
Abstract
Apoptosis is characterized by the programmed activation of specific biochemical pathways leading to the organized demise of cells. To date, aspects of the intracellular signaling machinery involved in this phenomenon have been extensively dissected and characterized. However, recent studies have elucidated a novel role for changes in the intracellular milieu of the cells as important modulators of the cell death program. Specially, intracellular ionic homeostasis has been reported to be a determinant in both the activation and progression of the apoptotic cascade. Several apoptotic insults trigger specific changes in ionic gradients across the plasma membrane leading to depolarization of the plasma membrane potential (PMP). These changes lead to ionic imbalance early during apoptosis. Several studies have also suggested the activation and/or modulation of specific ionic transport mechanisms including ion channels, transporters and ATPases, as mediators of altered intracellular ionic homeostasis leading to PMP depolarization during apoptosis. However, the role of PMP depolarization and of the changes in ionic homeostasis during the progression of apoptosis are still unclear. This review summarizes the current knowledge regarding the causes and consequences of PMP depolarization during apoptosis. We also review the potential electrogenic ion transport mechanisms associated with this event, including the net influx/efflux of cations and anions. An understanding of these mechamisms could lead to the generation of new therapeutic approaches for a variety of diseases involving apoptosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Programmed cell death or apoptosis, is a genetically encoded pathway that enables cells to undergo a highly regulated and organized death in response to specific signals. Apoptosis is involved in several physiological and pathophysiological states of the organism. For example, apoptosis is critical for the maintenance of normal tissue homeostasis (cell number) and is involved in the removal of cells during tissue development and remodeling, as well as during cell senescence and organism aging. Moreover, apoptosis occurs as a consequence of distinct pathologies, and its deregulation can lead to autoimmune, cancer and neurodegenerative diseases. This process is a ubiquitous, evolutionary highly conserved, cell death program that requires the specific activation of several signaling cascades, which ultimately lead to distinct biochemical and morphological alterations in the cell. These changes occur in different and sequentially organized stages. Early stages of apoptosis are characterized by the activation of caspases and endonucleases, phosphatidylserine externalization, cell shrinkage and nuclei condensation. Advanced stages of the cell death program are typified by plasma membrane blebbing or apoptotic body formation, DNA degradation, and finally cell fragmentation (Bortner & Cidlowski, 2002; Green, 2003).
The signaling machinery involved in apoptosis has been extensively studied and characterized. Recent studies have demonstrated a role for changes in the intracellular milieu of cells as important modulators and/or regulators of the cell death program. Changes in the intracellular ionic homeostasis of apoptotic cells have now been reported to occur in a variety of experimental paradigms. Moreover, it has been demonstrated that changes in the intracellular ionic homeostasis are important regulators for the progression of apoptosis (Yu, Canzoniero & Choi, 2001; Rizzuto et al., 2003; Bortner & Cidlowski, 2004). Recently, plasma membrane potential (PMP) depolarization has been reported to occur during early stages of apoptosis, but its consequences on the progression of the cell death program are less understood.
Ionic Homeostasis and Generation of Plasma Membrane Potential (PMP) in Cells
Ionic homeostasis is probably the most ancient of all the homeostatic mechanisms implicated in the maintenance, not only of cell physiology, but also of normal organ and body functions. In biological systems ions are not uniformly distributed, thus, concentrations in one compartment differ from those in other compartments. Major ionic gradients across the plasma membrane include sodium (Na+), potassium (K+), calcium (Ca2+) and chloride (Cl−). In both prokaryotic and eukaryotic cells, lipid membranes function as permeability barriers selective to ions, providing a mechanism for the steady-state maintenance of highly asymmetric concentrations of the major cations and anions across both plasma membranes and intracellular organelles. Thus, if there were no maintenance mechanisms, the concentrations of these ions inside and outside the cells would equilibrate, with deleterious effects on the overall cell homeostasis.
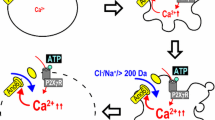
The maintenance of ionic concentration gradients involves both active and passive transport processes across the plasma membrane (Figure 1). Active transport of ions is mediated by proteins in the plasma membrane capable of pumping ions from one side of the membrane to the other against their concentration gradient. These mechanisms require energy from the hydrolysis of ATP (for the case of ATPases), or can use the driving force of another ion (coupled transporters). The Na+-K+ pump (or ATPase) is the primary ion transport mechanism involved in the maintenance of both Na+ and K+ concentration gradients across the plasma membrane. This pump is electrogenic and extrudes three Na+ for every two K+ pumped into the cell. Ion concentration gradients can also be maintained by their passive flux across the plasma membrane determined by the selective and distinct permeability of the membrane to various ions. The distinct permeability of the plasma membrane to different ions gives an electrical polarity across the lipid bilayer or membrane potential at rest that is present in all cell types. Most cell types have high resting permeability to K+, which together with the outwardly directed K+ gradient makes the interior of the cell electrically negative to the external solution. Thus in most cases PMP values are close to those of the actual Nernstian K+ potential. These permeability pathways are mediated by ion channels, and the movement of ions through these membrane pores does not require energy consumption but depends on the electrochemical gradient of each ion species (Wright, 2004).

Ionic homeostasis and generation of plasma membrane potential (PMP) in the cells. The electrical gradient in the cells or PMP at rest arises mainly from two physiological parameters: 1) the presence of large gradients for K+ and Na+ across the plasma membrane; and 2) the relative permeability of the plasma membrane to those ions. The maintenance of the gradient distribution of ions in the cells, and thus of the PMP, involves both active and passive transport mechanisms across the plasma membrane. From this, the Na+−K+ pump (or ATPase), which is the main ion transport mechanism involved in the maintenance of both Na+ and K+ concentration gradients, and the high permeability to K+ driven by the presence of background or leak conductances, mediated by the recently described K2P K+-channels (Kim, 2005), are the major determinants of PMP at rest (A). In a typical mammalian cell, major ionic gradients across the plasma membrane (B) are those of sodium (Na+), potassium (K+), calcium (Ca2+) and chloride (Cl-). Most biological membranes are, to varying degrees, permeable to K+ and Cl− ions. On the other hand, there is a reduced Na+ and Ca2+ permeability of most plasma membranes. The distinct permeability of the plasma membrane to different ions gives an electric polarity across the lipid bilayer. (C) Other transport mechanisms including ATPases (like the Ca2+-pump PMCA), transporters coupled to the driving force of another ion (like the Na+/Ca2+ exchanger NCX), the HCO3−Cl− exchanger, and the Na+−K+−Cl− and K+−CI− cotransporters) and ion channels (voltage-gated, ligan-gated or stress-activated ion channels) can also contribute to PMP at rest (by their tonic activity) or modulate it during different processes by their activation upon different stimuli. Due to the outwardly directed K+ and inwardly directed Na+ gradients maintained by the activity of the Na+−K+ ATPase, and high resting permeability to K+, the interior of the cell is electrically negative in relation to the external solution, thus in most cases PMP values are close to those of the actual Nernstian K+ potential (around ∼ −70 mV).
The movement of ions across the plasma membrane results in changes in electrical potential across the membrane. Such voltage changes have been reported to be primary signals that convey biological messages within the cell. Changes in the PMP occur during the normal physiology of the cells. Ion concentration gradients, ion transport activity and PMP reflect a triad whose regulation is critical for most homeostatic cellular functions. For example, the electrochemical gradient energy across the plasma membrane influences the transport of a vast array of nutrients in the cells and is the driving force in the movement of salt and water across cell membranes and between organ-based compartments. It is also essential in the signaling processes associated with coordinated movements of cells and organisms and is the basis of cognitive processes. In contrast, deregulated ionic imbalances are associated with pathological consequences, since ionic disturbances occur during apoptosis, ischemia and several channelopathies (Ronquist & Waldenstrom, 2003).
Changes in Cell Ionic Homeostasis and PMP During Apoptosis
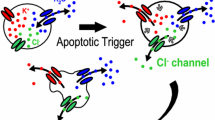
Changes in the PMP of the cells reflect movement of ions across the plasma membrane. Thus, the PMP is a consequence of the alteration in the distribution of ions. Alterations in the transmembrane gradients of several ions have been reported to influence the cell death program. Plasma membrane depolarization has been reported to occur in response to different apoptotic stimuli including receptor-induced, stress-induced and drug-induced apoptosis (Dallaporta et al., 1999; Detre et al., 2005; Bortner, Gomez-Angelats & Cidlowski, 2001; Mann et al., 2001; Mann & Cidlowski, 2001; Borzi et al., 2002; Dussmann et al., 2003b; Nolte et al., 2004; Scoltock & Cidlowski, 2004; Esteves et al., 2005). For example, increasing concentrations of Fas ligand result in concurrent increases in the number of cells that have a depolarized PMP (Fig. 2). Interestingly, a PMP hyperpolarization phenotype has been reported as one of the mechanisms by which Bcl-2 promotes apoptosis resistance (Gilbert et al., 1996; Williams et al., 2000); however, the role of PMP depolarization during apoptosis remains elusive. It is still unclear whether it is just an epiphenomenon associated with the ionic imbalance that occurs during apoptosis, or if there is a specific signaling role for the ionic changes associated with PMP depolarization during apoptosis. According to the electrochemical gradients across the plasma membrane of cells (Fig. 1), PMP depolarization can only arise from the net electrogenic inflow of Na+ and Ca2+ cations and / or the outflow of intracellular anions like Cl− or other intracellular organic anions (OA−). Plasma membrane potential depolarization during apoptosis has been associated with intracellular cation overload (Bortner et al., 2001; Mann et al., 2001; Nowak, 2002; Dussmann et al., 2003b; Nolte et al., 2004; Waring, 2005). This overload reflects an early rise in Na+ and Ca2+ that may account for the observed cellular depolarization (Fig. 3). Additionally, anion efflux has also been reported to mediate PMP depolarization during apoptosis (Nolte et al., 2004). Several reports have suggested the participation of a wide variety of ion transport proteins on the net intracellular cation overload and/or anion efflux associated with PMP depolarization during apoptosis, and are reviewed in the following section.

Plasma membrane potential (PMP) depolarization is an early hallmark during apoptosis. Changes in the PMP were measured by flow cytometry using DiBAC4(3). DiBAC4(3) is a negatively charged oxonal dye that freely crosses the plasma membrane. As the cell depolarizes more of this dye can enter the cell resulting in an increase in fluorescence. Fas ligand (FasL)-induced PMP depolarization was analyzed by fluorescence-activated cell sorting analysis, FACS, using a BD LSR II flow cytometer and BD FacsDiva Software (Becton Dickinson, San Diego, CA) for data analysis. Thirty minutes prior to each time of examination, Jurkat cells were preloaded with DiBAC4(3) at a final concentration of 150 nM, and the incubation was continued at 37°C, 7 % CO2 atmosphere. Immediately prior to flow cytometry examination, propidium iodide (PI) was added, and cells with increased PI fluorescence (i.e., loss of plasma membrane integrity) were discarded. Cells were analyzed at a cell density of 5 × 105 cells/ml, and in all cases, ten thousand cells were analyzed. DiBAC4(3) was excited using an Argon 488 laser and the fluorescence was detected with a 530/30 detector. For PI, cells were excited with an Argon 488 nm laser and emission was acquired with a 695/40 detector. A depolarized PMP is reflected by the appearance of a different population of cells with increased DiBAC4(3) (green) compared to control cells’ fluorescence (red). Frequency histograms of DiBAC4(3) fluorescence (upper panels) show that FasL induced the appearance of a population of cells with a depolarized PMP respective to control cells, in a concentration-dependent manner. We also analyzed the changes in DiBAC4(3) fluorescence in contour plots (lower panels) against changes in cell size. Cell size was determined as changes in the forward scatter pattern by exiting the cells with an Argon 488 nm laser. The forward-angle light scatter relates to cell diameter, i.e., cell shrinkage is reflected as a decrease in the amount of forward scatter light. We observed that the cells with a depolarized PMP (green) had a slight decrease in cell size reflected as a decrease in their forward scatter properties. The center of the quadrants is used as a reference for the mean DiBAC4(3) fluorescence and forward scatter signal of control cells.

Fas ligand (FasL)-induced apoptosis increases the intracellular concentration of sodium (Na+) and calcium (Ca2+) of cells. For the intracellular Na+ and Ca2+ measurements, cells were preloaded at 37°C, 7 % CO2 atmosphere with 5 μM (1 h) of SBFI-AM (Na+ fluorophore), or 2 μM (30 min) Fluo3-AM (Ca2+ fluorophore) prior to the time of examination. Fas ligand-induced changes in the intracellular concentration of these cations were analyzed by FACS. Immediately prior to flow cytometry examination, PI was added. For SBFI, cells were excited with a UV 350/360 laser and emission was acquired with a 440/40 detector. For Fluo3, cells were excited with an Argon 488 nm laser and emission was acquired with a 530/30 detector. Changes in the concentration of these cations are reflected by the appearance of different populations of cells with differences in SBFI or Fluo3 fluorescence, reflecting changes in the intracellular concentration of this ion. Frequency histograms of the fluorescence of these dyes show that an initial stage of FasL-induced apoptosis is characterized by an increase in the concentration of both Na+ and Ca2+ in the cells. This is reflected by the appearance of a population of cells with an increase in the concentration of these cations (blue population) compared to control cells (green population). A second population of cells (yellow) with a dramatic loss of intracellular cations, consists of cells in a late stage of the apoptotic program.
Ion Transport Pathways that Mediate Cation Overload During Apoptosis
PLASMA MEMBRANE ATPASES
The P-type ATPases comprise a nearly ubiquitous ion pump family with catalytic activities involved in diverse cellular homeostatic processes including the maintenance of osmotic balance and intracellular ionic composition (Apell, 2003). Two members of this group have been reported to be involved in the changes in intracellular cation overload that might be associated with PMP depolarization during apoptosis.
Plasma membrane Ca2+−ATPase (PMCA)
Plasma membrane Ca2+-ATPases are a subfamily of P-type ATPases, that extrude ionic Ca2+ across the plasma membrane against its electrochemical gradient (Lehotsky et al., 2002). Recent reports have shown a downregulation of the PMCA expression and activity during different pathological situations leading to apoptosis (Garcia et al., 2001). Furthermore, antisense-knockdown of the PMCA induces apoptosis in muscle cells (Sasamura et al., 2002). Overexpression of PMCA or its stimulation by growth factors has also been reported to protect cells from apoptosis (Garcia et al., 2001; Peluso, 2003). Interestingly, the PMCA is cleaved by caspase 3 during apoptosis, which impairs intracellular Ca2+ homeostasis and results in a further Ca2+ overload (Paszty et al., 2002; Schwab et al, 2002; Chami et al., 2003).
Sodium-Potassium Pump (Na+−K+ ATPase)
Alterations in the ionic gradient distribution of Na+ and K+ are early hallmarks of initial stages of the apoptotic program. Several reports have associated these phenomena with a reduction in the Na+−K+ ATPase activity during apoptosis (Tang, Cheng & Lin, 1996; Nobel et al., 2000; Bortner et al., 2001; Mann et al., 2001; Nowak, 2002; Orlov et al., 2003; Wang et al., 2003a, 2003b; Yu, 2003a; Arrebola et al., 2005). Accordingly, inhibition of the Na+/K+ ATPase with cardiac glycosides has been widely reported to induce PMP depolarization paralleled by cell toxicity and cell death (Chatterjee & Roy, 1965; Mason et al., 1971; Watabe et al., 1996; Olej et al., 1998; Kawazoe et al., 1999; Stelmashook et al., 1999; McConkey et al., 2000; Omar, Senatorov & Hu, 2000; Chueh et al., 2001; Kurosawa et al., 2001; Hennion et al, 2002; Schmiedt et al., 2002; Xiao et al., 2002a, 2002b; Huang et al., 2004; Esteves et al., 2005; Lang, Schulte & Schmiedt, 2005), or to enhance apoptosis induced by different stimuli in various model systems (Thevenod & Friedmann, 1999; Nobel et al., 2000; Penning et al., 2000; Verheye-Dua & Bohm, 2000; Bortner et al., 2001; Xiao et al., 2002a; Orlov et al., 2003; Esteves et al., 2005). Alterations in the expression and activity of the Na+-K+ ATPase have also been widely reported to occur during the pathogenesis of cardiovascular, neurological, renal and metabolic diseases, as well as during heavy metal-induced toxicity (Patrick & Hilton, 1979; Lees, 1991; Rose & Valdes, 1994; Chauhan, Lee & Siegel, 1997; Mishra & Delivoria-Papadopoulos, 1999; Thevenod & Friedmann, 1999; Ziegelhoffer et al., 2000; Kumar & Kurup, 2002; Rodrigo et al., 2002; Yu, 2003a; Cimen et al., 2004).
Recent reports have shown that degradation of the Na+-K+ ATPase occurs during apoptosis induced by death receptor activation (Bortner et al., 2001), glucocorticoids (Mann et al., 2001), and staurosporine (Dussmann et al., 2003a). Interestingly, apoptotic-resistant phenotypes of bcl-2–overexpressing cells (Gilbert et al., 1996; Gilbert & Knox, 1997), and of progesterone receptor-(Gonzalez Deniselle et al., 2002) and angiotensin receptor-mediated neuroprotection (Grammatopoulos et al., 2004), have been associated with an elevated Na+-K+ ATPase activity. The mechanisms and signals by which apoptosis modulates ATPase activity are still unclear. Since apoptosis is generally recognized to require intracellular ATP for its progression, increases in the intracellular ATP concentration occur under these circumstances (Eguchi et al., 1999; Nicotera & Melino, 2004; Zamaraeva et al., 2005). Thus, energy failure can likely be discarded as one of the mechanisms. Recent reports suggest the role of caspases (Mann et al., 2001; Dussmann et al., 2003a), protein kinases (Nowak, 2002; Wang & Yu, 2005), and reactive oxygen/nitrogen species (Chakraborti et al., 1998; Cimen et al., 2004; Rodrigo et al., 2002; Sen et al., 2004; Thevenod & Friedmann, 1999; Wang et al., 2003a) in the modulation of Na+-K+ ATPase expression and activity during apoptosis. But functional studies are needed to characterize the precise signaling pathways.
ION CHANNELS
Ion channels are integral membrane proteins that provide pores for the passive diffusion of ions across biological membranes that result in trans-membrane currents, thus being the main source of electrogenic ion flux pathways. They are normally classified depending on the ion species involved or according to their regulation of the gating or activation process. Ion channels participate in membrane potential maintenance, transduction of chemical signals to electric stimuli, and generation, coordination and propagation of electrical currents in excitable tissues. Other physiological functions ascribed to ion channels include the regulation of cell volume and pH, regulation of transepithelial transport of salt and water, hormone secretion and cell proliferation (Wehner et al., 2003; Wang, 2004). Recent studies have demonstrated the participation of several ion channels in apoptotic cell death (Lang et al., 2003a; Storey et al., 2003; Yu, 2003b; Okada et al., 2004; Wang, 2004; Remillard & Yuan, 2004). Additionally, deregulation of ion channel expression and function leads to diverse pathological conditions or channelopathies (Jentsch, Hubner & Fuhrmann, 2004; Waxman, 2001).
Voltage-gated Ion Channels
Voltage-gated Na+ channels (VGNC) have been widely studied in nerve and muscle cells where they mediate regenerative cell membrane depolarization and conduction of electrical signaling, and its deregulation has been associated with the appearance of hyperexcitability-derived diseases. They are also expressed in non-excitable cells including fibroblasts, osteoblasts, lymphocytes, glia, and metastatic cancer cells of epithelial origin, although their functions are less understood (Diss, Fraser & Djamgoz, 2004). The role of VGNC in apoptosis was suggested first by the observation that the VGNC activator, veratridine, induced apoptosis in neurons independently of the activation of voltage-gated Ca2+ channels (VGCC) (Dargent et al., 1996; Ulbricht, 1998; Koike & Ninomiya, 2000; Koike et al., 2000). These data implied a primary role of Na+ overload in the induction of apoptosis by PMP depolarization, although the mechanisms by which veratridine-induced Na+ overload induces apoptosis are still unclear. Other studies have also suggested a synergistic action of Ca2+ influx, reactive oxygen species generation, and p53 activation on veratridine-induced apoptosis that might further regulate the activation of the mitochondrial pathway of apoptosis and execution caspases (Callaway et al., 2001; Jordan et al., 2003; Banasiak, Burenkova & Haddad, 2004; Gomez-Lazaro et al., 2005). Several VGNC blockers have been reported to reduce brain damage and cell death during ischemia, hypoxia and traumatic brain injury (Taylor & Meldrum, 1995; Carter, 1998; Small, Morley & Buchan, 1999; Goldin, 2001; Banasiak et al., 2004) as well as to protect against apoptosis induced by ouabain (Xiao et al., 2002b) and death-receptor activation (Bortner & Cidlowski, 2003).
The voltage-gated family of Ca2+ channels (VGCC) is comprised of a large group of structurally related heterooligomers that couple cell excitability to intracellular signaling by permitting Ca2+ ions to enter thus producing transient intracellular Ca2+ signals (Miller, 2001). Thus, VGCC have also been implicated in initiating intracellular Ca2+-dependent events, such as contraction, secretion, synaptic transmission and gene expression. Voltage-gated Ca2+-channels have been shown to be activated during neurodegenerative disease- or aging- induced apoptosis (Mason et al., 1999; MacManus et al., 2000; Ho, Ortiz & Shea, 2001; Yagami et al., 2002, 2004; Otori et al., 2003; Ma et al., 2005), as well as during excitotoxicity- and ischemia-induced cell death (Kobayashi & Mori, 1998; Read, McCall & Gregg, 2002). Voltage-gated Ca2+ channel antagonists have been widely reported to prevent Ca2+ overload and apoptosis. Additionally, other types of apoptotic stimuli including cytokine withdrawal (Wang et al., 1999), lipid oxidation (Ares et al., 1997), cytotoxic agents (Kim et al., 2000a; Barone et al., 2004; Tanaka et al., 2004; Barone, Aguanno & D’Agostino, 2005) and phospholipase activation (Yagami et al., 2003b, 2003c, 2005) have also been shown to activate several types of VGCC whose inhibition protects from the progression of cell death. Apoptosis has also been correlated with the overexpression of the L-type VGCC alpha(1) subunits (Ba, Pang & Benishin, 2004; Grassi et al., 2004). The activation of VGCC during apoptosis has been associated with either changes in PMP, protein kinase activation or the generation of reactive oxygen species (MacManus et al., 2000; Cano-Abad et al., 2001; Yagami et al., 2003a; Ba et al., 2004; Grassi et al., 2004; Waring, 2005).
Non-selective Cation Channels
Non-selective cation channels are a diverse group of ion channels characterized by their relatively low selectivity between cation species. Their activity is modulated by various extracellular and intracellular signals. These nonselective cation channels are gated by diverse mechanisms, which can include voltage, cyclic nucleotides, ligands, reactive oxygen species and stretch. They contribute to depolarization of the membrane and in most cases to an increase in the intracellular Ca2+ concentration. The activation of NSCC in apoptosis has been widely reported in different cell types under various apoptotic stimuli (Gutierrez et al., 1999; Kim et al., 1999, 2006; Manion et al., 2000; Tapia-Vieyra & Mas-Oliva, 2001; Estacion & Schilling, 2002; Lang et al., 2003a,b,c; Jeulin, Dazy & Marano, 2002; Sook Han et al., 2003; Mukherjee et al., 2002; Sudhandiran & Shaha, 2003; Lang et al., 2004a,b; Lee, 2004; Mahta & Shaha, 2004). However, the molecular identities of the channels involved seem to vary in each case and usually according to the cell type studied.
The transient receptor potential (TRP) channels regulate the plasma membrane permeability of cells to a variety of ions in response to a wide diversity of stimuli. With the exception of the Ca2+-selective melastatin TRP channels (TRPM) and two members of the vanilloid receptor family of calcium-permeable channels (TRPV5 and TRPV6), which show a high selectivity for Ca2+, TRP channels are non-selective cation influx channels. TRP channels are activated by a variety of stimuli including intra- and extracellular ligands or signaling molecules, Ca2+-store depletion and mechanical or thermal stress. Thus, their activity has recently been shown to be involved in different physiological processes such as capacitative or store-operated Ca2+ entry (SOCE), sensory- and mechano-transduction (Voets & Nilius, 2003; Putney, 2005). Recent reports have also underlined the role of TRP channel activation in the induction of apoptosis. Activation of TRP channels in Drosophila leads to massive photoreceptor cell death (Yoon et al., 2000; Hong et al., 2002). Additionally, redox stress-induced and nicotinamide adenine dinucleotide-induced apoptosis have been suggested to depend on TRPM2 mediated Ca2+ influx (Sano et al., 2001; Hara et al., 2002; Zhang et al., 2003). Moreover, overexpression of TRPM7 in HEK293 has been reported to induce cell death (Yoon et al., 2000; Hong et al., 2002), and its activation by free radicals mediates neuronal death induced by oxygen-glucose deprivation (Aarts et al., 2003; Aarts & Tymianski, 2005). Apoptosis can also be induced by TRPV1 channel activation by particulate matter or capsaicin (Agopyan et al., 2003, 2004; Bodo et al., 2005).
Store-operated Ca2+ entry (SOCE) is a widespread phenomenon that represents the major mechanism of regulation of Ca2+ influx in non-excitable cells. Store-operated Ca2+ entry has been widely reported to induce apoptosis and has also been suggested to mediate glucocorticoid-induced cell death (Nam et al., 2003; Parekh & Putney, 2005), It has also been suggested that antiapoptotic effects of Bcl-2 are associated with the downregulation of SOCE channels, suggesting a pivotal role of SOCE on the progression of apoptosis (Vanden Abeele et al., 2002). A large number of studies have suggested the possibility that TRPC channels function as a source for SOCE (Parekh & Putney, 2005); however, this proposal has not been clearly established. SOCE associated with TRPC2 channel activation induces apoptosis triggered by the growth arrest and DNA damage-inducible gene (GADD153) overexpression, and acute K+ loss (Pigozzi et al., 2004). A recent study has shown that several genes involved in apoptosis are upregulated in cells with high levels of SOCE currents (Zagranichnaya et al., 2005).
Ion Transport Pathways that Mediate Anion Efflux During Apoptosis
Anion channels in the plasma membrane are permeable to anions such as iodide, bromide, nitrates, phosphates, and negatively charged amino acids. However, they are usually referred to as chloride (Cl−) channels since it is the most abundant anion and the predominant specie in all organisms. Chloride channel activity contributes to cell membrane potential, and maintains intracellular pH and cell volume. Chloride channels also play important roles in different physiological processes including epithelial transport and blood pressure regulation, muscle tone, cellular excitability, cell cycle and proliferation, and apoptosis (Okada et al., 2004). There are multiple families of chloride channels described by their electrophysiological and pharmacological properties (Nilius & Droogmans, 2003; Jentsch et al., 2005). Diverse apoptotic stimuli have been reported to activate Cl−-channels (Meng, Carruth & Weinman, 1997; Okada & Maeno, 2001; Porcelli et al., 2003; Dupere-Minier et al., 2004). In some cases, it has been suggested that Cl− channel modulation during apoptosis occurs by second messengers including intracellular Ca2+, ROS generation and kinases (Szabo et al., 1998; Nietsch et al., 2000; Kim, Kang & Lee, 2003; Shimizu, Numata & Okada, 2004). Moreover, Cl−-channel blockers have been widely reported to inhibit, with a distinct degree of potency and specificity, the progression of apoptosis (Fujita, Yanagisawa & Ishikawa, 1997; Szabo et al., 1998; Rasola et al., 1999; Maeno et al., 2000; Mizoguchi et al., 2002; Small, Tauskela & Xia, 2002; d’Anglemont de Tassigny et al., 2004; Myssina et al., 2004; Porcelli et al., 2004; Wei et al., 2004; Takahashi et al., 2005; Tanabe et al., 2005). Furthermore, media with reduced extracellular Cl− have also been shown to prevent the progression of apoptosis in a few model systems (Lang et al., 2004c; Tsukimoto et al., 2005). Electrophysiological studies have suggested that Cl−-channels activated during apoptosis have properties similar to the volume-regulated anion current (VRAC) that participates in volume recovery after cell swelling (Maeno et al., 2000; Souktani et al., 2000; d’Anglemont de Tassigny et al., 2004; Shimizu et al., 2004). However, the molecular identity of the Cl−-channels involved in the progression of apoptosis is still unclear. Recent reports have postulated the role of either the plasma membrane voltage-dependent anion channels (VDAC) (Elinder et al., 2005), the Ca2+-activated Cl−-channels (Schumann, Gardner & Raffin, 1993; Kim et al., 2003) or the voltage-gated Cl−-channels (Wei et al., 2004) as possible mechanisms of Cl− extrusion during apoptosis. Other reports have also suggested that Cl− efflux occurs during apoptosis associated with K+ loss and cell shrinkage or apoptotic volume decrease (AVD). According to the Cl− concentration distribution across the plasma membrane (Fig. 1), the opening of a passive Cl− flux pathway, like an anion channel, will drive an influx of Cl− down its electrochemical gradient. However, during AVD and cell volume regulation after cell swelling (RVD), the intracellular concentration of Cl− decreases (Arrebola et al., 2005; Zhou et al., 2005). Chloride loss in these cases has been postulated to be mediated by its extrusion coupled to the simultaneous loss of ionic K+ due to the pronounced voltage-mediated coupling between both K+ and Cl− conductance pathways, which should not contribute to a change in PMP (Wehner et al., 2003; Okada et al., 2004). Alternatively, it has been reported that during RVD an initial activation of K+ channels leads to a transient hyperpolarization, which then acts as the driving force for a sustained Cl− efflux and PMP depolarization (Jakab et al., 2002). However, this phenomenon has not been reported for AVD.
Role of Ion Movements Associated with PMP Depolarization in the Progression of Apoptosis
As discussed earlier, the ionic mediators of plasma membrane depolarization must involve either a net cation influx or a net anion efflux across the plasma membrane. In general, the effects of PMP depolarization on biological systems are associated with the changes in the intracellular concentration of the ionic species that mediate it. This is also the case for apoptosis, where the effects of PMP depolarization might be ion-species specific. This observation is supported by the fact that PMP depolarization under high extracellular K+ conditions is protective against apoptotic cell death (Chacon-Cruz et al., 1998; Lauritzen et al., 2003; Zhong et al., 2004; Johnson & D’Mello, 2005), although this effect has also been demonstrated to be related to the inhibition of K+ loss during apoptosis (Bortner, Hughes & Cidlowski, 1997; Yu et al., 1997; Thompson et al., 2001).
In contrast, PMP depolarization by either Na+ (Dargent et al., 1996; Jordan et al., 2000, 2002, 2003; Koike et al., 2000; Gomez-Lazaro et al., 2005; Banasiak et al., 2004), or Ca2+ ionophores (Gwag et al., 1999; Gil-Parrado et al., 2002; Zhu et al., 2002; Lang et al., 2003d) has been consistently reported to induce apoptosis.
Na+ OVERLOAD
In contrast to reports for Ca2+, evidence for a direct modulation of Na+ on enzyme activity or signaling protein function is scarce. However, there are data that suggest a direct role of Na+ ions on the regulation of apoptosis. For example, early phosphatidylserine exposure during apoptosis has been reported to be dependent on Na+, but not Ca2+ influx (Courageot et al., 2004). Sodium overload has also been demonstrated to modulate cytoskeleton organization (Chifflet et al., 2003, 2004) and activate the Rho-ROCK signaling pathways (Szaszi et al., 2005), which are necessary for apoptotic body formation (Croft et al., 2005).
An indirect effect of Na+ overload causing further ionic gradient imbalances during apoptosis might also modulate the activation of the apoptotic machinery. For example, sodium overload has been suggested to drive H+ entry via the Na+/H+ exchanger (Koike et al., 2000), which can further regulate the progression of apoptosis via acidification of the intracellular milieu. Additionally, intracellular Na+ increase has been shown to precede the activation of K+ efflux associated with cell shrinkage or apoptotic volume decrease (Bortner et al., 2001). A rise in the intracellular Na+ concentration has also been reported to directly activate G-protein–gated inwardly rectifying K+ channels but the role of these channels in apoptosis is unclear (Migheli et al., 1999). Sodium overload-induced plasma membrane depolarization has been reported to activate VGCC that mediates intracellular Ca2+ rise. Moreover, sodium overload can induce further intracellular Ca2+ increases by means of the activation of the Na+/Ca2+ exchanger which has been widely associated with apoptosis (Howes et al., 2003; Annunziato, Pignataro & Di Renzo, 2004; Elgel, Gursahani & Hadley, 2004). Finally, sodium overload has been recently reported to be necessary for cell shrinkage or AVD, but the mechanisms involved and implications are still elusive (Bortner & Cidlowski, 2003).
Ca2+ OVERLOAD
Ionic Ca2+ is a highly versatile intracellular signal that regulates numerous cellular processes. To date there are many examples describing how Ca2+ can directly regulate protein function by modulating either enzymatic activity or conformational changes (Berridge, Bootman & Roderick, 2003), and changes in intracellular Ca2+ clearly have a role in apoptotic cell death. Calcium overload has also been suggested to be the final common pathway of all types of cell death (Rizzuto et al., 2003). Intracellular Ca2+ increases have been suggested to act not only as apoptotic inducers, but also as regulators of the amplification loop of the death signal. In contrast, there is also evidence suggesting that apoptosis induced by different stimuli including death receptor activation, radiation and DNA damage is either Ca2+-independent (Jornot, Petersen & Junod, 1998; Rozental et al., 2004), or that it participates in only certain components of the cell death program (Scoltock et al., 2000).
Ionic Ca2+ has been reported to interact and modulate the apoptotic signaling machinery at different stages, and several studies have shown that cytosolic Ca2+ is elevated during both early and late stages of apoptosis (Tombal, Denmeade & Isaacs, 1999; Rizzuto et al., 2003). Cytosolic Ca2+ overload enhances mitochondrial Ca2+ uniport uptake that results in matrix swelling, mitochondrial depolarization and release of apoptogenic proteins. These effects have been suggested to be mediated by the direct opening of the permeability transition pore (PTP), the generation of ROS, cardiolipin peroxidation and activation of Ca2+-activated K+ channels (mitoKCa) (Hajnoczky, Davies & Madesh, 2003). Calcium overload may also activate apoptogenic effectors that control the cell death process independent from the mitochondria, by modulating the activity of kinases and phosphatases involved in apoptosis, such as calmodulin and calmodulin-dependent kinase II (Nutt et al., 2005; Wu et al., 2005). Furthermore, calcineurin, a Ca2+/calmodulin-dependent protein phosphatase has also been demonstrated to mediate the translocation of Bad (a Bcl-2 family member of the pro-apoptotic proteins) to the mitochondria. Another important mediator of Ca2+-dependent apoptosis is the family of Ca2+-dependent proteases, calpains. Calpains are cysteine proteases that act in a similar way as caspases and are activated from an inactive proenzyme by Ca2+-dependent autocatalytic cleavage. Calpains have been reported to mediate apoptosis by the further cleavage of various cellular apoptogenic proteins including caspases, calcineurin, Bcl-2 family members and X-linked inhibitors of apoptosis. Additional effectors of apoptosis mediated by cytosolic Ca2+-overload include DNases, nitric oxide synthases, phospholipases and transglutaminases (Orrenius, Zhivotovsky & Nicotera, 2003).
Cl−/ANION EFFLUX
Chloride/anion flux pathways have also been reported to modulate the progression of apoptosis. This has been studied primarily through the inhibitory effect of chloride channel blockers and reduced extracellular Cl− media on the apoptotic signaling cascade. However, the exact role of reduced intracellular Cl− concentration on the cell death program is far from being understood. Chloride/anion efflux is necessary for cell shrinkage or AVD during apoptosis (Szabo et al., 1998; Okada & Maeno, 2001; d’Anglemont de Tassigny et al., 2004; Okada et al., 2004; Porcelli et al.., 2004). However, the idea of AVD as a necessary signal for the activation of apoptosis (Orlov et al., 1996; Maeno et al., 2000; Friis et al., 2005) has been challenged by studies showing either the occurrence of apoptosis in the absence of cell shrinkage (Bortner & Cidlowski, 2003; Vereninov et al., 2004), or the inhibition of apoptosis by cell shrinkage (Gulbins et al., 1997; Uhlemann et al., 2000).
POSSIBLE rOLE OF VOLTAGE CHANGES
The role of PMP depolarization in the cell death program appears to depend on the ionic species implicated, however, one cannot also discard the existence of voltage-sensitive steps acting on the signaling cascade. Changes in membrane potential affect ion flux pathways with an intrinsic voltage sensor (ion channels), which alters intracellular chemical conditions and modulates a variety of biological processes. Evidence concerning the direct effect of the changes in PMP potential on enzyme activity or other signaling pathways is limited. A recent study has demonstrated the presence of an intrinsic voltage sensor in non-channel signaling proteins, which suggests that other enzymes, including apoptogenic signaling proteins, might also possess a similar mechanism (Murata et al., 2005). Apoptosis induced by different stimuli leads to the activation of voltage-activated ion channels, including VGCC and voltage-gated K+ channels that have been reported to be involved in the progression of apoptosis (Yu et al., 1997; Storey et al., 2003; Yu, 2003b; Remillard & Yuan, 2004; Wang, 2004). Electrical activity has also been demonstrated to regulate programmed cell death in neurons by activation of Na+ channels (Svoboda, Linares & Ribera, 2001). Other kinds of electric-mediated signaling have been described as well for ion channels functionally linked to membrane receptors involved in apoptosis (Arcangeli et al., 1993; Olivotto et al., 1996; Brassard et al., 1999; Lewis, Truong & Schwartz, 2002). Thus, it is plausible to hypothesize that other death receptors or membrane domains might employ similar mechanisms for signal transduction during apoptosis.
Concluding Remarks and Perspectives
Apoptosis is an evolutionarily conserved process involved in both physiological and pathophysiological phenomena. Despite a large number of studies dedicated to the elucidation of the signaling machinery involved in apoptosis, there are still many aspects of this process to be resolved. Changes in the intracellular milieu of the cells have been reported to be a determinant for the activation, modulation and progression of apoptotic cell death, and maintaining a normal ionic homeostasis may be an important inhibitory mechanism for apoptosis in cells. Ionic homeostasis regulation is a transcendental phenomenon for the normal physiology of all cell types, and accordingly, ionic homeostasis deregulation is a common hallmark of apoptosis. Particularly, PMP depolarization has been observed to be an initial feature of apoptosis associated with either cation overload or anion efflux; however, the interrelationship of these phenomena is still far from being understood. In this review, we have summarized the current knowledge and evidence about the role of electrogenic ion transport (including channels, transporters and ATPases) and PMP depolarization in apoptosis. Evidence suggests that direct PMP depolarization is able to trigger the cell death program in some cell types, as shown by the observation that direct activation of Na+ and Ca2+ ionophores induces apoptosis. These effects seem to be ion species- specific. Notwithstanding, and less studied is the possibility of intrinsic voltage-sensors in non-channel signaling proteins, as suggested by recent studies (Murata et al., 2005). In addition, the level of complexity is raised by the fact that there is evidence suggesting that not only changes in the concentration of ionic Ca2+, but also of Na+ and Cl−, might have direct effects on the signaling machinery of apoptosis. As summarized here, both cation overload and/or anion efflux associated with PMP depolarization can modulate or activate the apoptotic signaling machinery at different steps in the cell death program. However, current evidence is insufficient to make a clear synthesis of the pathways involved, and more studies are necessary to clarify the role of ionic imbalance and PMP depolarization in apoptosis.
Abbreviations
- AVD,:
-
apoptotic volume decrease;
- PMP,:
-
plasma membrane potential;
- RVD,:
-
regulatory volume decrease;
- VGNC,:
-
voltage-gated Na+ channel;
- VGCC, :
-
voltage-gated Ca2+ channels.
References
Aarts M., lihara K., Wei W.L., Xiong Z.G., Arundine M., Cerwinski W., MacDonald J.F., Tymianski M. 2003. A key role for TRPM7 channels in anoxic neuronal death. Cell. 115:863–877
Aarts, M.M., Tymianski, M. 2005. TRPMs and neuronal cell death. Pfluegers Arch. 451:243–249
Agopyan N., Bhatti T., Yu S., Simon S.A. 2003. Vanilloid receptor activation by 2- and 10-microm particles induces responses leading to apoptosis in human airway epithelial cells. Toxicol. Appl. Pharmacol. 192:21–35
Agopyan N., Head J., Yu S., Simon S.A. 2004. TRPV1 receptors mediate particulate matter-induced apoptosis. Am. Physiol. 286:L563–L572
Annunziato L., Pignataro G., Di Renzo G.F. 2004. Pharmacology of brain Na+/Ca2+ exchanger: from molecular biology to therapeutic perspectives. Pharmacol. Rev. 56:633–654
Apell H.J. 2003. Structure-function relationship in P-type ATPases—a biophysical approach. Rev. Physiol. Biochem. Pharmacol. 150:1–35
Arcangeli A., Becchetti A., Mannini A., Mugnai G., De Filippi P., Tarone G., Del Bene M.R., Barletta E., Wanke E., Olivotto M. 1993. Integrin-mediated neurite outgrowth in neuroblastoma cells depends on the activation of potassium channels. J. Cell Biol. 122:1131–1143
Ares M.P., Porn-Ares M.I., Thyberg J., Juntti-Berggren L., Berggren P.O., Diczfalusy U., Kallin B., Bjorkhem I., Orrenius S., Nilsson J. 1997. Ca2+ channel blockers verapamil and nifedipine inhibit apoptosis induced by 25-hydroxycholesterol in human aortic smooth muscle cells. J. Lipid Res. 38:2049–2061
Arrebola F., Zabiti S., Canizares F.J., Cubero M.A., Crespo P.V., Fernandez-Segura E. 2005. Changes in intracellular sodium, chlorine, and potassium concentrations in staurosporine-induced apoptosis. J. Cell Physiol. 204:500–507
Ba F., Pang P.K., Benishin C.G. 2004. The role of Ca2+ channel modulation in the neuroprotective actions of estrogen in beta-amyloid protein and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) cytotoxic models. Neurochem. Int. 45:31–38
Banasiak K.J., Burenkova O., Haddad G.G. 2004. Activation of voltage-sensitive sodium channels during oxygen deprivation leads to apoptotic neuronal death. Neuroscience. 126:31–44
Barone F., Aguanno S., D′Agostino A. 2005. Modulation of MAA-induced apoptosis in male germ cells: role of Sertoli cell P/Q-type calcium channels. Reprod. Biol. Endocrinol. 3:13
Barone F., Aguanno S., D′Alessio A., D′Agostino A. 2004. Sertoli cell modulates MAA-induced apoptosis of germ cells throughout voltage-operated calcium channels. FASEB J. 18:353–354
Berridge M.J., Bootman M.D., Roderick H.L. 2003. Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rey. Mol. Cell Biol. 4:517–529
Bodo E., Biro T., Telek A., Czifra G., Griger Z., Toth B.I., Mescalchin A., Ito T., Bettermann A., Kovacs L, Paus R. 2005. A hot new twist to hair biology: involvement of vanilloid receptor-1 (VR1/TRPV1) signaling in human hair growth control. Am. J. Pathol. 166:985–998
Bortner C.D., Cidlowski J.A. 2002. Apoptotic volume decrease and the incredible shrinking cell. Cell Death Differ. 9:1307–1310
Bortner C.D., Cidlowski J.A. 2003. Uncoupling cell shrinkage from apoptosis reveals that Na+ influx is required for volume loss during programmed cell death. J. Biol. Chem. 278:39176–39184
Bortner C.D., Cidlowski J.A. 2004. The role of apoptotic volume decrease and ionic homeostasis in the activation and repression of apoptosis. Pfluegers Arch. 448:313–318
Bortner C.D., Gomez-Angelats M., Cidlowski J.A. 2001. Plasma membrane depolarization without repolarization is an early molecular event in anti-Fas-induced apoptosis. J. Biol. Chem. 276:4304–4314
Bortner C.D., Hughes F.M., Jr., Cidlowski J.A. 1997. A primary role for K+ and Na+ efflux in the activation of apoptosis. J. Bio. Chem. 272:32436–32442
Borzi R.M., Mazzetti I., Magagnoli G., Paoletti S., Uguccioni M., Gatti R., Orlandini G., Cattini L., Facchini A. 2002. Growth-related oncogene alpha induction of apoptosis in osteoarthritis chondrocytes. Arthritis Rheum. 46:3201–3211
Brassard D.L., Maxwell E., Malkowski M., Nagabhushan T.L., Kumar C.C., Armstrong L. 1999. Integrin alpha(v)beta(3)-mediated activation of apoptosis. Exp. Cell Res. 251:33–45
Callaway J.K., Beart P.M., Jarrott B., Giardina S.F. 2001. Incorporation of sodium channel blocking and free radical scavenging activities into a single drug, AM-36, results in profound inhibition of neuronal apoptosis. Br. J. Pharmacol. 132:1691–1698
Cano-Abad M.F., Villarroya M., Garcia A.G., Gabilan N.H., Lopez M.G. 2001. Calcium entry through L-type calcium channels causes mitochondria disruption and chromaffin cell death. J. Biol. Chem. 276:39695–39704
Carter A.J. 19980. The importance of voltage-dependent sodium channels in cerebral ischaemia. Amino Acids 14:159–169
Chacon-Cruz E., Oelberg D.G., Davis P., Buescher E.S. 1998. Membrane depolarization and depletion of intracellular calcium stores are associated with delay of apoptosis in human neutrophils. J. Leukoc. Biol. 64:759–766
Chakraborti T., Ghosh S.K., Michael J.R., Batabyal S.K., Chakraborti S. 1998. Targets of oxidative stress in cardiovascular system. Mol. Cell Biochem. 187:1–10
Chami M., Ferrari D., Nicotera P., Paterlini-Brechot P., Rizzuto R. 2003. Caspase-dependent alterations of Ca2+ signaling in the induction of apoptosis by hepatitis B virus X protein. J. Biol. Chem. 278:31745–31755
Chatterjee M.L., Roy A.R. 1965. Toxic effects of ouabain on the isolated heart of reserpinised rabbit. Bull. Calcutta Sch. Trop. Med. 13:54–57
Chauhan N.B., Lee J.M., Siegel G.J. 1997. Na,K-ATPase mRNA levels and plaque load in Alzheimer’s disease. J. Mol. Neurosci. 9:151–166
Chifflet S., Correa V., Nin V., Justet C., Hernandez J.A. 2004. Effect of membrane potential depolarization on the organization of the actin cytoskeleton of eye epithelia. The role of adherens junctions. Exp. Eye Res. 79:769–777
Chifflet S., Hernandez J.A., Grasso S., Cirillo A. 2003. Nonspecific depolarization of the plasma membrane potential induces cytoskeletal modifications of bovine corneal endothelial cells in culture. Exp. Cell Res. 282:1–13
Chueh S.C., Guh J.H., Chen J., Lai M.K., Teng C.M. 2001. Dual effects of ouabain on the regulation of proliferation and apoptosis in human prostatic smooth muscle cells. dJJrol. 166:347–353
Cimen B., Turkozkan N., Seven I., Unlu A., Karasu C. 2004. Impaired Na+,K+- ATPase activity as a mechanism of reactive nitrogen species-induced cytotoxicity in guinea pig liver exposed to lipopolysaccharides. Mol. Cell. Biochem. 259:53–57
Courageot M.P., Lepine S., Hours M., Giraud F., Sulpice J.C. 2004. Involvement of sodium in early phosphatidylserine exposure and phospholipid scrambling induced by P2X7 purinoceptor activation in thymocytes. J. Biol. Chem. 279:21815–21823
Croft D.R., Coleman M.L., Li S., Robertson D., Sullivan T., Stewart C.L., Olson M.F. 2005. Actin-myosin-based contraction is responsible for apoptotic nuclear disintegration. J. Cell Biol. 168:245–255
Dallaporta B., Marchetti P., de Pablo MA, Maisse C., Due H.T., Metivier D., Zamzami N., Geuskens M., Kroemer G. 1999. Plasma membrane potential in thymocyte apoptosis. J. Immunol. 162:6534–6542
d’Anglemont de Tassigny A., Souktani R., Henry P., Ghaleh B., Berdeaux A. 2004. Volume-sensitive chloride channels (ICI,vol) mediate doxorubicin-induced apoptosis through apoptotic volume decrease in cardiomyocytes. Fundam. Clin. Pharmacol. 18:531–538
Dargent B., Arsac C., Tricaud N., Couraud F. 1996. Activation of voltage-dependent sodium channels in cultured cerebellar poffule cells induces neurotoxicity that is not mediated by glutamate release. Neuroscience 73:209–216
Detre, C., Kiss, E., Varga, Z., Ludanyi, K., Paszty, K., Enyedi, A., Kovesdi, D., Panyi, G., Rajnavolgyi, E., Matko, J. 2006. Death or survival: Membrane ceramide controls the fate and activation of antigen-specific T-cells depending on signal strength and duration. Cell Signal 18:294–306
Diss J.K., Fraser S.P., Djamgoz M.B. 2004. Voltage-gated Na+ channels: multiplicity of expression, plasticity, functional implications and pathophysiological aspects. Eur. Biophys. J. 33:180–193
Dupere-Minier G., Hamelin C., Desharnais P., Bernier J. 2004. Apoptotic volume decrease, pH acidification and chloride channel activation during apoptosis requires CD45 expression in HPB-ALL T cells. Apoptosis 9:543–551
Dussmann H., Kogel D., Rehm M., Prehn J.H. 2003a. Mitochondrial membrane permeabilization and superoxide production during apoptosis. A single-cell analysis. J. Biol. Chem. 278:12645–12649
Dussmann H., Rehm M., Kogel D., Prehn J.H. 2003b. Outer mitochondrial membrane permeabilization during apoptosis triggers caspase-independent mitochondrial and caspase-dependent plasma membrane potential depolarization: a single-cell analysis. J. Cell Sci. 116:525–536
Eguchi Y., Srinivasan A., Tomaselli K.J., Shimizu S., Tsujimoto Y. 1999. ATP-dependent steps in apoptotic signal transduction. Cancer Res. 59:2174–2181
Eigel B.N., Gursahani H., Hadley R.W. 2004. Na+/Ca2+ exchanger plays a key role in inducing apoptosis after hypoxia in cultured guinea pig ventricular myocytes. Am. J. Physiol 287:H1466–1475
Elinder F., Akanda N., Tofighi R., Shimizu S., Tsujimoto Y., Orrenius S., Ceccatelli S. 2005. Opening of plasma membrane voltage-dependent anion channels (VDAC) precedes caspase activation in neuronal apoptosis induced by toxic stimuli. Cell Death Differ. 12:1134–1140
Estacion M., Schilling W.P. 2002. Blockade of maitotoxin-induced oncotic cell death reveals zeiosis. BMC PhysioL 2:2
Esteves M.B., Marques-Santos L.F., Affonso-Mitidieri O.R., Rumjanek V.M. 2005. Ouabain exacerbates activation-induced cell death in human peripheral blood lymphocytes. An. Acad. BraS. Cienc. 77:281–292
Friis M.B., Friborg C.R., Schneider L., Nielsen M.B., Lambert I.H., Christensen ST., Hoffmann E.K. 2005. Cell shrinkage as a signal to apoptosis in NIH 3T3 fibroblasts. J. Physiol. 567:427–443
Fujita H., Yanagisawa A., Ishikawa K. 1997. Suppressive effect of a chloride bicarbonate exchanger inhibitor on staurosporine-induced apoptosis of endothelial cells. Heart Vessels. Suppl 12:84–88
Garcia M.L, Usachev Y.M., Thayer S.A., Strehler E.E., Windebank A.J. 2001. Plasma membrane calcium ATPase plays a role in reducing Ca2+ mediated cytotoxicity in PC12 cells. J. Neurosci Res. 64:661–669
Gilbert M., Knox S. 1997. Influence of Bcl-2 overexpression on Na+/K+- ATPase pump activity: correlation with radiation-induced programmed cell death. JCellPhysiol. 171:299–304
Gilbert M.S., Saad A.M., Rupnow B.A., Knox S.J. 1996. Association of BCL-2 with membrane hyperpolarization and radioresistance. J. Cell Physiol. 168:114–122
Gil-Parrado S., Fernandez-Montalvan A., Assfalg-Machleidt I., Popp O., Bestvater F., Holloschi A., Knoch T.A., Auerswald E.A., Welsh K., Reed J.C., Fritz H., Fuentes-Prior P., Spiess E., Salvesen G.S., Machleidt W. 2002. lonomycin-activated calpain triggers apoptosis. A probable role for Bcl-2 family members. J. Biol. Chem. 277:27217–27226
Goldin A.L. 2001. Resurgence of sodium channel research. AnnuRe&Physiol. 63:871–894
Gomez-Lazaro, M., Galindo, M.F., Fernandez-Gomez, F.J., Prehn, J.H., Jordan, J. 2005. Activation of p53 and the pro-apoptotic p53 target gene PUMA during depolarization-induced apoptosis of chromaffin cells. Exp. Neurol. 196:96–103
Gonzalez Deniselle M.C., Lopez Costa J.J., Gonzalez S.L., Labombarda F., Garay L., Guennoun R., Schumacher M., De Nicola A.F. 2002. Basis of progesterone protection in spinal cord neurodegeneration. J. Steroid Biochem. Mol. Biol. 83:199–209
Grammatopoulos T.N., Johnson V., Moore S.A., Andres R., Weyhenmeyer J.A. 2004. Angiotensin type 2 receptor neuroprotection against chemical hypoxia is dependent on the delayed rectifier K+ channel, Na+ Ca2+ exchanger and Na+K+ ATPase in primary cortical cultures. Neurosci. Res. 50:299–306
Grassi C., D′Ascenzo M., Torsello A., Martinotti G., Wolf F., Cittadini A., Azzena G.B. 2004. Effects of 50 Hz electromagnetic fields on voltage-gated Ca2+ channels and their role in modulation of neuroendocrine cell proliferation and death. Cell Calcium. 35:307–315
Green D.R. 2003. Overview: apoptotic signaling pathways in the immune system. Immunol Rev. 193:5–9
Gulbins E., Welsch J., Lepple-Wienhuis A., Heinle H., Lang F. 1997. Inhibition of Fas-induced apoptotic cell death by osmotic cell shrinkage. Biochem. Biophys. Res. Commun. 236:517–521
Gutierrez A.A., Arias J.M., Garcia L, Mas-Oliva J., Guerrero-Hernandez A. 1999, Activation of a Ca2+-permeable cation channel by two different inducers of apoptosis in a human prostatic cancer cell line. J. Physiol. 517:95–107
Gwag B.J., Canzoniero L.M., Sensi S.L., Demaro J.A., Koh J.Y., Goldberg M.P., Jacquin M., Choi D.W. 1999. Calcium ionophores can induce either apoptosis or necrosis in cultured cortical neurons. Neurosciencel 90:1339–1348
Hajnoczky G., Davies E., Madesh M. 2003. Calcium signaling and apoptosis. Biochem. Biophy. Rev. Commun. 304:445–454
Hara Y., Wakamori M., Ishii M., Maeno E., Nishida M., Yoshida T., Yamada H., Shimizu S., Mori E., Kudoh J., Shimizu N., Kurose H., Okada Y., Imoto K., Mori Y. 2002. LTRPC2 Ca+permeable channel activated by changes in redox status confers susceptibility to cell death. Mol. Cell. 9:163–173
Hennion J.P., el-Masri MA, Huff M.O., el-Mailakh R.S. 2002. Evaluation of neuroprotection by lithium and valproic acid against ouabain-induced cell damage. Bipolar Disord. 4:201–206
Ho R., Ortiz D., Shea T.B. 2001. Amyloid-beta promotes calcium influx and neurodegeneration via stimulation of L voltage-sensitive calcium channels rather than NMDA channels in cultured neurons. J. Alzheimers Dis. 3:479–483
Hong Y.S., Park S., Geng C., Baek K., Bowman J.D., Yoon J., Pak W.L. 2002. Single amino acid change in the fifth transmembrane segment of the TRP channel causes massive degeneration of photoreceptors. J. Biol. Chem. 277:33884–33889
Howes A.L., Arthur J.F., Zhang T., Miyamoto S., Adams J.W., Dorn I.G., Woodcock E.A., Brown J.H. 2003. Akt-mediated cardiomyocyte survival pathways are compromised by G alpha q-induced phosphoinositide 4,5-bisphosphate depletion. J BioLChem. 278:40343–40351
Huang Y.T., Chueh S.C., Teng C.M., Guh J.H. 2004. Investigation of ouabain-induced anticancer effect in human androgen-independent prostate cancer PC-3 cells. Biochem. Pharmacol. 67:727–733
Jakab M., Furst J., Gschwentner M., Botta G., Garavaglia M.L., Bazzini C., Rodighiero S., Meyer G., Eichmueller S., Woll E., Chwatal S., Ritter M., Paulmichl M. 2002. Mechanisms sensing and modulating signals arising from cell swelling. Cell Physiol Biochem. 12:235–258
Jentsch T.J., Hubner C.A., Fuhrmann J.C. 2004. Ion channels: function unravelled by dysfunction. Nat Cell BioL 6:1039–1047
Jentsch T.J., Poet M., Fuhrmann J.C., Zdebik A.A. 2005. Physiological functions of CLC Cl- channels gleaned from human genetic disease and mouse models. Annu. Rev. Physiol. 67:779–807
Jeulin C., Dazy A.C., Marano F. 2002. Effects of hydrogen peroxide and hydroxyl radicals on the cytosolic side of a non-selective cation channel in the cultured human bronchial epithelial cell line 16HBE14o. Pfluegers Arch. 443:574–583
Johnson K., D’Mello S.R. 2005. p21-Activated kinase-1 is necessary for depolarization-mediated neuronal survival. J. Neurosci. Res. 79:809–815
Jordan J., Galindo M.F., Calvo S., Gonzalez-Garcia C., Cena V. 2000. Veratridine induces apoptotic death in bovine chromaffin cells through superoxide production. Br. J. PharmacoL 137:1496–1504
Jordan J., Galindo M.F., Gonzalez-Garcia C., Cena V. 2003. Role and regulation of p53 in depolarization-induced neuronal death. Neuroscience 122:707–715
Jordan J., Galindo M.F., Tornero D., Benavides A., Gonzalez C., Agapito M.T., Gonzalez-Garcia C., Cena V. 2002. Superoxide anions mediate veratridine-induced cytochrome c release and caspase activity in bovine chromaffin cells. J. Pharmacol. 137:993–1000
Jornot L., Petersen H., Junod A.F. 1998. Hydrogen peroxide-induced DMA damage is independent of nuclear calcium but dependent on redox-active ions. Biochem. J. 335:85–94
Kawazoe N., Aiuchi T., Masuda Y., Nakajo S., Nakaya K. 1999. Induction of apoptosis by bufalin in human tumor cells is associated with a change of intracellular concentration of Na+ ions. J. Biochem. (Tokyo) 126:278–286
Kim A.H., Sheline C.T., Tian M., Higashi T., McMahon R.J., Cousins R.J., Choi D.W. 2000a. L-type Ca2+ channel-mediated Zn2+ toxicity and modulation by ZnT-1 in PC12 cells. Brain Res. 886:99–107
Kim D. 2005. Physiology and pharmacology of two-pore domain potassium channels. CurLPhanripes. 11:2717–2736
Kim J.A., Kang Y.S., Jung M.W., Kang G.H., Lee S.H., Lee Y.S. 2000b. Ca2+ influx mediates apoptosis induced by 4-aminopyridine, a K+ channel blocker, m HepG2 human hepatoblastoma cells. Pharmacology 60:74–81
Kim J.A., Kang Y.S., Jung M.W., Lee S.H., Lee Y.S. 1999. Involvement of Ca2+ influx in the mechanism of tamoxifen-induced apoptosis in HepG2 human hepatoblastoma cells. Cancer Lett 147:115–123
Kim J.A., Kang Y.S., Lee Y.S. 2003. Role of Ca2+ activated Cl− channels in the mechanism of apoptosis induced by cyclosporin A in a human hepatoma cell line. Biochem. Biophys. Res. Commun. 309:291–297
Kobayashi T., Mori Y. 1998. Ca2+ channel antagonists and neuroprotection from cerebral ischemia. Eur. J. Pharmacol. 363:1–15
Koike T., Ninomiya T. 2000. Alteration of veratridine neurotoxicity in sympathetic neurons during development in vitro. Neuroreport 11:151–155
Koike T., Tanaka S., Oda T., Ninomiya T. 2000. Sodium overload through voltage-dependent Na+ channels induces necrosis and apoptosis of rat superior cervical ganglion cells in vitro, Brain Res. Bull. 51:345–355
Kumar A.R., Kurup P.A. 2002. Inhibition of membrane Na+-K+ ATPase activity: a common pathway in central nervous system disorders. J. Assoc. Physicians India 50:400–406
Kurosawa M., Tani Y., Nishimura S., Numazawa S., Yoshida T. 2001. Distinct PKC isozymes regulate bufalin-induced differentiation and apoptosis in human monocytic cells. Am. J. Physiol. 280:C459–464
Lang F., Lang K.S., Wieder T., Myssina S., Birka C., Lang PA, Kaiser S., Kempe D., Duranton C., Huber S.M. 2003a. Cation channels, cell volume and the death of an erythrocyte. Pfluegers Arch. 447:121–125
Lang F., Lang P.A., Lang K.S., Brand V., Tanneur V., Duranton C., Wieder T., Huber S.M. 2004a. Channel-induced apoptosis of infected host cells-the case of malaria. Pfluegers Arch. 448:319–324
Lang H., Schulte B.A., Schmiedt R.A. 2005. Ouabain induces apoptotic cell death in type I spiral ganglion neurons, but not type II neurons. J. Assoc. Res. Otolaryngol. 6:63–74
Lang K.S., Duranton C., Poehlmann H., Myssina S., Bauer C., Lang F., Wieder T., Huber S.M. 2003b. Cation channels trigger apoptotic death of erythrocytes. Cell Death Differ. 10:249–256
Lang K.S., Myssina S., Brand V., Sandu C., Lang P.A., Berchtold S., Huber S.M., Lang F., Wieder T. 2004b. Involvement of ceramide in hyperosmotic shock-induced death of erythrocytes. Cell Death Differ. 11:231–243
Lang K.S., Myssina S., Lang P.A., Tanneur V., Kempe D.S., Mack A.F., Huber S.M., Wieder T., Lang F., Duranton C. 2004c. Inhibition of erythrocyte phosphatidylserine exposure by urea and Cl. Am. J. Physiol. 286:F1046–1053
Lang K.S., Myssina S., Tanneur V., Wieder T., Huber S.M., Lang F., Duranton C. 2003c. Inhibition of erythrocyte cation channels and apoptosis by ethylisopropylamiloride. Naunyn Schmiedebergs Arch. Pharmacol. 367:391–396
Lang PA., Kaiser S., Myssina S., Wieder T., Lang F., Huber S.M. 2003d. Role of Ca2+ activated K+ channels in human erythrocyte apoptosis. Am. J. Physiol. 285:C1553–1560
Lauritzen I., Zanzouri M., Honore E., Duprat F., Ehrengruber M.U., Lazdunski M., Patel A.J. 2003. K+ Independent cerebellar granule neuron apoptosis. Role of task leak K+ channels. J. Biol. Chem. 278:32068–32076
Lee Y.S. 2004. Mechanism of apoptosis induced by diazoxide, a K+ channel opener, in HepG2 human hepatoma cells. Arch. Pharm. Res. 27:305–313
Lees G.J. 1991. Inhibition of sodium-potassium-ATPase: a potentially ubiquitous mechanism contributing to central nervous system neuropathology. Brain Res. Rev. 16:283–300
Lehotsky J., Kaplan P., Murin R., Raeymaekers L. 2002. The role of plasma membrane Ca+ pumps (PMCAs) in pathologies of mammalians cells. Front. Biosci. 7:d53–84.
Lewis J.M., Truong T.N., Schwartz M.A. 2002. Integrins regulate the apoptotic response to DNA damage through modulation of p53. Proc. Natl. Acad. Sci. USA 99:3627–3632
Ma Z.G., Wang J., Jiang H., Xie J.X., Chen L. 2005. C31 enhances voltage-gated calcium channel currents in undifferentiated PC12 cells. Neurosci. Lett 382:102–105
MacManus A., Ramsden M., Murray M., Henderson Z., Pearson H.A., Campbell V.A. 2000. Enhancement of (45) Ca2+ influx and voltage-dependent Ca2+ channel activity by beta-amyloid-(1-40) in rat cortical synaptosomes and cultured cortical neurons. Modulation by the proinflammatory cytokine interleukin-1beta. J. Bio. Chem. 275:4713–4718
Maeno E., Ishizaki Y., Kanaseki T., Hazama A., Okada Y. 2000. Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc. Natl. Acad. Sci. USA 97:9487–9492
Manion M.K., Su Z., Villain M., Blalock J.E. 2000. A new type of Ca2+ channel blocker that targets Ca2+ sensors and prevents Ca2+ mediated apoptosis: Faseb J. 14:1297–1306
Mann C.L., Bortner C.D., Jewell C.M., Cidlowski J.A. 2001. Glucocorticoid-induced plasma membrane depolarization during thymocyte apoptosis: association with cell shrinkage and degradation of the Na+/K+-adenosine triphosphatase. Endocrinology 142: 5059–5068
Mann C.L., Cidlowski J.A. 2001. Glucocorticoids regulate plasma membrane potential during rat thymocyte apoptosis in vivo and in vitro. Endocrinology 142:421–429
Mason D.T., Zelis R., Lee G., Hughes J.L., Spann J.F., Jr., Amsterdam E.A. 1971. Current concepts and treatment of digitalis toxicity. Am. J. Cardiol. 27:546–559
Mason R.P., Leeds P.R., Jacob R.F., Hough C.J., Zhang K.G., Mason P.E., Chuang D.M. 1999. Inhibition of excessive neuronal apoptosis by the calcium antagonist amlodipine and antioxidants in cerebellar granule cells. J. Neurochem. 72:1448–1456
McConkey D.J., Lin Y., Nutt L.K., Ozel H.Z., Newman R.A. 2000. Cardiac glycosides stimulate Ca2+ increases and apoptosis in androgen-independent, metastatic human prostate adenocarcinoma cells. Cancer Res. 60:3807–3812
Mehta A., Shaha C. 2004. Apoptotic death in Leishmania donovani promastigotes in response to respiratory chain inhibition: complex II inhibition results in increased pentamidine cytotoxicity. J. Biol. Chem. 279:11798–11813
Meng X.J., Carruth M.W., Weinman S. 1997. Leukotriene D4 activates a chloride conductance in hepatocytes from lipopolysaccharide-treated rats. J. Clin. Invest. 99:2915–2922
Migheli A., Piva R., Casolino S., Atzori C., Dlouhy S.R., Ghetti B. 1999. A cell cycle alteration precedes apoptosis of granule cell precursors in the weaver mouse cerebellum. Am. J. Pathol. 155:365–373
Miller R.J. 2001. Rocking and rolling with Ca2+ channels. Trends Neurosci. 24:445–449
Mishra O.P., Delivoria-Papadopoulos M. 1999. Cellular mechanisms of hypoxic injury in the developing brain. Brain Res. Bull. 48:233–238
Mizoguchi K., Maeta H., Yamamoto A., Oe M., Kosaka H. 2002. Amelioration of myocardial global ischemia/reperfusion injury with volume-regulatory chloride channel inhibitors in vivo. Transplantation 73:1185–1193
Mukherjee S.B., Das M., Sudhandiran G., Shaha C. 2002. Increase in cytosolic Ca2+ levels through the activation of non-selective cation channels induced by oxidative stress causes mitochondrial depolarization leading to apoptosis-like death in Leishmania donovani promastigotes. J. Biol. Chem. 277:24717–24727
Murata Y., Iwasaki H., Sasaki M., Inaba K., Okamura Y. 2005. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature. 435:1239–1243
Myssina S., Lang P.A., Kempe D.S., Kaiser S., Huber S.M., Wieder T., Lang F. 2004. Clythannel blockers NPPB and niflumic acid blunt Ca2+-induced erythrocyte ‘apoptosis’. Cell Physiol. Biochem. 14:241–248
Nam J.H., Yoon S.S., Kirn T.J., Uhm D.Y., Kirn S.J. 2003. Slow and persistent increase of [Ca2+]c in response to ligation of surface IgM in WEHI-231 cells. FEBSLett 535:113–118
Nicotera P., Melino G. 2004. Regulation of the apoptosis-necrosis switch. Oncogene, 23:2757–2765
Nietsch H.H., Roe M.W., Fiekers J.F., Moore A.L., Lidofsky S.D. 2000. Activation of potassium and chloride channels by tumor necrosis factor alpha. Role in liver cell death. J. Biol. Chem. 275:20556–20561
Nilius B., Droogmans G. 2003. Amazing chloride channels: an overview. Acta Physiol. Scand 177:119–147
Nobel C.S., Aronson J.K., Dobbelsteen D.J., Slater A.F. 2000. Inhibition of Na+/K+-ATPase may be one mechanism contributing to potassium efflux and cell shrinkage in CD95-induced apoptosis. Apoptosis 5:153–163
Nolte F., Friedrich O., Rojewski M., Fink R.H., Schrezenmeier H., Korper S. 2004. Depolarisation of the plasma membrane in the arsenic trioxide (As2O3)-and anti-CD95-induced apoptosis in myeloid cells. FEBS Lett. 578:85–89
Nowak G. 2002. Protein kinase C-alpha and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells. J. Biol. Chem. 277:43377–43388
Nutt L.K., Margolis S.S., Jensen M., Herman C.E., Dunphy W.G., Rathmell J.C., Kornbluth S. 2005. Metabolic regulation of oocyte cell death through the CaMKII-mediated phosphorylation of caspase-2. Cell 123:89–103
Okada Y., Maeno E. 2001. Apoptosis, cell volume regulation and volume-regulatory chloride channels. Comp Biochem Physiol A Mol Integr Physiol. 130:377–383
Okada Y., Maeno E., Shimizu T., Manabe K., Mori S., Nabekura T. 2004. Dual roles of plasmalemmal chloride channels in induction of cell death. Pfluegers Arch. 448:287–295
Olej B., dos Santos N.F., Leal L, Rumjanek V.M. 1998. Ouabain induces apoptosis on PHA-activated lymphocytes. Biosci Rep. 18:1–7
Olivotto M., Arcangeli A., Carla M., Wanke E. 1996. Electric fields at the plasma membrane level: a neglected element in the mechanisms of cell signalling. Bioessays. 18:495–504
Omar A.I., Senatorov V.V., Hu B. 2000. Ethidium bromide staining reveals rapid cell dispersion in the rat dentate gyrus following ouabain-induced injury. Neuroscience. 95:73–80
Orlov S.N., Dam T.V., Tremblay J., Hamet P. 1996. Apoptosis in vascular smooth muscle cells: role of cell shrinkage. Biochem. Biophys. Res. Commun. 221:708–715
Orlov S.N., Pchejetski D.V., Sarkissian S.D., Adarichev V., Taurin S., Pshezhetsky A.V., Tremblay J., Maximov G.V., deBlois D., Bennett M.R., Hamet P., 2003. [3H]-thymidine labelling of DMA triggers apoptosis potentiated by E1 A-adenoviral protein. Apoptosis. 8:199–208
Orrenius S., Zhivotovsky B., Nicotera P. 2003. Regulation of cell death: the calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 4:552–565
Otori Y, Kusaka S., Kawasaki A., Morimura H., Miki A., Tano Y. 2003. Protective effect of nilvadipine against glutamate neurotoxicity in purified retinal ganglion cells. Brain Res. 961:213–219
Parekh A.B., Putney J.W., Jr. 2005. Store-operated calcium channels. Physiol Rev. 85:757–810
Paszty K., Verma A.K., Padanyi R., Filoteo A.G., Penniston J.T., Enyedi A. 2002. Plasma membrane Ca2+ ATPase isoform 4b is cleaved and activated by caspase-3 during the early phase of apoptosis. J. Biol. Chem. 277:6822–6829
Patrick J., Hilton P.J. 1979. Characterization of sodium-transport disorders in disease: different effects upon sodium and potassium of changes in the sodium pump and in membrane permeability. Clin. Sci. (Lond). 57:289–293
Peluso J.J. 2003. Basic fibroblast growth factor (bFGF) regulation of the plasma membrane calcium ATPase (PMCA) as part of an anti-apoptotic mechanism of action. Biochem. Pharmacol. 66:1363–1369
Penning L.C., Denecker G., Vercammen D., Declercq W., Schipper R.G., Vandenabeele P. 2000. A role for potassium in TNF-induced apoptosis and gene-induction in human and rodent tumour cell lines. Cytokine 12:747–750
Pigozzi D., Tombal B., Ducret T., Vacher P., Gailly P. 2004. Role of store-dependent influx of Ca2+ and efflux of K+ in apoptosis of CHO cells. Cell Calcium 36:421–430
Porcelli A.M., Ghelli A., Zanna C., Valente P., Ferroni S., Rugolo M. 2003. Staurosporine induces apoptotic volume decrease (AVD) in ECV304 cells. Am. N. Y. Acad. Sci. 1010:342–346
Porcelli A.M., Ghelli A., Zanna C., Valente P., Ferroni S., Rugolo M. 2004. Apoptosis induced by Staurosporine in ECV304 cells requires cell shrinkage and upregulation of Cl- conductance. Cell Death Differ. 11:655–662
Putney J.W. 2005. Physiological mechanisms of TRPC activation. Pflugers Arch. 451:29–34
Rasola A., Farahi Far D., Hofman P., Rossi B. 1999. Lack of internucleosomal DMA fragmentation is related to Cl− efflux impairment in hematopoietic cell apoptosis. FASEB. 13:1711–1723
Read D.S., McCall M.A., Gregg R.G. 2002. Absence of voltage-dependent calcium channels delays photoreceptor degeneration in rd mice. Exp Eye Res. 75:415–420
Remillard C.V., Yuan J.X. 2004. Activation of K+ channels: an essential pathway in programmed cell death. Am. J. Physiol. 286:L49–67
Rizzuto R., Pinton P., Ferrari D., Chami M., Szabadkai G., Magalhaes P.J., Di Virgilio F., Pozzan T. 2003. Calcium and apoptosis: facts and hypotheses. Oncogenel 22:8619–8627
Rodrigo R., Trujillo S., Bosco C., Orellana M., Thielemann L., Araya J. 2002. Changes in (Na + K)-adenosine triphosphatase activity and ultrastructure of lung and kidney associated with oxidative stress induced by acute ethanol intoxication. Chest 121:589–596
Ronquist G., Waldenstrom A. 2003. Imbalance of plasma membrane ion leak and pump relationship as a new aetiological basis of certain disease states. J. Intern. Med. 254:517–526
Rose A.M., Valdes R., Jr. 1994. Understanding the sodium pump and its relevance to disease, Clin. Chem. 40:1674–1685
Rozental R., Faharani R., Yu Y., Johnson J.M., Chan S.O., Chiu F.C. 2004. Sodium butyrate induces apoptosis in MSN neuroblastoma cells in a calcium independent pathway. Neurochem. Res. 29:2125–2134
Sano Y., Inamura K., Miyake A., Mochizuki S., Yokoi H., Matsushime H., Furuichi K. 2001. Immunocyte Ca2+ influx system mediated by LTRPC2. Science. 293:1327–1330
Sasamura S., Furukawa K., Shiratori M., Motomura S., Ohizumi Y. 2002. Antisense-inhibition of plasma membrane Ca2+ pump induces apoptosis in vascular smooth muscle cells. Jpn. J. Pharmacol. 90:164–172
Schmiedt R.A., Okamura H.O., Lang H., Schulte B.A. 2002. Ouabain application to the round window of the gerbil cochlea: a model of auditory neuropathy and apoptosis. J. Assoc. Res. Otolaryngol. 3:223–233
Schumann M.A., Gardner P., Raffin T.A. 1993. Recombinant human tumor necrosis factor alpha induces calcium oscillation and calcium-activated chloride current in human neutrophils. The role of calcium/calmodulin-dependent protein kinase. J. Biol. Chem. 268:2134–2140
Schwab B.L, Guerini D., Didszun C., Bano D., Ferrando-May E., Fava E., Tarn J., Xu D., Xanthoudakis S., Nicholson D.W., Carafoli E., Nicotera P. 2002. Cleavage of plasma membrane calcium pumps by caspases: a link between apoptosis and necrosis. Cell Death Differ. 9:818–831
Scoltock A.B., Bortner C.D., St J.B.G., Putney J.W., Jr., Cidlowski J.A. 2000, A selective requirement for elevated calcium in DMA degradation, but not early events in anti-Fas-induced apoptosis, J. Biol. Chem. 275:30586–30596
Scoltock A.B., Cidlowski J.A.. 2004. Activation of intrinsic and extrinsic pathways in apoptotic signaling during UV-C-induced death of Jurkat cells: the role of caspase inhibition. Exp. Cell Res. 297:212–223
Sen N., Das B.B., Ganguly A., Mukherjee T., Bandyopadhyay S., Majumder H.K. 2004. Camptothecin-induced imbalance in intracellular cation homeostasis regulates programmed cell death in unicellular hemoflagellate Leishmania donovani. J. Biol. Chem. 279:52366–52375
Shimizu T., Numata T., Okada Y. 2004. A role of reactive oxygen species in apoptotic activation of volume-sensitive Cl(-) channel. Proc. Natl. Acad. Sci USA 101:6770–6773
Small D.L, Morley P., Buchan A.M. 1999. Biology of ischemic cerebral cell death. Prog. Cardiovasc. Dis. 42:185–207
Small D.L., Tauskela J., Xia Z. 2002. Role for chloride but not potassium channels in apoptosis in primary rat cortical cultures. Neurosci. Lett. 334:95–98
Sook Han M., Shin K.J., Kim Y.H, Kim S.H, Lee T., Kim E., Ho Ryu S., Suh P.G. 2003. Thiram and ziram stimulate non-selective cation channel and induce apoptosis in PC12 cells. Neurotoxicology. 24:425–434
Souktani R., Berdeaux A., Ghaleh B., Giudicelli J.F., Guize L., Le Heuzey J.Y., Henry P. 2000. Induction of apoptosis using sphingolipids activates a chloride current in Xenopus laevis oocytes. Am. J. Physiol. 279:C158–165
Stelmashook E.V., Weih M., Zorov D., Victorov I., DirnagI U., Isaev N. 1999. Short-term block of Na+/K+-ATPase in neuro-glial cell cultures of cerebellum induces glutamate dependent damage of granule cells. FEBS Lett 456:41–44
Storey N.M., Gomez-Angelats M., Bortner C.D., Armstrong D.L., Cidlowski J.A. 2003. Stimulation of Kv1.3 potassium channels by death receptors during apoptosis in Jurkat T lymphocytes. J. Biol. Chem. 278:33319–33326
Sudhandiran G., Shaha C. 2003. Antimonial-induced increase in intracellular Ca2+ through non-selective cation channels in the host and the parasite is responsible for apoptosis of intracellular Leishmania donovani amastigotes. J. Biol. Chem. 278:25120–25132
Svoboda K.R., Linares A.E., Ribera A.B. 2001. Activity regulates programmed cell death of zebrafish Rohon-Beard neurons. Development 128:3511–3520
Szabo I., Lepple-Wienhues A., Kaba K.N., Zoratti M., Gulbins E., Lang F. 1998. Tyrosine kinase-dependent activation of a chloride channel in CD95-induced apoptosis in T lymphocytes. Proc. Natl. Acad. Sci. USA 95:6169–6174
Szaszi K., Sirokmany G., Di Ciano-Oliveira C., Rotstein O.D., Kapus A. 2005. Depolarization induces Rho-Rho kinase-mediated myosin light chain phosphorylation in kidney tubular cells. Am. J. Physiol. 289:C673–685
Takahashi N., Wang X., Tanabe S., Uramoto H., Jishage K., Uchida S., Sasaki S., Okada Y. 2005. CIC-3-independent sensitivity of apoptosis to Cl− channel blockers in mouse cardiomyocytes. Cell Physiol. Biochem. 15:263–270
Tanabe S., Wang X., Takahashi N., Uramoto H., Okada Y. 2005. HCO -3 /– independent rescue from apoptosis by stilbene derivatives in rat cardiomyocytes. FEBS Lett. 579:517–522
Tanaka T., Nangaku M., Miyata T., Inagi R., Ohse T., Ingelfinger J.R., Fujita T. 2004. Blockade of calcium influx through L-type calcium channels attenuates mitochondrial injury and apoptosis in hypoxic renal tubular cells. J. Am. Soc. Nephrol. 15:2320–2333
Tang M.J., Cheng Y.R., Lin H.H. 1996. Role of apoptosis in growth and differentiation of proximal tubule cells in primary cultures. Biocherm. Biophys. Res. Commun. 218:658–664
Tapia-Vieyra J.V., Mas-Oliva J. 2001. Apoptosis and cell death channels in prostate cancer. Arch. Med. Res. 32:175–185
Taylor C.P., Meldrum B.S. 1995. Na+ channels as targets for neuroprotective drugs. Trends Pharmacol. Sci. 16:309–316
Thevenod F., Friedmann J.M. 1999. Cadmium-mediated oxidative stress in kidney proximal tubule cells induces degradation of Na+/K+-ATPase through proteasomal and endo-/lysosomal proteolytic pathways. FASEB J. 13:1751–1761
Thompson G.J., Langlais C., Cain K., Conley E.G., Cohen G.M. 2001. Elevated extracellular K+ inhibits death-receptor- and chemical-mediated apoptosis prior to caspase activation and cytochrome c release. Biochem. J. 357:137–145
Tombal B., Denmeade S.R., Isaacs J.T. 1999. Assessment and validation of a microinjection method for kinetic analysis of [Ca2+]i in individual cells undergoing apoptosis. Cell Calcium 25:19–28
Tsukimoto M., Harada H., Ikari A., Takagi K. 2005. Involvement of chloride in apoptotic cell death induced by activation of ATP-sensitive P2X7 purinoceptor. J. Biol. Chem. 280:2653–2658
Uhlemann A.C, Muller C,, Madlung J., Gulbins E., Lang F., 2000. Inhibition of CD95/Fas-induced DMA degradation by osmotic cell shrinkage. Cell. Physiol. Biochem. 10:219–228
Ulbricht W. 1998. Effects of veratridine on sodium currents and fluxes. Rev. Biochem. Pharmacol. 133:1–54
Vanden Abeele F, Skryma R, Shuba Y, Van Coppenolle F, Slomianny C, Roudbaraki M, Mauroy B, Wuytack F, Prevarskaya N. 2002. Bcl-2-dependent modulation of Ca2+ homeostasis and store-operated channels in prostate cancer cells. Cancer Cell 1:169–179
Vereninov A.A, Goriachaia T.S, Matveev V.V, Moshkov A.V, Rozanov lu M, Sakuta G.A, Shirokova A.V, lurinskaia V.E. 2004. [Cell shrinkage during apoptosis is not obligatory. Apoptosis of U937 cells induced by staurosporine and etoposide]. Tsitologiia 46:609–619
Verheye-Dua FA, Bohm L. 2000. Influence of apoptosis on the enhancement of radiotoxicity by ouabain. Strahlenther. Onkol. 176:186–191
Voets T., Nilius B. 2003. TRPs make sense. J. Membrane. Biol. 192:1–8
Wang L, Bhattacharjee A., Zuo Z, Hu F., Honkanen R.E., Berggren P.O., Li M. 1999. A low voltage-activated Ca2+ current mediates cytokine-induced pancreatic beta-cell death. Endocrinology 140: 1200–1204
Wang X.Q., Xiao A.Y., Sheline C., Hyrc K., Yang A., Goldberg M.P., Choi D.W., Yu S.P. 2003a. Apoptotic insults impair Na+, K+-ATPase activity as a mechanism of neuronal death mediated by concurrent ATP deficiency and oxidant stress. J.Cell Sci. 116:2099–2110
Wang X.Q., Xiao A.Y., Yang A., LaRose L., Wei L., Yu S.P. 2003b. Block of Na+, K+ ATPase and induction of hybrid death by 4-aminopyridine in cultured cortical neurons. J. Pharmacol. Exp. Ther. 305:502–506
Wang X.Q, Yu S.P. 2005. Novel regulation of Na, K-ATPase by Src tyrosine kinases in cortical neurons. J. Neurochem. 93:1515–1523
Wang Z. 2004. Roles of K+ channels in regulating tumour cell proliferation and apoptosis. Pfluegers Arch. 448:274–286
Waring P. 2005. Redox active calcium ion channels and cell death. Arch Biochem Biophys. 434:33–42
Watabe M, Masuda Y, Nakajo S, Yoshida T, Kuroiwa Y, Nakaya K. 1996. The cooperative interaction of two different signaling pathways in response to bufalin induces apoptosis in human leukemia U937 cells. J. Biol. Chem. 271:14067–14072
Waxman S.G. 2001, Transcriptional channelopathies: an emerging class of disorders. Nat. Rev. Neurosci. 2:652–659
Wehner F., Olsen H., Tinel H., Kinne-Saffran E., Kinne R.K. 2003. Cell volume regulation: osmolytes, osmolyte transport, and signal transduction. Rev. Physiol. Biochem. Pharmacol. 148:1–80
Wei L, Xiao A.Y, Jin C, Yang A., Lu Z.Y, Yu S,P. 2004. Effects of chloride and potassium channel blockers on apoptotic cell shrinkage and apoptosis in cortical neurons. Pfluegers Arch. 448:325–334
Williams S.S., French J.N., Gilbert M., Rangaswami A.A., Walleczek J., Knox S.J. 2000. Bcl-2 overexpression results in enhanced capacitative calcium entry and resistance to SKF-96365-induced apoptosis. Cancer Res. 60:4358–4361
Wright S.H. 2004. Generation of resting membrane potential. Ady. Physiol. Educ. 28:139–142
Wu X, Ahn E.Y., McKenna MA, Yeo H., McDonald J.M. 2005. Fas binding to calmodulin regulates apoptosis in osteoclasts. J. Biol. Chem. 280:29964–29970
Xiao A.Y., Wang X.Q., Yang A., Yu S.P. 2002a. Slight impairment of Na+, K+-ATPase synergistically aggravates ceramide- and beta-amyloid-induced apoptosis in cortical neurons. Brain Res. 955:253–259
Xiao A.Y., Wei L, Xia S., Rothman S., Yu S.P. 2002b. Ionic mechanism of ouabain-induced concurrent apoptosis and necrosis in individual cultured cortical neurons. J. Neurosci. 22:1350–1362
Yagami T., Ueda K., Asakura K., Nakazato H., Hata S., Kuroda T., Sakaeda T., Sakaguchi G., Itoh N., Hashimoto Y., Hori Y. 2003a. Human group IIA secretory phospholipase A2 potentiates Ca2+ influx through L-type voltage- sensitive Ca2+ channels in cultured rat cortical neurons. J. Neurochem. 85:749–758
Yagami T., Ueda K., Asakura K., Okamura N., Sakaeda T., Sakaguchi G., Itoh N., Hashimoto Y., Nakano T., Fujimoto M. 2003b. Effect of Gas6 on secretory phospholipase A(2)-IIA-induced apoptosis in cortical neurons. Brain Res. 985:142–149
Yagami T., Ueda K., Asakura K., Sakaeda T., Hata S., Kuroda T., Sakaguchi G., Itoh N., Hashimoto Y., Hori Y. 2003c. Porcine pancreatic group IB secretory phospholipase A2 potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels. Brain Res. 960:71–80
Yagami T., Ueda K., Asakura K., Sakaeda T., Nakazato H., Kuroda T., Hata S., Sakaguchi G., Itoh N., Nakano T., Kambayashi Y., Tsuzuki H. 2002. Gas6 rescues cortical neurons from amyloid beta protein-induced apoptosis. Neuropharmacology 43: 1289–1296
Yagami T., Ueda K., Sakaeda T., Itoh N., Sakaguchi G., Okamura N., Hori Y., Fujimoto M. 2004. Protective effects of a selective L-type voltage-sensitive calcium channel blocker, S-312-d, on neuronal cell death. Biochem. Pharmacol. 67:1153–1165
Yagami T., Ueda K., Sakaeda T., Okamura N., Nakazato H., Kuroda T., Hata S., Sakaguchi G., Itoh N., Hashimoto Y., Fujimoto M. 2005. Effects of an endothelin B receptor agonist on secretory phospholipase A2-IIA-induced apoptosis in cortical neurons. Neuropharmacology 48:291–300
Yoon J., Ben-Ami H.C., Hong Y.S., Park S., Strong L.L., Bowman J., Geng C., Baek K., Minke B., Pak W.L. 2000. Novel mechanism of massive photoreceptor degeneration caused by mutations in the trp gene of Drosophila. J. Neurosci. 20:649–659
Yu S.P. 2003a. Na+, K+-ATPase: the new face of an old player in pathogenesis and apoptotic/hybrid cell death. Biochem. Pharmacol. 66:1601–1609
Yu S.P. 2003b. Regulation and critical role of potassium homeostasis in apoptosis. Prog. Neurobiol. 70:363–386
Yu S.P., Canzoniero L.M., Choi D.W. 2001. Ion homeostasis and apoptosis. Curr. Opin. Cell Biol. 13:405–411
Yu S.P., Yeh C.H., Sensi S.L, Gwag B.J., Canzoniero L.M., Farhangrazi Z.S., Ying H.S., Tian M., Dugan L.L., Choi D.W. 1997. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science 278:1 14–1 17
Zagranichnaya T.K., Wu X., Danos A.M., Villereal M.L. 2005. Gene expression profiles in HEK-293 cells with low or high store-operated calcium entry: can regulatory as well as regulated genes be identified? Physio. Genomics 21: 14–33
Zamaraeva M.V., Sabirov R.Z., Maeno E., Ando-Akatsuka Y., Bessonova S.V., Okada Y. 2005. Cells die with increased cytosolic ATP during apoptosis: a bioluminescence study with intracellular luciferase. Cell Death Differ. 12:1390–1397
Zhang W., Chu X., long Q., Cheung J.Y., Conrad K., Masker K., Miller B.A. 2003. A novel TRPM2 isoform inhibits calcium influx and susceptibility to cell death. J. Biol. Chem. 278:16222-16229
Zhong J., Deng J., Huang S., Yang X., Lee W.H. 2004. High K+ and IGF-1 protect cerebellar granule neurons via distinct signaling pathways. J. Neurosci. Res. 75:794–806
Zhou J.G., Ren J.L, Qiu Q.Y., He H., Guan Y.Y. 2005. Regulation of intracellular Cl-concentration through volume-regulated CIC-3 chloride channels in A10 vascular smooth muscle cells. J. Biol. Chem. 280:7301–7308
Zhu D.M., Tibbies H.E., Vassilev A.O., Uckun P.M. 2002, SYKand LYN mediate B-cell receptor-independent calcium-induced apoptosis in DT-40 lymphoma B-cells. Leuk. Lymphoma 43:2165–2170
Ziegelhoffer A., Kjeldsen K., Bundgaard H., Breier A., Vrbjar N., Dzurba A. 2000. Na,K-ATPase in the myocardium: molecular principles, functional and clinical aspects. Gen. Physiol. Biophys. 19:9–47
Acknowledgment
We appreciate the input of Dr. James W. Jr. Putney.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Franco, R., Bortner, C. & Cidlowski, J. Potential Roles of Electrogenic Ion Transport and Plasma Membrane Depolarization in Apoptosis. J Membrane Biol 209, 43–58 (2006). https://doi.org/10.1007/s00232-005-0837-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00232-005-0837-5