
Abstract
Modulation of stem cell proliferation is a crucial aspect of neural developmental biology and regenerative medicine. To investigate the effect of optical stimulation on neural stem cell proliferation, cells transduced with channelrhodopsin-2 (ChR2) were used to analyze changes in cell proliferation and cell cycle distribution after light stimulation. Blue light significantly inhibited cell proliferation and affected the cell cycle, which increased the percentage of cells in G1 phase and reduced the percentage in S phase. It is likely that the influence of blue light on cell proliferation and the cell cycle was mediated by membrane depolarization, which induced accumulation of p21 and p27 proteins. Our data provide additional specific evidence that membrane depolarization may inhibit neural stem cell proliferation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Optogenetics is a rapidly evolving field of technology that allows optical control of genetically targeted biological systems at high temporal and spatial resolution (Miesenbock 2011; Rein and Deussing 2011; Yawo et al. 2013). Although optogenetics was originally applied to manipulate neuronal activity by controlling ion flux, it is also widely applicable to other types of cells, such as stem cells (Stroh et al. 2011; Toettcher et al. 2011; Tonnesen et al. 2011; Weick et al. 2010). Developments in optogenetics have recently been extended to stem cell research and regenerative medicine (Stroh et al. 2011; Weick et al. 2010). Stem cells can be photosensitive by genetic induction of channelrhodopsins (ChR). Channelrhodopsin-2 (ChR2) is a rapidly gated blue-light-sensitive cation channel suitable for noninvasive control of ion flux and alternating membrane potential (Nagel et al. 2003; Zhang et al. 2006).
Neural stem cells (NSCs) are multipotent, uncommitted cells with the ability to proliferate, self-renew, and differentiate into cells of all neural lineages (Flax et al. 1998). The development and maturation of neural cells are regulated by membrane ion channel activity (Spitzer 2006). Membrane potential oscillation is tightly correlated with cell proliferation-related events, such as DNA synthesis, and hyperpolarized membrane levels block cells residing in the G1 phase of the cell cycle from entering the S phase of DNA synthesis, thus inhibiting mitosis (Sundelacruz et al. 2009). Changes in ion channel activity have been detected in proliferating glial progenitors in the brain (Momose-Sato et al. 2007). Moreover, membrane depolarization or hyperpolarization has also been reported in modulating neural developmental processes (Deisseroth et al. 2004; Wang et al. 2003). Blockade of K+ channels may inhibit neural progenitor cell proliferation by regulating the accumulation of the cyclin-dependent kinase inhibitors, p27(Kip1) and p21(CIP1) (Ghiani et al. 1999; Knutson et al. 1997). In addition, transcriptional repression of Bmp2 by p21Waf1/Cip1 links quiescence of neural stem cell to stemness (Porlan et al. 2013). Here, we investigated the effect of optogenetic-induced ion flux change on the proliferation of neural stem cells and whether the optogenetic-induced ion flux modulates cell proliferation of NSCs through p21 and p27 pathways.
Materials and Methods
C17.2 Neural Stem Cell Culture
The C17.2 neural stem cell line was cultured in standard DMEM containing 10 % fetal calf serum (FCS), 5 % horse serum, and 2 mM glutamine as previously described (Snyder et al. 1992).
Transduction of Neural Stem Cells with ChR2
A lentivirus carrying the ChR2-green fluorescent protein (GFP) fusion gene under the control of the ubiquitin C (UbC) promoter was generated (Boyden et al. 2005). Viruses were concentrated via ultracentrifugation and re-dissolved in PBS at 1:1,000 of the original volume. C17.2 neural stem cell was transduced with lentivirus (MOI = 5) in the presence of 5 μg/ml polybrene followed by incubation for 24 hours. Transduction efficiency was evaluated via fluorescent microscopy 1 week after transduction. To obtain a highly and homogenously expressing ChR2 colony, cells were sorted using fluorescence-activated cell sorting (FACS), and a subpopulation consisting of the top 1 % of GFP-expressing cells was collected. One cell per well was seeded in a 96-well plate, and the single-cell-derived colony was collected, identified, and proliferated.
Immunostaining of Cultured Cells
Cells were fixed with 4 % paraformaldehyde for 30 minutes. The fixed cells were washed three times with PBS. The cells were then permeabilized and blocked (10 % bovine serum albumin (BSA)/0.3 %Triton/PBS) for 60 min and incubated in primary antibody solution at 4 °C overnight. Cells were washed three times and incubated with secondary antibody at room temperature for 1 hour. Cells were washed three times with PBS, and 4′,6-diamidino-2-phenylindole (DAPI) was added (1:100,000) at the final washing step. Coverslips were mounted using anti-quenching fluoromount. Primary antibodies included mouse anti-Ki67 (1:400; Chemicon, USA) and mouse anti-nestin (1:200; Chemicon, USA).
Optical Stimulation
Light stimulation was achieved using a custom 470 nm light-emitting diode (LED) device (Thorlabs, USA). For whole-cell patch-clamp recording, light was delivered using a standard 40× water immersion microscope objective and a digitally operated shutter. Light intensity was modulated using a potentiometer ranging from 0.1 to 10 mW/mm2. The inward current peak was calculated at different light intensities. For optogenetic stimulation of cultured ChR2-C17.2 neural stem cells, optical stimulation was conducted in the incubator using the LED light source. All regions of the culture plate were stimulated with blue light for 5 min per hour (10 Hz, 5 min/hour duration, and 10 mW/mm2 light intensity). At the end of each experiment, the coverslips were removed from the plates and immediately fixed with paraformaldehyde and stained for Ki67, and sister cells were prepared for cell counting. Additionally, we prepared cells exposed to different light intensities (low intensity of 0.1 mW, medium intensity of 1 mW, and high intensity of 10 mW) for western blot analysis.
Whole-Cell Patch-Clamp Recording
Cells were plated on glass coverslips in the dish. Cells were continuously perfused with external solution containing (in mM): 145 NaCl, 5 KCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose with pH 7.4 adjusted with NaOH and 295–320 mOsm in osmolarity. The membrane currents were recorded using an Axopatch 200B patch amplifier (Axon Instruments, USA) and digitized with a Digidata 1440 A/D converter and Clampex 10.0 software (Molecular Devices, USA). Recording pipettes were pulled from borosilicate glass tubing with a horizontal puller (P-97, Sutter Instruments, USA), and these pipettes typically had a resistance of 3.0–5.0 MΩ when filled with pipette solution containing (in mM) 140 KCl, 10 EGTA, 10 HEPES, and 5 MgATP, with pH 7.3 adjusted with KOH and 290–310 mOsm in osmolarity. After establishment of the whole-cell configuration, the capacitance compensation and series resistance compensation were adjusted before recording the membrane signals. Of the series resistance, 70–80 % was compensated electronically. Signals were filtered at 2 kHz and digitized at 10 kHz. A leak subtraction protocol was performed. Light was delivered via a standard 40× water immersion microscope objective, using a digitally operated shutter.
Cell Proliferation Assay and Viability Test
To perform proliferation assays, 0.5 × 104 cells were seeded into a single well of a 24-well plate. Neural stem cells were treated with blue light stimulation, and medium was changed daily. Cells in each well were collected at 48 h post-stimulation for cell counting. Cells treated without light were used as the control. Numbers of viable and nonviable cells were determined by the trypan blue exclusion assay.
Flow Cytometry
Cells were collected by trypsinization for subsequent cell cycle analysis. For analysis of DNA content, cells were fixed in 70 % ethanol, rehydrated in PBS, treated for 30 min with RNase A (1 mg/ml), and treated for 5 min with propidium iodide (PI; 1 mg/ml). Fluorescence intensity was determined by flow cytometry using a FACSCanto (BD Pharmingen, USA). Data acquisition was performed with CellQuest software (BD Pharmingen, USA), and the percentage of G0/G1, S, and G2/M phase cells was calculated with the MODFIT software program. To detect apoptosis, cells were stained and analyzed with PI- and APC-conjugated annexin V using an APC Annexin V Apoptosis kit (BD Biosciences, USA) according to the manufacturer’s protocol.
Protein Extraction and Western Blot Analysis
Cells were harvested and proteins were extracted using ice-cold RIPA. Protein aliquots were stored at −80 °C. For western blot analysis, proteins were separated in a 12 % SDS-polyacrylamide gel. After electrophoresis, the gel was electrophoretically transferred onto PVDF membranes (Millipore, USA) and blocked for 1 h at 37 °C with 5 % non-fat milk in Tris-buffered saline containing 0.1 % Tween-20 (TBST). Incubation with primary antibodies was performed at 4 °C overnight. Primary antibodies were used at the following dilutions: anti-p27 (Abcam, USA) at 1:2000, anti-p21 (Abcam, USA) at 1:2,000, and anti-GAPDH (Abcam, USA) at 1:10,000. After primary antibody probing, membranes were washed in TBST, and incubated with HRP-conjugated secondary antibody (Dako, Denmark) at 1:5,000 for 60 min at room temperature. After further washing, protein expression was detected by enhanced chemiluminescent (ECL) substrate (Pierce, Thermo Fisher Scientific, USA) and protein bands were visualized by film exposure. GAPDH was used as an internal control. The immunoreactive density was analyzed by Quantity One (Bio-Rad, USA).
Statistical Analysis
All quantitative values are presented as the mean ± SEM. Statistical comparisons were made using the unpaired Student’s t test with SPSS (version 15) software when appropriate. P < 0.05 was considered to be statistically significant.
Results
Functional Expression of ChR2 in C17.2 Neural Stem Cells
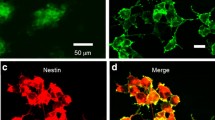
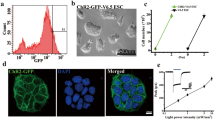
To assess the application potential of optogenetics in stem cells, C17.2 neural stem cells were infected with a ChR2-GFP-lentivirus under the control of the Ubiquitin C (UbC) promoter. One week post-transduction with lentivirus, 20 % of the C17.2 neural stem cells expressed ChR2-GFP, and after sorting for the top 1 % based on GFP fluorescence intensity (Fig. 1a), a single cell could form a neural stem cell clone (Fig. 1b). In addition, immunostaining showed that the C17.2 ChR2-neural stem cells expressed the neural stem cell marker, nestin (Fig. 1c). The C17.2 ChR2-neural stem cells showed typical inward photocurrents evoked by illumination with blue light under voltage clamp mode (470 nm with 200 ms pulse duration), and steady-state photocurrents showed little inactivation (Fig. 1d).

Functional expression of ChR2 in C17.2 neural stem cells. a A gate enclosing 1 % of cells with the strongest GFP fluorescence was applied for FACS sorting. b Micrograph of C17.2 ChR2-neural stem cell clone from one cell after sorting. c Fluorescent staining of cells expressing ChR2-GFP co-localized with nestin. d Electrophysiological recording of light (wave length of 473 nm) of various intensities stimulating evoked inward currents in C17.2 ChR2-neural stem cells in voltage clamp mode (light stimulation is indicated by the blue bar). Inset representative ChR2 photocurrents (Color figure online)
Optical Stimulation Inhibits C17.2 ChR2-Neural Stem Cell Proliferation
Subsequently, we tested if the blue light-induced membrane depolarization affected the proliferation of neural stem cells. Application of blue light stimulation caused a decrease in proliferation of C17.2 ChR2-neural stem cells according to cell counts at 48 h in comparison to the control (wt/no light) (Fig. 2a, b). We also found that the percentage of Ki67-positive cells in the light group was lower than the no light control group (Fig. 2c, d). The blue light stimulation had no obvious effect on wild-type C17.2 neural stem cell proliferation (Fig. 2a, b).

Optogenetic modulation of C17.2 ChR2-neural stem cell proliferation. a, b Quantitative analysis of the number of C17.2 ChR2-neural stem cells treated with blue light stimulation at 48 h. Optical stimulation significantly inhibited proliferation. The histograms were normalized revealing an optically driven decreased proliferation rate. Light stimulation had no effect on C17.2 neural stem cells. c, d Expression of Ki67 after a 48 h treatment of optical stimulation. Immunostaining images showed the downregulated expression of Ki67 in C17.2 ChR2-neural stem cells after treatment with blue light. Histogram chart showing the expression of Ki67 after stimulation. (n = 6; values are mean ± SEM. *P < 0.05 versus control group in this figure and the following figures) (Color figure online)
Optical Stimulation Does Not Affect the Survival of C17.2 ChR2-Neural Stem Cells
To further identify if blue light stimulation could affect survival of the C17.2 ChR2-neural stem cells, the percentage of viable cells was analyzed by the trypan blue exclusion assay. Light stimulation did not influence C17.2 ChR2-neural stem cell viability (Fig. 3c). We then used flow cytometry to assay the apoptosis rate between the two groups, and there was no significant difference between the two groups (Fig. 3a, b, d). These results indicated that the light-induced membrane depolarization may affect the signaling pathway involved in cell proliferation.

Effects of optical stimulation on the viability of C17.2 ChR2-neural stem cells. a, b Flow cytometric analysis showed apoptosis of neural stem cells in the light and no light groups. c Histogram showing that optical stimulation did not decrease the viability of cells when compared with that of the no light control. d Summary graphs showing that optical stimulation did not induce obvious apoptosis when compared with that of the no light control
Optical Stimulation Causes G1 Phase Accumulation and S Phase Reduction
The cell cycle distribution of C17.2 ChR2-neural stem cell after light stimulation is summarized in Fig. 4a. Application of light stimulation resulted in a significant reduction in the fraction of cells in S phase and accumulation of cells in G0/G1 phase. While the percentage of cells in G0/G1 phase in the no light control group was 38.57 ± 4 %, blue light stimulation increased the percentage to 52.69 ± 5 % (P < 0.05 vs. control). In addition, the proportion of cells in S phase decreased from 48.56 ± 3 % in control C17.2 ChR2-neural stem cells to 33.4 ± 4 % in blue light stimulation groups (P < 0.05 vs. control) (Fig. 4b).

Effect of optical stimulation on the cell cycle distribution of C17.2 ChR2-neural stem cells. a Representative raw traces of cell cycle analysis of different groups by the Modfit program. b Blue light treatment increased the percentage of cells in G0/G1 phase and decreased the percentage of cells in S phase (Color figure online)
Optical-Induced Membrane Depolarization Attributes to Cell Proliferation Inhibition
We next explored the mechanism of optical stimulation on neural stem cell proliferation. In the current clamp mode, the blue light evoked membrane depolarization of C17.2 ChR2-neural stem cells, because ions fluxed into the cells during light stimulation (Fig. 5a). We analyzed the expression level of p27Kip1 and p21CIP1, which are two members of the Kip/CIP family known to regulate cell proliferation and terminal differentiation in a variety of cell types. After 48 h of treatment, the blue light significantly increased both p27Kip1 and p21CIP1 expression levels in a dose-dependent manner as the light intensity increased (Fig. 5b), thereby suggesting that optical-induced membrane depolarization blocked neural stem cell proliferation by accumulation of cdkis, p27Kip1 and p21CIP1.

Optical-induced membrane depolarization attributes to C17.2 ChR2-neural stem cell proliferation inhibition. a Representative traces of membrane depolarization from current clamp mode recordings of C17.2 ChR2-neural stem cells during a 200 ms 470 nm blue light stimulus. b Western blot showing the expression of p21 and p27 in C17.2 ChR2-neural stem cells after different intensities of optical stimulation (low intensity of 0.1 mW, medium intensity of 1 mW, and high intensity of 10 mW). c, d Bar graph revealing light intensity-dependent upregulation of the expression of p21 and p27 after light stimulation (Color figure online)
Discussion
By using optogenetic techniques, we found an inhibitory effect of membrane depolarization on neural stem cell proliferation in the present study. As the field of optogenetics is emerging, it provides a direct and stringent means of probing the organization of neural circuits and of identifying the neural substrates of behavior. Moreover, optogenetics is widely applicable for other types of cells and biological systems (Miesenbock 2011).
Stem cells exhibit unique electrophysiological profiles in their undifferentiated state (Biagiotti et al. 2006; Cai et al. 2004; Cho et al. 2002; Heubach et al. 2004). Membrane ion channel activity is involved in neural development (Spitzer 2006). For instance, release of GABA from differentiating neuroblasts depolarizes their progenitor cells in the postnatal mouse subventricular zone and limits proliferation (Liu et al. 2005). Modulation of voltage-gated ion channels may modify the proliferative state of cortical oligodendrocyte progenitor cells (Ghiani et al. 1999). Proliferation of astrocytes and Schwann cells is also inhibited by membrane depolarization or by K+ channel blockers (Chiu 1991; Li et al. 2008; MacFarlane and Sontheimer 2000). Knutson et al. demonstrated that membrane depolarization induced by high extracellular K+ inhibits O-2A glial progenitor cell proliferation and that, this inhibitory effect is reversible (Ghiani et al. 1999). Retinoic acid (RA), which is an agent important for modulating embryonic neural development, may also inhibit neural precursor cell proliferation by inhibiting K+ channel function (Ghiani et al. 1999). Membrane depolarization could also influence the differentiation of NSCs as a recent study has suggested that prolonged membrane depolarization may enhance midbrain dopamine neuron differentiation (He et al. 2011). However, nearly all these studies have studied the effect of membrane depolarization on neural cell behavior by exerting high extracellular K+. Our data extend the previous analyses of cell membrane voltage to neural stem cells by using the new optogenetic technique. Neural stem cells or ESCs can be made photosensitive by genetic modification with ChRs and enabled to analyze proliferation or differentiate change by depolarization with rhythmic photo-stimulation (Stroh et al. 2011; Tonnesen et al. 2011; Weick et al. 2010). Optical stimulation-induced depolarization inhibited proliferation of neural stem cells in our study. Moreover, this effect was not due to a toxicity response because no apoptosis increase or viability change was observed. This effect may be due to the induction of cell cycle inhibitory proteins by light stimulation.
Changes of the extracellular environment may affect proliferation of most cell types (Roovers and Assoian 2000). Cell cycle progression is tightly regulated by classes of cyclin-dependent kinases (cdks) and cyclin-dependent kinase inhibitor (cdkis) families (Ross 1996). The cell cycle flow cytometric assay showed increased G1 phase cells and decreased S phase cells in our study, which, indicated that membrane depolarization causes a block in G1 phase of the neural stem cell cycle and prevents G1–S transition. However, the mechanism of the effect of membrane potential changes on cell cycle and proliferation is not fully demonstrated. This mechanism might involve the coordination of genes involved in cell cycle progression. Induction or failure to down-regulate p27Kip1 and p21Cip1, which are two members of the cdkis Kip/CIP family, has been repeatedly linked to inactivation of cyclin E-cdk2 and G1 phase arrest (Coqueret 2003; Fang et al. 1996; Lloyd et al. 1997; Sewing et al. 1997; Woods et al. 1997). Cell membrane depolarization induces G1 arrest in the OP cell cycle through accumulation of the cyclin-dependent kinase inhibitors, p27(Kip1) and p21(CIP1) (Ghiani et al. 1999). Thus, we analyzed the involvement of p27Kip1 and p21Cip1 in the G1 phase accumulation of the cell cycle, and we found an upregulation of these two proteins after optical stimulation. Interestingly, one study has found that hyperpolarization of the membrane potential and progression through G1 are functionally linked (Lau et al. 2011). Therefore, the dynamic changes of the membrane potential are involved in cell cycle progression, and if the balance is disturbed, the orderly cell cycle progression would be blocked.
In conclusion, by employing optogenetic techniques, we have provided more evidence that cell membrane potential plays an important role in the regulation of neural stem cell proliferation and cell cycle progression. Our data and other previous studies demonstrate that intracellular signal transduction pathways associated with the activation of different receptor systems and distinct membrane channels converge on two cdkis, namely, p27Kip1 and p21CIP1, which regulate proliferation and differentiation in a variety of cell lineages, including C17.2 neural stem cells.
References
Biagiotti T, D’Amico M, Marzi I, Di Gennaro P, Arcangeli A, Wanke E, Olivotto M (2006) Cell renewing in neuroblastoma: electrophysiological and immunocytochemical characterization of stem cells and derivatives. Stem Cells 24:443–453
Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K (2005) Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci 8:1263–1268
Cai J, Cheng A, Luo Y, Lu C, Mattson MP, Rao MS, Furukawa K (2004) Membrane properties of rat embryonic multipotent neural stem cells. J Neurochem 88:212–226
Chiu S (1991) Functions and distribution of voltage-gated sodium and potassium channels in mammalian Schwann cells. Glia 4:541–558
Cho T, Bae JH, Choi HB, Kim SS, McLarnon JG, Suh-Kim H, Kim SU, Min CK (2002) Human neural stem cells: electrophysiological properties of voltage-gated ion channels. Neuroreport 13:1447–1452
Coqueret O (2003) New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol 13:65–70
Deisseroth K, Singla S, Toda H, Monje M, Palmer TD, Malenka RC (2004) Excitation-neurogenesis coupling in adult neural stem/progenitor cells. Neuron 42:535–552
Fang F, Orend G, Watanabe N, Hunter T, Ruoslahti E (1996) Dependence of cyclin E-CDK2 kinase activity on cell anchorage. Science 271:499–502
Flax JD, Aurora S, Yang C, Simonin C, Wills AM, Billinghurst LL, Jendoubi M, Sidman RL, Wolfe JH, Kim SU (1998) Engraftable human neural stem cells respond to development cues, replace neurons, and express foreign genes. Nat Biotechnol 16:1033–1039
Ghiani CA, Yuan X, Eisen AM, Knutson PL, DePinho RA, McBain CJ, Gallo V (1999) Voltage-activated K+ channels and membrane depolarization regulate accumulation of the cyclin-dependent kinase inhibitors p27(Kip1) and p21(CIP1) in glial progenitor cells. J Neurosci 19:5380–5392
He XB, Yi SH, Rhee YH, Kim H, Han YM, Lee SH, Lee H, Park CH, Lee YS, Richardson E, Kim BW (2011) Prolonged membrane depolarization enhances midbrain dopamine neuron differentiation via epigenetic histone modifications. Stem Cells 29:1861–1873
Heubach JF, Graf EM, Leutheuser J, Bock M, Balana B, Zahanich I, Christ T, Boxberger S, Wettwer E, Ravens U (2004) Electrophysiological properties of human mesenchymal stem cells. J Physiol 554:659–672
Knutson P, Ghiani CA, Zhou JM, Gallo V, McBain CJ (1997) K+ channel expression and cell proliferation are regulated by intracellular sodium and membrane depolarization in oligodendrocyte progenitor cells. J Neurosci 17:2669–2682
Lau YT, Wong CK, Luo J, Leung LH, Tsang PF, Bian ZX, Tsang SY (2011) Effects of hyperpolarization-activated cyclic nucleotide-gated (HCN) channel blockers on the proliferation and cell cycle progression of embryonic stem cells. Pflugers Arch 461:191–202
Li M, Wang B, Lin W (2008) Cl-channel blockers inhibit cell proliferation and arrest the cell cycle of human ovarian cancer cells. Eur J Gynaecol Oncol 29:267–271
Liu X, Wang Q, Haydar TF, Bordey A (2005) Nonsynaptic GABA signaling in postnatal subventricular zone controls proliferation of GFAP-expressing progenitors. Nat Neurosci 8:1179–1187
Lloyd AC, Obermüller F, Staddon S, Barth CF, McMahon M, Land H (1997) Cooperating oncogenes converge to regulate cyclin/cdk complexes. Genes Dev 11:663–677
MacFarlane SN, Sontheimer H (2000) Changes in ion channel expression accompany cell cycle progression of spinal cord astrocytes. Glia 30:39–48
Miesenbock G (2011) Optogenetic control of cells and circuits. Annu Rev Cell Dev Biol 27:731–758
Momose-Sato Y, Sato K, Kinoshita M (2007) Spontaneous depolarization waves of multiple origins in the embryonic rat CNS. Eur J Neurosci 25:929–944
Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P, Bamberg E (2003) Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci USA 100:13940–13945
Porlan E, Morante-Redolat JM, Marqués-Torrejón MÁ, Andreu-Agulló C, Carneiro C, Gómez-Ibarlucea E, Soto A, Vidal A, Ferrón SR, Fariñas I (2013) Transcriptional repression of Bmp2 by p21Waf1/Cip1 links quiescence to neural stem cell maintenance. Nat Neurosci 16:1567–1575
Rein ML, Deussing JM (2011) The optogenetic (r)evolution. Mol Genet Genom
Roovers K, Assoian RK (2000) Integrating the MAP kinase signal into the G 1 phase cell cycle machinery. Bioessays 22:818–826
Ross ME (1996) Cell division and the nervous system: regulating the cycle from neural differentiation to death. Trends Neurosci 19:62–68
Sewing A, Wiseman B, Lloyd AC, Land H (1997) High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1. Mol Cell Biol 17:5588–5597
Snyder EY, Deitcher DL, Walsh C, Arnold-Aldea S, Hartwieg EA, Cepko CL (1992) Multipotent neural cell lines can engraft and participate in development of mouse cerebellum. Cell 68:33–51
Spitzer NC (2006) Electrical activity in early neuronal development. Nature 444:707–712
Stroh A, Tsai HC, Wang LP, Zhang F, Kressel J, Aravanis A, Santhanam N, Deisseroth K, Konnerth A, Schneider MB (2011) Tracking stem cell differentiation in the setting of automated optogenetic stimulation. Stem Cells 29:78–88
Sundelacruz S, Levin M, Kaplan DL (2009) Role of membrane potential in the regulation of cell proliferation and differentiation. Stem Cell Rev 5:231–246
Toettcher JE, Voigt CA, Weiner OD, Lim WA (2011) The promise of optogenetics in cell biology: interrogating molecular circuits in space and time. Nat Methods 8:35–38
Tonnesen J, Parish CL, Sorensen AT, Andersson A, Lundberg C, Deisseroth K, Arenas E, Lindvall O, Kokaia M (2011) Functional integration of grafted neural stem cell-derived dopaminergic neurons monitored by optogenetics in an in vitro Parkinson model. PLoS One 6:e17560
Wang DD, Krueger DD, Bordey A (2003) GABA depolarizes neuronal progenitors of the postnatal subventricular zone via GABAA receptor activation. J Physiol 550:785–800
Weick JP, Johnson MA, Skroch SP, Williams JC, Deisseroth K, Zhang SC (2010) Functional control of transplantable human ESC-derived neurons via optogenetic targeting. Stem Cells 28:2008–2016
Woods D, Parry D, Cherwinski H, Bosch E, Lees E, McMahon M (1997) Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol Cell Biol 17:5598–5611
Yawo H, Asano T, Sakai S, Ishizuka T (2013) Optogenetic manipulation of neural and non-neural functions. Dev Growth Differ 55:474–490
Zhang F, Wang LP, Boyden ES, Deisseroth K (2006) Channelrhodopsin-2 and optical control of excitable cells. Nat Methods 3:785–792
Acknowledgments
This research was supported by the National Natural Science Foundation of China (NSFC 8113001) and (973 Project 2013CB967002).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Wang, S.J., Weng, C.H., Xu, H.W. et al. Effect of Optogenetic Stimulus on the Proliferation and Cell Cycle Progression of Neural Stem Cells. J Membrane Biol 247, 493–500 (2014). https://doi.org/10.1007/s00232-014-9659-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00232-014-9659-7