evolutionary genomics and systems biology, Wiley, Hoboken (in press)
Swofford DL (2003) PAUP*: phylogenetic analysis using parsimony (*and other methods), Version 4.0b10. Sinauer Associates, Sunderland
Szymanski M, Barciszewska MZ, Erdmann VA, Barciszewski J (2002) 5S ribosomal RNA database. Nucleic Acids Res 30:176–178
Szymanski M, Barciszewska MZ, Erdmann VA, Barciszewski J (2003) 5 S rRNA: structure and interactions. Biochem J 371:641–651
Tanaka T, Kikuchi Y (2001) Origin of the cloverleaf shape of transfer RNA—the double-hairpin model: implication for the role of tRNA intron and the long extra loop. Viva Origino 29:134–142
Teixido J, Altamura S, Londei P, Amils R (1989) Structural and functional exchangeability of 5S RNA species from the eubacterium E. coli and the thermoacidophilic archaebacterium Sulfolobus solfataricus. Nucleic Acids Res 17:845–851
Villanueva E, Luehrsen KR, Gibson J, Delihas N, Fox GE (1985) Phylogenetic origins of the plant mitochondrion based on a comparative analysis of 5S ribosomal RNA sequences. J Mol Evol 22:46–52
Wang M, Caetano-Anollés G (2006) Global phylogeny determined by the combination of protein domains in proteomes. Mol Biol Evol 23:2444–2454
Wang M, Caetano-Anollés G (2009) The evolutionary mechanics of domain organization in proteomes and the rise of modularity in the protein world. Structure 17:66–78
Wang M, Yafremava LS, Caetano-Anollés D, Mittenthal JE, Caetano-Anollés G (2007) Reductive evolution of architectural repertoires in proteomes and the birth of the tripartite world. Genome Res 17:1572–1585
Weiner AM, Maizels N (1987) tRNA-like structures tag the 3′ ends of genomic RNA molecules for replication: implications for the origin of protein synthesis. Proc Natl Acad Sci USA 84:7383–7387
Widmann J, Di Giulio M, Yarus M, Knight R (2005) tRNA creation by hairpin duplication. J Mol Evol 61:24–535
Woese CR (1969) The biological significance of the genetic code. Prog Mol Subcell Biol 1:5–46
Woese CR (1998) The universal ancestor. Proc Natl Acad Sci USA 95:6854–6859
Woese CR, Kandler O, Wheelis ML (1990) Towards a natural system of organisms: proposal for domains Archaea, Bacteria and Eucarya. Proc Natl Acad Sci USA 95:6854–6859
Wong JT-F, Chen J, Mat W-K, Ng S-K, Xue H (2007) Polyphasic evidence delineating the root of life and roots of biological domains. Gene 403:39–52
Wool IG (1986) Studies of the structure of eukaryotic (mammalian) ribosomes. In: Hardesty B, Kramer G (eds) Structure function and genetics of ribosomes. Springer, New York, pp 391–411
Xue H, Tong K-L, Marck C, Grosjean H, Wong JT-F (2003) Transfer RNA paralogs: evidence for genetic code-amino acid biosynthesis coevolution and an archaeal root of life. Gene 310:59–66
Xue H, Ng S-K, Tong K-L, Wong JT-F (2005) Congruence of evidence for a Methanopyrus-proximal root of life based on transfer RNA and aminoacyl-tRNA synthetase genes. Gene 360:120–130
Yonath A (2002) The search and its outcome: high-resolution structures of ribosomal particles from mesophilic, thermophilic, and halophilic bacteria at various functional states. Annu Rev Biophys Biomol Struct 31:257–273
Yusupov MM, Yusupov GZ, Baucom A, Lieberman K, Earnest TN, Cate JHD, Noller HF (2001) Crystal structure of the ribosome at 5.5 Å resolution. Science 292:883–896
Zhaxybayeva O, Lapierre P, Gogarten JP (2005) Ancient gene duplications and the root(s) of the tree of life. Protoplasma 227:53–64
Acknowledgments
We thank Ajith Harish for help with 3D mappings, Minglei Wang for calculating nd values, and Hee Shin Kim, Ajith Harish, Minglei Wang, Liudmila Yafremava, Kyung Mo Kim, and Jay Mittenthal for helpful discussions. This study was supported by National Science Foundation Grants MCB-0343126 and MCB-0749836, the Critical Research Initiative of the University of Illinois, and the United Soybean Board. Any opinions, findings, and conclusions and recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the funding agencies. Both authors designed and performed the experiments, analyzed the data, and wrote the article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Sun, FJ., Caetano-Anollés, G. The Evolutionary History of the Structure of 5S Ribosomal RNA. J Mol Evol 69, 430–443 (2009). https://doi.org/10.1007/s00239-009-9264-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-009-9264-z