
Abstract
Activation of ion channels and pores are essential steps during regulated cell death. Channels and pores participate in execution of apoptosis, necroptosis and other forms of caspase-independent cell death. Within the program of regulated cell death, these channels are strategically located. Ion channels can shrink cells and drive them towards apoptosis, resulting in silent, i.e. immunologically unrecognized cell death. Alternatively, activation of channels can induce cell swelling, disintegration of the cell membrane, and highly immunogenic necrotic cell death. The underlying cell death pathways are not strictly separated as identical stimuli may induce cell shrinkage and apoptosis when applied at low strength, but may also cause cell swelling at pronounced stimulation, resulting in regulated necrosis. Nevertheless, the precise role of ion channels during regulated cell death is far from being understood, as identical channels may support regulated death in some cell types, but may cause cell proliferation, cancer development, and metastasis in others. Along this line, the phospholipid scramblase and Cl−/nonselective channel anoctamin 6 (ANO6) shows interesting features, as it participates in apoptotic cell death during lower levels of activation, thereby inducing cell shrinkage. At strong activation, e.g. by stimulation of purinergic P2Y7 receptors, it participates in pore formation, causes massive membrane blebbing, cell swelling, and membrane disintegration. The LRRC8 proteins deserve much attention as they were found to have a major role in volume regulation, apoptotic cell shrinkage and resistance towards anticancer drugs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Regulated cell death
Regulated cell death is no longer characterized only by apoptotic cell death. Apoptosis is just a particular form of regulated cell death that is required to remove billions of aged or harmed cells per day from the human body, which need to be replaced by fresh ones. Cell removal occurs unrecognized, i.e. without stirring up immune mechanisms and without inducing inflammation. There are, however, other types of regulated cell death, which are very immunogenic, thus inducing pronounced inflammation and/or repair responses in the host. Due to its phenotypic appearance including cell swelling, swollen organelles, transparent cytoplasm, and loss of membrane integrity, this type of cell death was called programmed cell necrosis or necroptosis [1]. Thus, genetically encoded forms of cell death comprise both apoptosis and regulated necroptosis. Necroptosis is different from accidental cell death which is caused by microenvironmental disturbances, ischemia (Oncosis) or noxious influences [2]. While an organism benefits from apoptosis by getting rid of unwanted or damaged cells without alerting the immune system of the entire organism, necroptosis, often induced by identical insults but of higher strength, may serve as an organismal warning system [1]. Despite the rather different morphologic appearances of apoptosis and necroptosis, both mechanisms of cell death require the transport of ions, organic osmolytes, and water. The contribution of these transport proteins and underlying mechanisms will be summarized in the present review.
Caspase-dependent regulated cell death
Caspase-dependent regulated cell death has been a synonym for apoptosis and is characterized by cytoplasmic condensation, nuclear pyknosis due to chromatin condensation, karyorrhexis with nuclear fragmentation, membrane blebbing, DNA laddering, and the formation of apoptotic bodies [3–7]. Apoptosis is regarded as the prototype of genetically controlled cell death and has been extensively studied. Its physiological role and pathological consequences are well recognized. It is executed by effector caspases-3, -6, and -7. It may be induced as intrinsic apoptosis by opening of mitochondrial permeability transition pore (MPTP), BCL-2-associated X protein (BAX), and BCL-2 antagonist BAK. It leads to mitochondrial outer membrane permeabilization (MOMP) and loss of mitochondrial transmembrane potential, with a stop in ATP synthesis. Pro-apoptotic proteins such as cytochrome c, SMAC/DIABLO, apoptosis-inducing factor (AIF) and others are released into the cytosol. Cytochrome C, apaf-1 binding, and apoptosome formation activate initiator caspase-9 [6, 8, 9]. More details will be supplied by other excellent reviews in this special edition.
Extrinsic apoptosis is triggered by ligand binding to death receptors (FAS ligand to CD95, TNF to TNF-R, or TRAIL to TRAIL-R) and activation of the initiator caspase-8 and -10 [10]. Receptor-mediated apoptosis may also be initiated by so-called dependence receptors, a class of receptors that mediate apoptosis in the absence of their ligands, but support cell survival after ligand binding [11]. As a third mechanism, endoplasmic reticulum stress caused by disturbed glycosylation, misfolded proteins, perturbed calcium homeostasis or glucose deprivation, provokes a so-called unfolded protein response that may lead to apoptosis, but can also induce necrosis. This mechanism requires the presence of caspase-12 [12]. Apoptosis often takes place in conjunction with autophagy, a process of degradation of unnecessary or dysfunctional cellular components through formation of a autophagolysosome [6, 13]. Anoikis, also a form of regulated cell death, is induced in anchorage-dependent cells detaching from the surrounding extracellular matrix [14]. Such a mechanism is found for exfoliated epithelial cells of the gastrointestinal tract [15]. Independent of the precise mechanism underlying apoptosis, cell death will always lead to a non-inflammatory, non-immunogenic “silent” removal of cells. This is in contrast to highly immunogenic pyroptosis, which is another caspase-dependent process. However, unlike apoptosis which uses caspase-3, caspase-7, caspase-8 or caspase-9, pyroptosis relies on caspase-11 and also includes downstream caspase-1 activity [16–19]. Further details and recent findings on the critical role of the downstream protein gasdermin D will be presented in [20, 21] and a separate article of this special edition.
Role of Ca2+ signaling and Ca2+ channels
Apart from ion channels directly controlling cell volume, plasma membrane, and organelle-localized ion channels that control intracellular Ca2+ levels cause sustained Ca2+ increase and trigger apoptosis [22–24]. Among them are ER-localized Ca2+ release channels such as IP3 and ryanodine receptors, and the PM-localized store operated Ca2+ influx channels Orai1/Stim as well as the transient receptor potential channels TRPC1,3,6, TRPM2,7,8, TRPML2, TRPP5, and TRPV1,2,4 (for references see [25]). Noteworthy, the TRP channels TRPV4, TRPM3, and TRPM7 have been demonstrated to be directly activated by cell swelling [26, 27]. Furthermore, there is evidence for a role of additional mechanosensitive TRP channels [28]. Interestingly, caspase-dependent cleavage of TRPM7 releases a kinase domain from the ion channel and regulates compartmentalization of the Fas receptor and Fas-related signaling [29]. The contribution of these channels to apoptosis and the role of other cation channels such as purinergic P2X7 receptors, voltage-gated calcium channels [9, 30, 31], and Na+ channels has been reviewed elsewhere and will not be further discussed here [9, 25].
Although intracellular Ca2+ is well recognized as an important factor during apoptosis, it remains rather sketchy how increase in Ca2+ and consecutive cell shrinkage induce the apoptotic process. Certainly Ca2+-activated K+ channels play a central role, as outlined in the next paragraph. However, a large number of additional contributing factors have been identified, among them mitogen-activated protein kinases (MAPKs), in particular p38 MAPK. MAPKs are targeted by upstream small G proteins Rho and Rac. Downstream targets comprise mitochondrial protein Bcl-2 and p53. Further signaling cascades, as well as the contribution of reactive oxygen species (ROS), phospholipase A2 (PLA2) and the arachidonic acid pathway, integrin receptors, tyrosine kinase receptors, WNK, and focal adhesion kinases have been excellently reviewed elsewhere [25, 32].
K+ channels in apoptosis
Despite some ongoing controversy [25, 33], it appears that cell shrinkage is the hallmark and initial step during apoptosis. This is indicated by countless observations, which have been excellently summarized [25]. The so-called apoptotic volume decrease (AVD) that occurs under isosmotic conditions is believed to share common mechanisms with the regulatory volume decrease (RVD), which is a reshrinkage of the cell volume after increase of intracellular osmolarity or after exposure to hypotonic bath solution [25, 32, 34–39]. Cellular loss of K+ is the primary cause for cells to shrink, followed by activation of caspases and nucleases [7, 40]. Apoptotic cells transiently activate mechanisms to counteract shrinkage, by increasing ion uptake through Na+/H+ exchange via NHE1, the Na+/2Cl−/K+ cotransporter NKCC1, and cation channels [25].
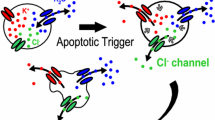
Cell shrinkage is regularly observed during apoptosis, while both shrinkage and apoptosis are inhibited by uptake of ions through cotransporters (e.g. Na+/K/2Cl− cotransport NKCC1) or activation of cation channels, a common mechanism for malignomas to escape from apoptosis [25, 36, 41, 42]. As an example, cell shrinkage, massive membrane blebbing, and subsequent swelling with apoptotic/necroptotic cell death have been observed recently in macrophages during ATP-binding to P2X7 receptors. ATP largely increased intracellular Ca2+ and activated the Ca2+-activated phospholipid scramblase/Cl− channel anoctamin 6 (ANO6), which will be discussed further below (Fig. 1) [43]. Anoctamins are also activated by triggering intrinsic apoptosis with staurosporine. We found that cells overexpressing ANO6, ANO9 or ANO10 demonstrate larger whole cell currents when exposed to staurosporine [44, 45]. In Jurkat T-lymphocytes expressing endogenous receptor/caspase-activated phospholipid scramblase Xkr8 [46, 47], ANO6, and ANO10, we found that induction of extrinsic apoptosis with TNF activate whole cell currents that were inhibited by siRNA-knockdown of Xkr8, ANO6, and ANO10. Moreover, the Cl− channel inhibitor NPPB (50 µM) inhibited whole cell currents activated through P2X7, staurosporine or TNF (Kunzelmann et al., unpublished results).

Role of anoctamins in cell death. Model for activation of anoctamin 6 (ANO6) by stimulation of purinergic P2X7 receptors with ATP in macrophages. Stimulation of P2X7 with ATP leads to sudden cell shrinkage (at moderate Ca2+ increase), which quickly changes into massive blebbing and cell swelling with a subsequent disintegration of the plasma membrane at increasing intracellular Ca2+ concentrations [43]. At low Ca2+ concentration, ANO6 is relatively selective for Cl− but changes into a nonselective channel/pore at larger cytosolic Ca2+ concentrations [43, 118]
Shrinkage results from a loss of KCl and cell water [25, 42, 48–50]. Of particular importance is the activation of K+ channels. Very different types of K+ channels have been implicated in activation of apoptosis, like voltage-gated Kv channels, ATP-regulated KATP channels, two-pore K+ channels, several types of Ca2+-dependent K+ channels and others [25, 32, 51]. Activation of K+ channels during apoptosis appears cell type specific, and, surprisingly, in some cells even counteracts apoptosis (for review see [25]. Loss of cytosolic potassium due to activation of K+ channels is essential, or at least supportive to drive cells into apoptosis [25, 37, 52–56]. The contribution of Ca2+-activated K+ channels, Ca2+ influx, activation of phospholipase A2, and ceramide formation has been well elaborated in erythrocytes, for which the term eryptosis was coined [25, 57]. Although the concept of cellular K+ loss in apoptosis is generally accepted, it has also been proposed that a decrease in intracellular K+ concentration is not obligatory for apoptosis [58]. Nevertheless, the variety of K+ channels that have been implicated in apoptosis suggests that in principle any type of K+ channel can support apoptosis, but it is not entirely clear how this can operate as a specific switch driving cells into regulated cell death.
Cl− channels in apoptosis
VRAC
Regulated cell shrinkage (regulatory volume decrease; RVD) provides a mechanism to keep cell volume constant whenever an osmotic disequilibrium (increase in intracellular osmolytes or decrease in extracellular osmolarity) favors influx of free water [32]. RVD and apoptotic cell shrinkage (apoptotic volume decrease; AVD) are thought to share the same underlying Cl− channel activity [36]. This Cl− channel has been described phenomenologically as volume-activated anion channel (VRAC) and is an essential component of the apoptotic cell death machinery. The terms VRAC, VSOR (volume sensitive outwardly rectifying anion channel) or volume-sensitive organic osmolyte and anion channel (VSOAC) were coined for anion currents detected in whole cell and single channel patch clamp recordings under extracellular hypotonicity. These currents demonstrate outward rectification, more or less time-dependent inactivation at strongly depolarizing clamp voltages, intermediate single-channel conductance, low-field strength anion selectivity (Eisenman type 1 halide permeability sequence) and a number of other characteristic features [59–63]. The estimated pore size is around 11 Å, allowing transport of anions as well as ATP and osmolytes, i.e. amino acids like taurine, aspartate, glutamate [64–67].
The Nilius team elegantly demonstrated that it is not the size of the swollen cell, but rather the intracellular ionic strength, that is detected by a still unknown mechanism that is transmitted as a signal to activate VRAC [68–70]. Also, other mechanisms have been shown to activate VRAC, like increase in intracellular Ca2+ by stimulation of purinergic and bradykinin receptors. Importantly, Ca2+ signaling occurs in Ca2+ nanodomains, rather than a global cytosolic Ca2+ increase. Moreover, ROS and phosphorylation were proposed to activate the channel [71–75], while swelling-independent activation by GTPγS [76] and sphingosin-1-phosphate generated by binding of bacterial LPS [65, 77] could lead to activation of VRAC during necroptotic cell death, as discussed below. A straightforward molecular concept involving a particular signaling cascade that leads to activation of VRAC is currently not available. Rather there is rather a bewildering number of contributing factors, including the small G proteins Rho/Rock, MAP kinase and phosphoinositide 3-kinase (PI3) kinase, ROS, membrane cholesterol, the actin cytoskeleton, tyrosine kinases and others. These factors have been reviewed extensively [32, 67, 78]. Taken together, a drop in intracellular ionic strength that activates PLA2, which cleaves PM phospholipids to lysophospholipids thereby changing plasma membrane tension and channel activity, could be crucial factors leading to activation of VRAC. Such a concept has been proposed recently and accommodates most of the previous observations related to VRAC [79] (Fig. 2). Activation of VRAC is further modified by additional factors outlined above, which are also highly relevant during apoptosis. The contribution to VRAC, ANO6 and other anoctamins such as ANO10 is not entirely clear [80]. Anoctamins may control compartmentalized Ca2+ signals and could function as osmosensors, thereby transferring swelling signals to the actual VRAC, which contains LRRC8A as an essential component [45, 81, 82]. Anoctamins may also operate as plasma membrane ion channels and thereby control cell volume [79, 83]. We found that anoctamins can be coimmunoprecipitated when overexpressed together with LRRC8A, but do not coimmunoprecipitate with an LRRC8A mutant lacking the LRR domain [45]. Notably, VRAC is inhibited by a broad range of compounds, which all inhibit Ca2+-activated anoctamin Cl− channels [67, 84, 85].

Activation of volume regulated anion currents. Model for activation of volume activated anion channels (VRAC). Intracellular Ca2+ is regarded as a modulatory factor for VRAC activity and volume regulation (RVD), but evidence was also provided that RVD and VRAC necessarily require Ca2+ [79]. A hypotonic bath solution will induce cell swelling by water influx through aquaporin (AQP) water channels, which leads to a drop in intracellular ionic strength and activation of Ca2+-insensitive phospholipase A2 (PLA2). Ca2+ moves into the cells through transient receptor potential (TRP) channels, most likely TRPC1 and TRPM7, and is released from endoplasmic reticulum (ER) Ca2+ stores, which may activate additional Ca2+-sensitive PLA2. PLA2 cleaves phospholipids (PL) in the plasma membrane and in the ER membrane to lysophospholipids (LPL), thereby increasing plasma membrane tension, which activates VRAC leading to regulatory volume decrease (RVD)
LRRC8
VRAC’s molecular nature remained enigmatic until recently, when LRRC8A, a member of the Leucine-Rich Repeat–Containing 8 family of 5 proteins (LRRC8A-E) was detected as an indispensable protein component of VRAC [86, 87]. VRAC channels/currents require LRRC8A and at least one additional LRRC8 isoform. LRRC8A-E may form hexameric channels as they are homologues to the pannexin family of ion channels [88]. Further evidence for LRRC8A-E as VRAC was provided recently by demonstrating transport of glutamate and taurine by LRRC8A and by showing a downregulation of LRRC8A in cisplatin-resistant cells [89, 90]. Moreover, loss of the subunits LRRC8A and LRRC8D leads to cellular resistance to platinum-based anti-cancer drugs [91].
Agammaglobulinemia was detected in a patient expressing a truncated form of LRRC8. These results indicated that LRRC8 is responsible for the B cell deficiency observed in this patient and that LRRC8 is required for B cell development [92]. Moreover, a mouse knockout model exists for LRRC8A. Notably, LRRC8A-/- mice have an enhanced mortality in utero. Newborn knockout animals appear inconspicuous at birth, but later show enhanced mortality and several defects such as growth retardation, hydronephrosis, sterility, epidermal hyperkeratosis, vacuolar renal tubular cells and others [93]. Experiments were performed to demonstrate the channel pore-forming character of LRRC8A: cysteine substitution experiments indicated a shift in the anion selectivity of the channel, suggesting that LRRC8A forms part of the conductance pathway [87]. Time-dependent current-inactivation at strongly depolarizing clamp voltage in patch clamp experiments appears to be a biophysical hallmark of VRAC. The degree of current inactivation was depending on the composition of LRRC8 isoforms, which, again, provides fairly good evidence that LRRC8 proteins indeed form the channel pore [86, 87]. Strangely, overexpression of LRRC8A did not further increase swelling-activated whole cell currents in cultured mammalian cells, but rather inhibited VRAC. Our laboratory made similar observations when VRAC was activated in LRRC8A-overexpressing HEK293 and HeLa cells (Kunzelmann et al., unpublished results). This may suggest that additional components of VRAC are still missing and/or that a proper stoichiometric relation of LRRC8 subunits is essential to build VRAC.
In a recent study, we found that expression of LRRC8A in Xenopus oocytes did not inhibit but induced a VRAC, which clearly supports the role of LRRC8A for volume-regulated anion currents [45]. Also, expression of anoctamins such as ANO6 and ANO10 induced VRAC, while additional expression of LRRC8A in ANO10 expressing oocytes did not further augment VRAC [45, 79]. The putative anoctamin family of Ca2+-activated Cl− channels and phospholipid scramblases (ANO1-ANO10) will be discussed in more detail later in this review. Noteworthy, we found that both LRRC8A and ANO10 could be coimmunoprecipitated, when overexpressed in HEK293 cells [45]. Because ANO10 is an intracellular protein controlling intracellular Ca2+ signals, we proposed a model by which ANO10 may tether Ca2+ sources (endoplasmic reticulum) to LRRC8A, similar to ANO1 [82]. It is speculated that anoctamins may be components of a VRAC/ICl-swell complex [67]. At any rate, experiments clearly suggest that VRAC represented by the LRRC8 channel complex requires increase in compartmentalized intracellular Ca2+ to be activated by cell swelling.
CFTR
The Cl− channel cystic fibrosis transmembrane conductance regulator (CFTR) has been reported to induce apoptosis. CFTR’s role in intracellular acidification [94–96] and formation of ceramide-rich membrane rafts [97, 98] were made responsible for its pro-apoptotic effects. Thus, the function of acid sphingomyelinase and clearance of the bacterium Pseudomonas aeruginosa depends on CFTR function and is impaired in CF [97–99]. Consequently, inhibitors of acid sphingomyelinase were shown to normalize pulmonary ceramide and inflammation in cystic fibrosis [100, 101]. Moreover, a pro-inflammatory but anti-apoptotic phenotype was shown to underlie the susceptibility to acute pancreatitis in cystic fibrosis (CF) [102, 103]. Cells expressing normal CFTR were shown to be more sensitive to oxidative stress-induced apoptosis than cells expressing defective CFTR [104–106]. Glutathione transport by CFTR appears to have a major role in this [107–110]. Along the same line, CFTR was also demonstrated to be essential for ROS-mediated autophagy [111]. CFTR has repeatedly been reported to operate as a volume-regulated Cl− channel [109, 110, 112]. Impaired cell volume regulation has been observed in intestinal crypt epithelia of cystic fibrosis mice [113, 114]. Subsequent studies indicated that CFTR affects volume regulation by autocrine ATP release [115]. Moreover, CFTR has been shown to protect hypoxic myocardial tissue from reperfusion damage, by reducing necrotic cell swelling [116].
Anoctamins (TMEM16 proteins)
The purpose of apoptosis is the non-inflammatory removal of cells by engulfment through phagocytosing cells (macrophages, epithelial cells), which requires a so-called eat-me signal [117]. The “eat-me” signal is mainly given by exposure of phosphatidylserine (PS) to the outer plasma membrane leaflet. This is realized by ANO6 (TMEM16F), which operates as a Ca2+-regulated channel for Cl− and cations such as Ca2+, and as a phospholipid scramblase, thereby exposing PS [44, 117–125]. ANO6 belongs to the family of Ca2+-activated ion channels and phospholipid scramblases [84]. It is probably identical with the ubiquitous outwardly rectifying Cl− channel ORCC (ICOR, ORDIC) that causes apoptotic cell shrinkage and cell death [43, 44, 126]. For ANO6 to be activated, influx of extracellular Ca2+ is required [120, 126].
The founder member ANO1 (TMEM16A) is a Ca2+-regulated Cl− channel that was also shown to support cellular volume regulation [83, 126]. ANO1 is activated during cell swelling due to ATP release and autocrine binding to purinergic receptors [83]. ANO1 was also demonstrated to mediate alpha-hemolysin-induced shrinkage of erythrocytes and to shrink cells of the choroid plexus by modulating water flux through interaction with TRPV4 channels [127, 128]. Finally, developmental volume changes of mouse cochlear cells and volume regulation of rat articular chondrocytes may also depend on ANO1 [129, 130]. A recent X-ray analysis of a fungal TMEM16/anoctamin protein (nhTMEM16) provides an idea how these proteins may operate as phospholipid scramblases and ion channels [131]. Also ANO4, ANO7, and ANO9 operate as Ca2+-dependent phospholipid scramblases [47]. We found that all of these anoctamins induce Ca2+-activated Cl− currents when expressed in HEK293 cells [118]. However, activation of Cl− currents appears to require lower intracellular Ca2+ levels compared to phospholipid scrambling. Cl− currents were activated through stimulation of purinergic receptors and 0.5–1 µM of the Ca2+ ionophore ionomycin, while scrambling was not detectable upon ATP stimulation and required 3–10 µM ionomycin [47, 118, 123]. This is likely to correspond to the cell shrinkage preceding phospholipid scrambling [132].
Expression of ANO10 has been correlated to VRAC and RVD [133]. A genome-wide association study (GWAS) demonstrated that seropositivity for anti-Borrelia antibodies was correlated to the single nucleotide polymorphism R263H in ANO10 [45]. It was found that ANO10 produces volume-regulated Cl−currents (IHypo) in Xenopus oocytes, HEK293 cells, lymphocytes, and macrophages, where it supports migration and phagocytosis of spirochetes. The R263H variant was shown to be inhibitory on IHypo, RVD and intracellular Ca2+ signals, thereby delaying spirochete clearance and increasing adaptive immunity. Thus, ANO10 seems to have an important role in the innate immune defense against Borrelia infection, by operating as volume-regulated anion channel or as a regulator of VRAC [45].
Other anion channels in apoptotic cell death
Bestrophin, another type of Ca2+-activated Cl− channel, is also regulated by cell volume, but its role for apoptotic cell death remains obscure [134]. Other Cl− channels have been discussed in the context of cellular volume regulation and AVD [62, 135–137]. These channels, however, have been discarded as VRAC, and also the contribution of the voltage-dependent Cl− channels ClC2 and ClC-3 is disputed [138–143]. Notably, VRAC/AVD was shown to be activated by reactive oxygen species (ROS). As endosomally located ClC-3 was shown to be relevant for ROS production, it may produce VRAC currents indirectly by increasing ROS [144]. Finally, large conductance voltage-dependent anion channels (VDAC), located in the mitochondrial membrane, have been suggested as volume-regulated channels activated during apoptotic cell death [145, 146]. However, its role as plasma membrane localized anion channel remains controversial.
Ion channels, apoptosis, and cancer
K+ channels as a target in cancer
Apoptosis is essential to maintain normal tissue homeostasis by controlling cell turnover. Excessive or reduced execution of regulated cell death by inappropriate activation of ion channels may lead to neurological disorders, inflammation, and tissue reperfusion damage during heart attack, or graft rejection during transplantation, autoimmune disease, and cancer, respectively. Ion channels, particularly K+ channels, may be used as therapeutic targets, e.g. to induce apoptosis in cancer cells. However, similar types of K+ channels that can induce apoptosis in some cell types were shown to be upregulated in cancer cells [147–151]. Assessment of ion channel activity in cancer has been discussed earlier [152]. However, other non-conducting mechanisms, such as recruitment of signaling cascades by K+ channels, may have additional impact on cell cycle progression and proliferation [150].
Activation of Cl− channels to induce apoptosis
Cl− channels might be a another target to induce apoptosis of cancer cells, since pro-proliferative effects have been reported for ClC-3 and anoctamins, while CLCA proteins are accessory proteins and do not form Cl− channels. Little is known about the role of intracellular CLIC channels [153–155]. However, intracellular ion channels may also have a role during cancer, which has been reviewed previously [156]. Downregulation of VRAC has been shown to be involved in multidrug resistance [157, 158]. The resistance towards the cytotoxic drug cisplatin, normally inducing AVD and apoptosis, is paralleled by a loss of VRAC activity [158–160]. ANO6 was shown to form an outwardly rectifying Cl− channel activated during apoptotic stimulation and cell death [44]. Notably, cisplatin-induced caspase-3 activity was markedly reduced after knockdown of ANO6 expression [126].
Inhibition of Cl− channels to reduce proliferation
Inhibition of other Cl− channels has been shown to reduce proliferation in cancer cells. ANO1 is strongly upregulated in gastrointestinal stromal tumors (GIST) and serves as a robust clinical marker [161–166]. Extracellular signal regulated kinase 1, 2 (Erk1, 2), and cyclin D activity is upregulated in ANO1-overexpressing tumor cells, while inhibition of ANO1 was shown to lower Erk1, 2, and cyclin D activity [162]. Notably, ANO1 was also downregulated by inhibition of histone deacetylase [163, 167]. Moreover, ANO1 supports cell migration, metastasis and is correlated with poor outcome in patients with head and neck cancer HNSCC [168–170]. Studies are now focusing on ANO1 inhibitors for the treatment of GIST [171–173]. Meanwhile, enhanced expression of ANO1 has also been demonstrated for other types of hyperplasia, including those of breast, prostate, and GI tract [163, 174].
Targeting intracellular Ca2+ levels
Many of the ion channels described above require intracellular Ca2+ to operate. Targeting intracellular Ca2+ levels is, therefore, another way to control channel activity and cell proliferation. However, Ca2+ signaling and the related (transient receptor potential) TRP channels have an ambiguous role, since they control both apoptosis and proliferation. Thus, cytosolic and mitochondrial Ca2+ overload is clearly pro-apoptotic, while TRP channels, voltage-gated Ca2+ channels, Ca2+ transporters, and Ca2+-binding/storage proteins support proliferation and are overexpressed in cancer [175, 176]. In their review, Prevarskaya and coworkers nicely describe how Ca2+ concentrations in the cytosol, endoplasmic reticulum, and mitochondria become unbalanced in cancer, and how spatio-temporal patterns of Ca2+ signaling are set to pro-proliferative activity [175]. Moreover, as most ion channels show a broad and no cancer-specific expression, no universal ion channel population has been identified so far that could be targeted in cancer cells to induce apoptosis. Thus, it is difficult to target these channels without affecting the function of healthy tissues. Finally, the lack of specific drugs and numerous alternative splice products, e.g. in the case of ANO1 and ANO6, imposes further caveats.
Pyroptosis and the role ion channels
Pyroptosis or caspase-11-dependent cell death is inherently inflammatory, in contrast to non-inflammatory apoptosis. It is triggered by various pathological stimuli, such as stroke, heart attack or cancer, and during microbial infections [16]. Pyroptosis is characterized by rapid rupture of the plasma membrane. Host- and microorganism-derived danger signals are sensed by cells and can induce pyroptosis. Activation of Toll-like receptors and Nod-like danger receptors induce production of a number of inflammatory cytokines, such as tumor necrosis factor (TNF), IL-6, IL-8, and interferons. In infected host macrophages, pyroptosis was shown to be induced by activation of plasma membrane pores, which leads to osmotic lysis of the cell [177]. Along this line, it was shown that Salmonella invasin SipB induces regulated cell death in macrophages by binding to caspase-1 [178]. Very recent reports illuminate the signaling pathway downstream of caspase 11 and demonstrate how caspase-11 executes pyroptosis via caspase-1 and activation of the non-canonical NLRP3 inflammasome. Thus, caspase-11 cleaves gasdermin D, a protein with previously unknown function, whose amino-terminal fragment promotes both pyroptosis and NLRP3-dependent activation of caspase-1 [20, 179]. The formation of caspase 1-dependent pores in the plasma membrane leads to cytokine release by macrophages. Thus, release of the cytokines IFNα, IFNβ, TNF, IL-12, IL-6, IL-8, IL-18, IL-1β, activation of the bacterial type III secretion system, and cell lysis by caspase 1-dependent pore formation are hallmarks of pyroptosis [177, 180].
The membrane pores lead to a loss of cellular ionic gradients with the consequence of water influx, cell swelling, and osmotic cell lysis. Similar to apoptosis, also in pyroptosis potassium efflux is a common cellular response and apparently necessary to activate caspase-1. Thus, preventing K+ efflux has been shown to block activation of caspase 1 [181, 182]. The myriads of inflammatory factors can trigger multiple intracellular signaling pathways that each may potentially contribute to activation of membrane ion channels [183]. However, the essential distal mechanisms triggering cell death by formation of membrane pores with an apparent size of 1.1–2.4 nm still remain unidentified. In addition, bacterial pathogens may invade host cells and elicit cell lysis by pore-forming toxins [184–186]. Taken together, molecular characterization and understanding of caspase-1-activated pores should be a major step forward in understanding pyroptosis.
Ion channels during necroptosis and other forms of caspase-independent regulated cell death
Necroptosis
Several types of caspase-independent cell death exist with necroptosis currently being the best examined cell death pathway [187]. In contrast to apoptosis, necroptosis is characterized by cell swelling with subsequent decay of the cell and annexin V/PI—positivity [6, 188]. Cell swelling may not always be easily detectable as suggested from continuous recordings of holographic images (Fig. 3). In fact, volumes calculated from the holographic images indicated very little initial volume increase in the range of 10–15 % with even subsequent cell shrinkage. However, when calculating cell volume changes from membrane capacitance measured in patch clamp experiments, which is proportional to the membrane surface area, cell volumes roughly doubled during necroptotic cell death, due to multiple protrusions, invaginations, and cellular deformations (Kunzelmann et al., unpublished results).

Cell appearances during necroptotic cell death of NIH3T3 cells. a Holographic images (HoloMonitor, I&L Biosystems) of NIH3T3 cells exposed to the necroptotic cocktail TSZ (10 ng/ml TNF, 5 µM of the SMAC mimetic birinapant, and 25 µM of the pan-caspase inhibitor Z-VAD). Note the decay of the cells after 6 h. The vertical scale bar indicates cell height ranging form 0 to 15,71 µm. b FACS analysis of TSZ-treated NIH3T3 cells indicating PS scrambling (annexin V binding) and membrane permeabilization (PI)
Necrosis was considered exclusively as a passive form of cell death resulting from cellular stress. However, it became clear that necrosis can occur in a regulated manner by both physiological and pathological stimuli. Thus, regulated necrosis or necroptosis is an alternative form of regulated cell death. It is activated when apoptosis is inhibited during host defense against infection and during inflammation [189]. Receptor-interacting protein kinase 3 or RIPK3 is the major controller that switches between necroptosis and apoptosis [190]. It has been proposed that activated RIPK3 phosphorylates and recruits mixed lineage kinase domain like (MLKL), a pseudokinase, to the plasma membrane, thereby inducing membrane leakage and release of inflammatory mediators [191, 192]. However, the detailed cellular mechanisms, particularly those acting distal from pMLKL, are poorly understood. Unmasking these molecular events is of high priority as necroptosis takes place in many inflammatory diseases [193]. For example, staphylococcus aureus toxin-induced necroptosis is the major mechanism of lung damage caused by staphylococcus aureus infection. It can be inhibited by interfering with RIPK1-RIPK3-MLKL function, illuminating the significance of this pathway as novel drug target [194].
Role of Ca2+
Within the process of necroptosis, calcium appears to be a major factor. Ca2+ may enter the cell through store-operated Ca2+ influx channels (SOCE), nonselective TRP cation channels, and acid-sensing ion channels (ASICs). ROS-activated TRPC3/4 and TRPM2/7 channels may, in addition, participate in Na+ and Ca2+ influx [6, 195–200]. Necroptosis is characterized by a drop in intracellular ATP levels and oxidative stress/ROS that leads to energy failure thus compromising energy-dependent transport and mitochondrial function [6]. Moreover, TRPC channels such as TRPC3, 6, and 7 are activated due to Ca2+ release from the endoplasmic reticulum and modulation by diacylglycerol [6]. Volume regulation and volume recovery from cell swelling were shown to be compromised during necrosis. VRAC requires intracellular ATP to operate and is, therefore, not functioning under hypoxia and ATP-depletion [63, 201, 202].
The drop in intracellular ATP levels also compromises the function of the Na+/K+-ATPase, leading to accumulation of intracellular Na+. Moreover, reduced Ca2+ pumping by SERCA and plasma membrane Ca2+-ATPase (PMCA) contributes to necrotic Ca2+ increase [6]. It has been reported that moderate increase in intracellular Ca2+ to approximately 200–400 nM triggers apoptosis, while a concentration above 1 μM is associated with necrosis [203]. However, own unpublished studies indicate only moderate increase in intracellular Ca2+ during necroptosis, when induced by application of TNF, caspase inhibitor, and SMAC mimetics (Kunzelmann et al., unpublished results). Moreover, enzymes are activated such as PLA2, PLC, and PLD, which break down membrane phospholipids. Downstream products of the cyclooxygenase, lipoxygenase, and epoxygenase pathways are strongly inflammatory, which leads to further tissue damage. These inflammatory factors increase capillary permeability, thereby enabling migration of immune cells into damaged regions, attracted by released immunogenic factors.
Channel forming by pMLKL?
The final step during necroptosis requires RIPK3-dependent phosphorylation of MLKL. It has been reported that phospho-MLKL forms a homotrimer through its amino-terminal coiled-coil domains that allows translocation to the cell plasma membrane during TNF-induced necroptosis [204, 205]. One study demonstrates Ca2+ influx through TRPM7 channels [204]. The authors show current/voltage relationships from necroptotic HT29 cells. These relatively small currents, however, are outwardly rectifying, which suggests predominant activation of a Cl− conductance [204]. In fact, one would assume that swelling requires a pathway for outward Cl− currents (influx of Cl−), while outflow of Cl− antagonizes necroptosis/necrosis, as demonstrated recently in heart muscle cells [206]. It is discussed that Ca2+ may kill cells by amyloid-mediated membrane disruption [204]. According to this, Ca2+ supports amyloid-triggered fiber growth that leads to removal of lipids from the bilayer through a detergent-like mechanism thereby inducing disruption of the plasma membrane [207]. Another study claims membrane translocation of pMLKL into lipid rafts. Apparently, a four-α-helix bundle of the MLKL amino acids 1–130 is sufficient to trigger necroptosis. The team claims that the plasma membrane MLKL complex itself acts as cation influx channel or, alternatively, may activate other proteins to increase Na+ influx, osmotic pressure, and membrane rupture [205]. A four-helix bundle structure of the MLKL N-terminal region has been determined by nuclear magnetic resonance spectroscopy. In this study, membrane insertion and membrane leakiness have been shown by reconstitution of MLKL in liposomes [208]. The authors propose opening of the four-helix bundle by RIPK3-mediated phosphorylation. Nevertheless, the contribution of such a Na+ permeable “bundle channel” remains to be demonstrated. Moreover, own unpublished results show little evidence for cation influx and intracellular Ca2+ levels rise only moderately during TNF-induced necroptosis (Kunzelmann et al., unpublished results). As necroptosis shows a rise in cytosolic Ca2+ and increase in reactive oxygen species (ROS) along with intracellular acidification, it is entirely possible that other/additional ion channels are involved. Cells may swell upon activation of MLKL, but this awaits proper cell volume measurements along with comprehensive patch clamp analysis of underlying currents. As it stands, there is certainly room for alternative mechanisms.
Mitochondrial leakage in necroptosis?
A quite different mechanism for necroptotic cell death was proposed recently [209]. The Bcl-2 family members Bax and Bak induce mitochondrial outer membrane permeabilization (MOMP) to mediate apoptotic cell death. As Bax/Bac-dependent MOMP was also observed during necrosis, the team demonstrates that Bax/Bak oligomerization and opening of the mitochondrial permeability transition pore are required for necroptotic cell death. Cells/tissues deficient for Bax/Bak were resistant to necroptotic cell death. The team also observed translocation of pMLKL to mitochondria rather than the plasma membrane. Thus, mitochondrial leakage, and not a plasma membrane pore, may underlie necroptotic cell death, and thus necroptosis, parthanatos and MPT-RN (discussed below) may share similar mechanisms [209]. However, as demonstrated in another study, TNF-induced necroptosis does still occur in cells depleted of mitochondria [210]. The results indicate that mitochondrial ROS production accompanies, but does not cause RIPK3-dependent necroptotic cell death.
Necroptosis or apoptosis?
It is not uncommon that a death stimulus activates both pathways, by triggering initial apoptosis which changes later into necrosis. Identical stimuli may lead to apoptosis at low dose, but may cause necroptosis at higher dose [1]. Such a response has been observed earlier through stimulation of purinergic P2X7 receptors [43, 211, 212]. We identified ANO6 (TMEM16F) as the underlying membrane-localized ion channel. Initial activation of ANO6 by mild increase of intracellular Ca2+ (to approximately 1 µM) induces relatively Cl−-selective currents and cell shrinkage. As intracellular Ca2+ levels further increase to high local concentrations (10–100 µM), the channel becomes increasingly non-selective and “pore-like” currents appear that massively swell cells [43]. ATP induced initial transient cell shrinkage preceding massive membrane blebbing, which was then followed by strong cell swelling and necroptotic cell death [43]. Under such severe conditions, cells die in a fashion that elicits a host response by alerting and activating the immune and nervous system.
Necrosis is characterized by a disturbance of intracellular Ca2+ homeostasis, and ANO6 itself may contribute to the rise of intracellular Ca2+, as it has been also identified as Ca2+ permeable channel [124]. Notably, anoctamin 1 and closely related transmembrane channel-like (TMC) proteins were found to control intracellular Ca2+ signaling [213–216]. In summary, intracellular Ca2+ levels and differential activation of ion channels may participate in the decision whether a cell undergoes apoptotic or necroptotic cell death.
Other forms of caspase-independent regulated cell death
There are additional mechanisms for caspase-independent regulated cell death, although very little is known regarding the underlying ion currents: (1) Ferroptosis is induced by toxic iron-catalyzed reactive oxygen species (ROS) [187, 217]. Ferroptosis has been shown to be involved in synchronized renal tubular cell death [218]. (2) NETosis is a NADPH oxidase (NOX)-dependent and, therefore, ROS-dependent mechanism of cell death that is specific to leukocytes [187, 219, 220]. NETosis is a process that leads to neutrophil extracellular traps (NETs), whose main components are DNA, antimicrobial peptides, and neutrophil proteins. These regulated cell death pathways have in common an excessive production of ROS leading to oxidative stress, damage of intracellular molecules and organelles and, ultimately, necrosis. ROS also oxidize double bonds in polyunsaturated fatty acids of membrane phospholipids. Lipid oxidation can lead to loss of integrity of both plasma membrane and intracellular membranes. ROS will also break disulfide bonds in proteins, thereby possibly affecting ion channel function [1, 221]. Volume regulatory channels such as VRAC are compromised under these conditions and can, therefore, not antagonize cell swelling. Indeed, under these conditions other Cl− channels may become essential for volume regulation, such as CFTR. This has been demonstrated in the heart, where cardiac CFTR protects heart muscle cells from reperfusion damage after myocardial infarction [116].
Finally, other forms of caspase-independent regulated cell death such as (3) parthanatos und (4) mitochondrial permeability transition-mediated regulated necrosis (MPT-RN) have in common more or less persistent mitochondrial outer membrane permeabilization (MOMP) [193, 222, 223]. A number of proteins have been demonstrated to participate in mitochondrial permeabilization such as voltage-dependent anion channels (VDAC), Bax-oligomerization and the adenine nucleotide translocator (ANT), while members of the Bcl-2 family of proteins are antagonistic regulators. These mechanisms are not compulsively ROS-dependent and do not rely on activation of plasma membrane ion channels. However, it might be more a general rule than an exception that both plasma membrane-localized channels and mitochondrial pores are activated simultaneously. This has been demonstrated for ischemia–reperfusion injury, which uses two independent pathways of regulated cell death, one of them being MPT-RN [223].
Closing remarks
Apoptosis and regulated necrosis may be initiated in response to very similar types of insults. The outcome, apoptosis or caspase-independent cell death, may often depend just on dosage or intensities of the insult. Stimulation of TNF receptors can lead to both types of cell death. Present work indicates that cells will move toward necroptosis when apoptosis is inhibited [6, 224]. Necroptosis appears as the ancient way of dying, while apoptosis seems phylogenetically advanced [225]. It is possible that necroptotic cell death has been underestimated, due to the lack of simple detection assays [226, 227]. At any rate, both pathways require activation of ion channels in either plasma membrane or intracellular ER/mitochondrial membranes and may lead to formation of large nonselective pores.
There is a considerable crosstalk between apoptotic and necroptotic cell death pathways and components of both may be activated simultaneously [6, 43, 211]. Increases in intracellular Ca2+, ROS and ceramide have been reported as major players during necrosis; however, all three signals are equally well known to induce apoptosis [228]. Regulated cell death may be a continuum, with apoptosis and necroptosis representing two extremes of biochemically overlapping death pathways. As an interesting example, activation of P2X7 receptors leads to initial pro-apoptotic cell shrinkage with subsequent large necroptotic cell swelling and plasma membrane disintegration [43]. It appears entirely possible that identical ion channels may contribute to either form of regulated cell death.
References
Zong WX, Thompson CB (2006) Necrotic death as a cell fate. Genes Dev 20:1–15
Galluzzi L, Kepp O, Krautwald S, Kroemer G, Linkermann A (2014) Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol 35:24–32
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35:495–516
Williams GT (1991) Programmed cell death: apoptosis and oncogenesis. Cell 65:1097–1098
Cohen JJ, Duke RC, Fadok VA, Sellins KS (1992) Apoptosis and programmed cell death in immunity. Annu Rev Immunol 10:267–293
Henriquez M, Armisen R, Stutzin A, Quest AF (2008) Cell death by necrosis, a regulated way to go. Curr Mol Med 8:187–206
Gulbins E, Jekle A, Ferlinz K, Grassme H, Lang F (2000) Physiology of apoptosis. Am J Physiol Renal Physiol 279:F605–F615
Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397:441–446
Kondratskyi A, Kondratska K, Skryma R, Prevarskaya N (2014) Ion channels in the regulation of apoptosis. Biochim Biophys Acta 1848:2532–2546
Nagata S (1997) Apoptosis by death factor. Cell 88:355–365
Mehlen P, DE Bredesen (2011) Dependence receptors: from basic research to drug development. Sci Signal 4:mr2
Adams JM (2003) Ways of dying: multiple pathways to apoptosis. Genes Dev 17:2481–2495
Ryter SW, Mizumura K, Choi AM (2014) The impact of autophagy on cell death modalities. Int J Cell Biol. 2014:502676
Frisch SM, Screaton RA (2001) Anoikis mechanisms. Curr Opin Cell Biol 13:555–562
Bertrand K (2011) Survival of exfoliated epithelial cells: a delicate balance between anoikis and apoptosis. J Biomed Biotechnol 2011:534139
Bergsbaken T, Fink SL, Cookson BT (2009) Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7:99–109
Kayagaki N, Warming S, Lamkanfi M, Vande WL, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479:117–121
Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, Monack DM (2012) Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490:288–291
Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, Forsberg LS, Carlson RW, Dixit VM (2013) Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341:1246–1249
Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM (2015) Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526:666–671
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526:660–665
Berridge MJ, Bootman MD, Lipp P (1998) Calcium—a life and death signal. Nature 395:645–648
Fang KM, Chang WL, Wang SM, Su MJ, Wu ML (2008) Arachidonic acid induces both Na+ and Ca2+ entry resulting in apoptosis. J Neurochem 104:1177–1189
Green DR, Reed JC (1998) Mitochondria and apoptosis. Science 281:1309–1312
Lang F, Hoffmann EK (2012) Role of ion transport in control of apoptotic cell death. Compr Physiol 2:2037–2061
Harteneck C, Reiter B (2007) TRP channels activated by extracellular hypo-osmoticity in epithelia. Biochem Soc Trans 35:91–95
Numata T, Shimizu T, Okada Y (2007) TRPM7 is a stretch- and swelling-activated cation channel involved in volume regulation in human epithelial cells. Am J Physiol Cell Physiol 292:C460–C467
Plant TD (2014) TRPs in mechanosensing and volume regulation. Handb Exp Pharmacol 223:743–766
Desai BN, Krapivinsky G, Navarro B, Krapivinsky L, Carter BC, Febvay S, Delling M, Penumaka A, Ramsey IS, Manasian Y, Clapham DE (2012) Cleavage of TRPM7 releases the kinase domain from the ion channel and regulates its participation in Fas-induced apoptosis. Dev Cell 22:1149–1162
Dubois C, Vanden Abeele F, Prevarskaya N (2013) Targeting apoptosis by the remodelling of calcium-transporting proteins in cancerogenesis. FEBS J 280:5500–5510
Joseph SK, Hajnoczky G (2007) IP3 receptors in cell survival and apoptosis: Ca2+ release and beyond. Apoptosis 12:951–968
Hoffmann EK, Lambert IH, Pedersen SF (2009) Physiology of cell volume regulation in vertebrates. Physiol Rev 89:193–277
Orlov SN, Model MA, Grygorczyk R (2013) CrossTalk opposing view: the triggering and progression of the cell death machinery can occur without cell volume perturbations. J Physiol 591:6123–6125
Lang F, Busch GL, Ritter M, Volkl H, Waldegger S, Gulbins E, Haussinger D (1998) Functional significance of cell volume regulatory mechanisms. Physiol Rev 78:247–306
Stutzin A, Hoffmann EK (2006) Swelling-activated ion channels: functional regulation in cell-swelling, proliferation and apoptosis. Acta Physiol (Oxf) 187:27–42
Okada Y, Maeno E, Shimizu T, Dezaki K, Wang J, Morishima S (2001) Receptor-mediated control of regulatory volume decrease (RVD) and apoptotic volume decrease (AVD). J Physiol 532:3–16
Maeno E, Ishizaki Y, Kanaseki T, Hazama A, Okada Y (2000) Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc Natl Acad Sci USA 97:9487–9492
Strange K, Emma F, Jackson PS (1996) Cellular and molecular physiology of volume-sensitive anion channels. Am J Physiol 270:C711–C730
Eggermont J, Trouet D, Carton I, Nilius B (2001) Cellular function and control of volume-regulated anion channels. Cell Biochem Biophys 35:263–274
Montague JW, Bortner CD, Hughes FM Jr, Cidlowski JA (1999) A necessary role for reduced intracellular potassium during the DNA degradation phase of apoptosis. Steroids 64:563–569
Bortner CD, Hughes FM Jr, Cidlowski JA (1997) A primary role for K+ and Na+ efflux in the activation of apoptosis. J Biol Chem 272:32436–32442
Poulsen KA, Andersen EC, Hansen CF, Klausen TK, Hougaard C, Lambert IH, Hoffmann EK (2010) Deregulation of apoptotic volume decrease and ionic movements in multidrug-resistant tumor cells: role of chloride channels. Am J Physiol Cell Physiol 298:C14–C25
Ousingsawat J, Wanitchakool P, Kmit A, Romao AM, Jantarajit W, Schreiber S, Kunzelmann K (2015) Anoctamin 6 mediates effects essential for innate immunity downstream of P2X7-receptors in macrophages. Nat Commun 6:6245
Martins JR, Faria D, Kongsuphol P, Reisch B, Schreiber R, Kunzelmann K (2011) Anoctamin 6 is an essential component of the outwardly rectifying chloride channel. Proc Natl Acad Sci USA 108:18168–18172
Hammer C, Wanitchakool P, Sirianant L, Papiol S, Monnheimer M, Faria D, Ousingsawat J, Schramek N, Schmitt C, Margos G, Michel A, Kraiczy P, Pawlita M, Schreiber R, Schulz TF, Fingerle V, Tumani H, Ehrenreich H, Kunzelmann K (2015) A coding variant of ANO10, affecting volume regulation of macrophages, is associated with Borrelia seropositivity. Mol Med 21:26–37
Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S (2014) Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 344:1164–1168
Suzuki J, Fujii T, Imao T, Ishihara K, Kuba H, Nagata S (2013) Calcium-dependent phospholipid scramblase activity of TMEM16 family members. J Biol Chem 288:13305–13316
Bortner CD, Cidlowski JA (2004) The role of apoptotic volume decrease and ionic homeostasis in the activation and repression of apoptosis. Pflugers Arch 448:313–318
Hughes FM Jr, Cidlowski JA (1999) Potassium is a critical regulator of apoptotic enzymes in vitro and in vivo. Adv Enzyme Regul 39:157–171
Okada Y (2004) Ion channels and transporters involved in cell volume regulation and sensor mechanisms. Cell Biochem Biophys 41:233–258
Hoffmann EK (2011) Ion channels involved in cell volume regulation: effects on migration, proliferation, and programmed cell death in non adherent EAT cells and adherent ELA cells. Cell Physiol Biochem 28:1061–1078
Beauvais F, Michel L, Dubertret L (1995) Human eosinophils in culture undergo a striking and rapid shrinkage during apoptosis. Role of K+ channels. J Leukoc Biol 57:851–855
Yurinskaya V, Goryachaya T, Guzhova I, Moshkov A, Rozanov Y, Sakuta G, Shirokova A, Shumilina E, Vassilieva I, Lang F, Vereninov A (2005) Potassium and sodium balance in U937 cells during apoptosis with and without cell shrinkage. Cell Physiol Biochem 16:155–162
Szabo I, Lepple-Wienhues A, Kaba KN, Zoratti M, Gulbins E, Lang F (1998) Tyrosine kinase-dependent activation of a chloride channel in CD95-induced apoptosis in T lymphocytes. Proc Natl Acad Sci USA 95:6169–6174
Wang Z (2004) Roles of K+ channels in regulating tumour cell proliferation and apoptosis. Pflugers Arch 448:274–286
Burg ED, Remillard CV, Yuan JX (2006) K+ channels in apoptosis. J Membr Biol 209:3–20
Lang F, Ritter M, Gamper N, Huber SM, Fillon S, Tanneur V, Lepple-Wienhues A, Szabo I, Gulbins E (2000) Cell volume in the regulation of cell proliferation and apoptotic cell death. Cell Physiol Biochem 10:417–428
Borjesson SI, Englund UH, Asif MH, Willander M, Elinder F (2011) Intracellular K+ concentration decrease is not obligatory for apoptosis. J Biol Chem 286:39823–39828
Cahalan MD, Lewis RS (1988) Role of potassium and chloride channels in volume regulation by T lymphocytes. Soc Gen Physiol Ser 43:281–301
Hazama A, Okada Y (1988) Ca2+ sensitivity of volume-regulatory K+ and Cl− channels in cultured human epithelial cells. J Physiol 402:687–702
Nilius B, Oike M, Zahradnik I, Droogmans G (1994) Activation of a Cl− current by hypotonic volume increase in human endothelial cells. J Gen Physiol 103:787–805
Nilius B, Eggermont J, Voets T, Buyse G, Manolopoulos V, Droogmans G (1997) Properties of volume-regulated anion channels in mammalian cells. Prog Biophys Mol Biol 68:69–119
Jackson PS, Morrison R, Strange K (1994) The volume-sensitive organic osmolyte-anion channel VSOAC is regulated by nonhydrolytic ATP binding. Am J Physiol 267:C1203–C1209
Droogmans G, Prenen J, Eggermont J, Voets T, Nilius B (1998) Voltage-dependent block of endothelial volume-regulated anion channels by calix[4]arenes. Am J Physiol 275:C646–C652
Burow P, Klapperstuck M, Markwardt F (2014) Activation of ATP secretion via volume-regulated anion channels by sphingosine-1-phosphate in RAW macrophages. Pflugers Arch
Jackson PS, Strange K (1993) Volume-sensitive anion channels mediate swelling-activated inositol and taurine efflux. Am J Physiol 265:C1489–C1500
Pedersen SF, Klausen TK, Nilius B (2015) The identification of VRAC (Volume Regulated Anion Channel): an amazing Odyssey. Acta Physiol (Oxf) 213:868–881
Nilius B, Prenen J, Voets T, Eggermont J, Droogmans G (1998) Activation of volume-regulated chloride currents by reduction of intracellular ionic strength in bovine endothelial cells. J Physiol 506:353–361
Sabirov RZ, Prenen J, Tomita T, Droogmans G, Nilius B (2000) Reduction of ionic strength activates single volume-regulated anion channels (VRAC) in endothelial cells. Pflugers Arch 439:315–320
Voets T, Droogmans G, Raskin G, Eggermont J, Nilius B (1999) Reduced intracellular ionic strength as the initial trigger for activation of endothelial volume-regulated anion channels. Proc Natl Acad Sci USA 96:5298–5303
Mongin AA, Kimelberg HK (2005) ATP regulates anion channel-mediated organic osmolyte release from cultured rat astrocytes via multiple Ca2+-sensitive mechanisms. Am J Physiol Cell Physiol 288:C204–C213
Akita T, Fedorovich SV, Okada Y (2011) Ca2+ nanodomain-mediated component of swelling-induced volume-sensitive outwardly rectifying anion current triggered by autocrine action of ATP in mouse astrocytes. Cell Physiol Biochem 28:1181–1190
Akita T, Okada Y (2011) Regulation of bradykinin-induced activation of volume-sensitive outwardly rectifying anion channels by Ca2+ nanodomains in mouse astrocytes. J Physiol 589:3909–3927
Varela D, Penna A, Simon F, Eguiguren AL, Leiva-Salcedo E, Cerda O, Sala F, Stutzin A (2010) P2X4 activation modulates volume-sensitive outwardly rectifying chloride channels in rat hepatoma cells. J Biol Chem 285:7566–7574
Varela D, Simon F, Riveros A, Jorgensen F, Stutzin A (2004) NAD(P)H oxidase-derived H2O2 signals chloride channel activation in cell volume regulation and cell proliferation. J Biol Chem 279:13301–13304
Voets T, Manolopoulos V, Eggermont J, Ellory C, Droogmans G, Nilius B (1998) Regulation of a swelling-activated chloride current in bovine endothelium by protein tyrosine phosphorylation and G proteins. J Physiol 506:341–352
Burow P, Markwardt F (2014) When S1P meets ATP. Channels (Austin) 8:385–386
Akita T, Okada Y (2014) Characteristics and roles of the volume-sensitive outwardly rectifying (VSOR) anion channel in the central nervous system. Neuroscience 275:211–231
Sirianant L, Ousingsawat J, Wanitchakool P, Schreiber R, Kunzelmann K (2015) Cellular volume regulation by anoctamin 6: Ca2+, phospholipase A2, osmosensing. Pflügers Arch. (in press)
Walters EA, Rome L, Luke RG, Galla JH (1991) Absence of a regulatory role of angiotensin II in acute chloride-depletion alkalosis in rats. Am J Physiol 261:F741–F745
Kunzelmann K, Cabrita I, Wanitchakool P, Ousingsawat J, Sirianant L, Benedetto R, Schreiber R (2016) Ca2+ signaling—a common link to diverse functions of anoctamins. Pflügers Arch. (in press)
Jin X, Shah S, Liu Y, Zhang H, Lees M, Fu Z, Lippiat JD, Beech DJ, Sivaprasadarao A, Baldwin SA, Zhang H, Gamper N (2013) Activation of the Cl− channel ANO1 by Localized calcium signals in nociceptive sensory neurons requires coupling with the IP3 receptor. Sci Signal 6:ra73
Almaca J, Tian Y, AlDehni F, Ousingsawat J, Kongsuphol P, Rock JR, Harfe BD, Schreiber R, Kunzelmann K (2009) TMEM16 proteins produce volume regulated chloride currents that are reduced in mice lacking TMEM16A. J Biol Chem 284:28571–28578
Pedemonte N, Galietta LJ (2014) Structure and function of TMEM16 proteins (anoctamins). Physiol Rev 94:419–459
Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK, Oh U (2008) TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 455:1210–1215
Voss FK, Ullrich F, Munch J, Lazarow K, Lutter D, Mah N, Andrade-Navarro MA, von Kries JP, Stauber T, Jentsch TJ (2014) Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344:634–638
Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, Reinhardt J, Orth AP, Patapoutian A (2014) SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 157:447–458
Abascal F, Zardoya R (2012) LRRC8 proteins share a common ancestor with pannexins, and may form hexameric channels involved in cell–cell communication. BioEssays 34:551–560
Hyzinski-Garcia MC, Rudkouskaya A, Mongin AA (2014) LRRC8A protein is indispensable for swelling-activated and the ATP-induced release of excitatory amino acids in rat astrocytes. J Physiol 592:4855–4862
Sorensen BH, Thorsteinsdottir UA, Lambert IH (2014) Acquired cisplatin resistance in humane ovarian cancer A2780 cells correlates with shift in Taurine homeostasis and ability to volume regulate. Am J Physiol Cell Physiol 307:C1071–C1080
Planells-Cases R, Lutter D, Guyader C, Gerhards NM, Ullrich F, Elger DA, Kucukosmanoglu A, Xu G, Voss FK, Reincke SM, Stauber T, Blomen VA, Vis DJ, Wessels LF, Brummelkamp TR, Borst P, Rottenberg S, Jentsch TJ (2015) Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. EMBO J e201592409
Sawada A, Takihara Y, Kim JY, Matsuda-Hashii Y, Tokimasa S, Fujisaki H, Kubota K, Endo H, Onodera T, Ohta H, Ozono K, Hara J (2003) A congenital mutation of the novel gene LRRC8 causes agammaglobulinemia in humans. J Clin Invest 112:1707–1713
Kumar L, Chou J, Yee CS, Borzutzky A, Vollmann EH, von Andrian UH, Park SY, Hollander G, Manis JP, Poliani PL, Geha RS (2014) Leucine-rich repeat containing 8A (LRRC8A) is essential for T lymphocyte development and function. J Exp Med 211:929–942
Gottlieb RA, Dosanjh A (1996) Mutant cystic fibrosis transmembrane conductance regulator inhibits acidification and apoptosis in C127 cells: possible relevance to cystic fibrosis. Proc Natl Acad Sci USA 93:3587–3591
Barriere H, Poujeol C, Tauc M, Blasi JM, Counillon L, Poujeol P (2001) CFTR modulates programmed cell death by decreasing intracellular pH in Chinese hamster lung fibroblasts. Am J Physiol 281:C810–C824
Noe J, Petrusca D, Rush N, Deng P, VanDemark M, Berdyshev E, Gu Y, Smith P, Schweitzer K, Pilewsky J, Natarajan V, Xu Z, Obukhov AG, Petrache I (2009) CFTR regulation of intracellular pH and ceramides is required for lung endothelial cell apoptosis. Am J Respir Cell Mol Biol 41:314–323
Grassme H, Jendrossek V, Riehle A, von Kurthy G, Berger J, Schwarz H, Weller M, Kolesnick R, Gulbins E (2003) Host defense against Pseudomonas aeruginosa requires ceramide-rich membrane rafts. Nat Med 9:322–330
Yu H, Zeidan YH, Wu BX, Jenkins RW, Flotte TR, Hannun YA, Virella-Lowell I (2009) Defective acid sphingomyelinase pathway with Pseudomonas aeruginosa infection in cystic fibrosis. Am J Respir Cell Mol Biol 41:367–375
Teichgraber V, Ulrich M, Endlich N, Riethmuller J, Wilker B, Oliveira-Munding CC, Van Heeckeren AM, Barr ML, von Kurthy G, Schmid KW, Weller M, Tummler B, Lang F, Grassme H, Doring G, Gulbins E (2008) Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat Med 14:382–391
Becker KA, Riethmuller J, Luth A, Doring G, Kleuser B, Gulbins E (2010) Acid sphingomyelinase inhibitors normalize pulmonary ceramide and inflammation in cystic fibrosis. Am J Respir Cell Mol Biol 42:716–724
Nahrlich L, Mainz JG, Adams C, Engel C, Herrmann G, Icheva V, Lauer J, Deppisch C, Wirth A, Unger K, Graepler-Mainka U, Hector A, Heyder S, Stern M, Doring G, Gulbins E, Riethmuller J (2013) Therapy of CF-patients with amitriptyline and placebo—a randomised, double-blind, placebo-controlled phase IIb multicenter, cohort-study. Cell Physiol Biochem 31:505–512
Dimagno MJ, Lee SH, Hao Y, Zhou SY, McKenna BJ, Owyang C (2005) A proinflammatory, antiapoptotic phenotype underlies the susceptibility to acute pancreatitis in cystic fibrosis transmembrane regulator (–/–) mice. Gastroenterology 129:665–681
Rottner M, Kunzelmann C, Mergey M, Freyssinet JM, Martinez MC (2007) Exaggerated apoptosis and NF-kappaB activation in pancreatic and tracheal cystic fibrosis cells. FASEB J. 21:2939–2948
Jungas T, Motta I, Duffieux F, Fanen P, Stoven V, Ojcius DM (2002) Glutathione levels and BAX activation during apoptosis due to oxidative stress in cells expressing wild-type and mutant cystic fibrosis transmembrane conductance regulator. J Biol Chem 277:27912–27918
Rottner M, Tual-Chalot S, Mostefai HA, Andriantsitohaina R, Freyssinet JM, Martinez MC (2011) Increased oxidative stress induces apoptosis in human cystic fibrosis cells. PLoS One 6:e24880
Rubera I, Duranton C, Melis N, Cougnon M, Mograbi B, Tauc M (2013) Role of CFTR in oxidative stress and suicidal death of renal cells during cisplatin-induced nephrotoxicity. Cell Death Dis 4:e817
Linsdell P, Hanrahan JW (1998) Glutathione permeability of CFTR. Am J Physiol 275:C323–C326
Hudson VM (2001) Rethinking cystic fibrosis pathology: the critical role of abnormal reduced glutathione (GSH) transport caused by CFTR mutation. Free Radic Biol Med 30:1440–1461
L’hoste S, Chargui A, Belfodil R, Duranton C, Rubera I, Mograbi B, Poujeol C, Tauc M, Poujeol P (2009) CFTR mediates cadmium-induced apoptosis through modulation of ROS level in mouse proximal tubule cells. Free Radic Biol Med 46:1017–1031
L’hoste S, Chargui A, Belfodil R, Corcelle E, Duranton C, Rubera I, Poujeol C, Mograbi B, Tauc M, Poujeol P (2010) CFTR mediates apoptotic volume decrease and cell death by controlling glutathione efflux and ROS production in cultured mice proximal tubules. Am J Physiol Renal Physiol 298:F435–F453
Okiyoneda T, Barriere H, Bagdany M, Rabeh WM, Du K, Hohfeld J, Young JC, Lukacs GL (2010) Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 329:805–810
Barriere H, Belfodil R, Rubera I, Tauc M, Poujeol C, Bidet M, Poujeol P (2003) CFTR null mutation altered cAMP-sensitive and swelling-activated Cl− currents in primary cultures of mouse nephron. Am J Physiol Renal Physiol 284:F796–F811
Valverde MA, O`Briens JA, Sepulveda FV, Ratcliff RA, Evans MJ, Colledge WH (1995) Impaired cell volume regulation in intestinal crypt epithelia of cystic fibrosis. Proc Natl Acad Sci 92:9038–9041
Gawenis LR, Franklin CL, Simpson JE, Palmer BA, Walker NM, Wiggins TM, Clarke LL (2003) cAMP inhibition of murine intestinal Na/H exchange requires CFTR-mediated cell shrinkage of villus epithelium. Gastroenterology 125:1148–1163
Braunstein GM, Roman RM, Clancy JP, Kudlow BA, Taylor AL, Shylonsky VG, Jovov B, Peter K, Jilling T, Ismailov II, Benos DJ, Schwiebert LM, Fitz JG, Schwiebert EM (2001) Cystic fibrosis transmembrane conductance regulator facilitates ATP release by stimulating a separate ATP release channel for autocrine control of cell volume regulation. J Biol Chem 276:6621–6630
Uramoto H, Okada T, Okada Y (2012) Protective role of cardiac CFTR activation upon early reperfusion against myocardial infarction. Cell Physiol Biochem 30:1023–1038
Suzuki J, Umeda M, Sims PJ, Nagata S (2010) Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468:834–838
Tian Y, Schreiber R, Kunzelmann K (2012) Anoctamins are a family of Ca2+ activated Cl− channels. J Cell Sci 125:4991–4998
Grubb S, Poulsen KA, Juul CA, Kyed T, Klausen TK, Larsen EH, Hoffmann EK (2013) TMEM16F (Anoctamin 6), an anion channel of delayed Ca2+ activation. J Gen Physiol 141:585–600
Shimizu T, Iehara T, Sato K, Fujii T, Sakai H, Okada Y (2013) TMEM16F is a component of a Ca2+-activated Cl− channel but not a volume-sensitive outwardly rectifying Cl− channel. Am J Physiol Cell Physiol 304:C748–C759
Kunzelmann K, Nilius B, Owsianik G, Schreiber R, Ousingsawat J, Sirianant L, Wanitchakool P, Bevers EM, Heemskerk JW (2014) Molecular functions of anoctamin 6 (TMEM16F): a chloride channel, cation channel or phospholipid scramblase? Pflügers Arch 466:407–414
Malvezzi M, Chalat M, Janjusevic R, Picollo A, Terashima H, Menon AK, Accardi A (2013) Ca2+-dependent phospholipid scrambling by a reconstituted TMEM16 ion channel. Nat. Commun. 4:2367
Kmit A, van Kruchten R, Ousingsawat J, Mattheij NJ, Senden-Gijsbers B, Heemskerk JW, Bevers EM, Kunzelmann K (2013) Calcium-activated and apoptotic phospholipid scrambling induced by Ano6 can occur independently of Ano6 ion currents. Cell Death Dis 25(4):e611
Yang H, Kim A, David T, Palmer D, Jin T, Tien J, Huang F, Cheng T, Coughlin SR, Jan YN, Jan LY (2012) TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell 151:111–122
Szteyn K, Schmid E, Nurbaeva MK, Yang W, Munzer P, Kunzelmann K, Lang F, Shumilina E (2012) Expression and functional significance of the Ca-activated Cl− channel ANO6 in dendritic cells. Cell Physiol Biochem 30:1319–1332
Juul CA, Grubb S, Poulsen KA, Kyed T, Hashem N, Lambert IH, Larsen EH, Hoffmann EK (2014) Anoctamin 6 differs from VRAC and VSOAC but is involved in apoptosis and supports volume regulation in the presence of Ca. Pflugers Arch 466:1899–1910
Landoure G, Zdebik AA, Martinez TL, Burnett BG, Stanescu HC, Inada H, Shi Y, Taye AA, Kong L, Munns CH, Choo SS, Phelps CB, Paudel R, Houlden H, Ludlow CL, Caterina MJ, Gaudet R, Kleta R, Fischbeck KH, Sumner CJ (2010) Mutations in TRPV4 cause Charcot-Marie-Tooth disease type 2C. Nat Genet 42:170–174
Takayama Y, Shibasaki K, Suzuki Y, Yamanaka A, Tominaga M (2014) Modulation of water efflux through functional interaction between TRPV4 and TMEM16A/anoctamin 1. FASEB J. 28:2238–2248
Yi E, Lee J, Lee CJ (2013) Developmental role of anoctamin-1/TMEM16A in Ca2+-dependent volume change in supporting cells of the mouse cochlea. Exp Neurobiol 22:322–329
Ponce A, Jimenez-Pena L, Tejeda-Guzman C (2012) The role of swelling-activated chloride currents [I(CL, swell)] in the regulatory volume decrease response of freshly dissociated rat articular chondrocytes. Cell Physiol Biochem 30:1254–1270
Brunner JD, Lim NK, Schenck S, Duerst A, Dutzler R (2014) X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature 516:207–212
Elliott JI, Higgins CF (2003) IKCa1 activity is required for cell shrinkage, phosphatidylserine translocation and death in T lymphocyte apoptosis. EMBO Rep 4:189–194
Owsianik G, Prenen J, Hermans C, Eggermont J, Nilius B (2010) Functional characterization of TMEM16 anion channels. FASEB J (Abstract) 608:12
Milenkovic A, Brandl C, Milenkovic VM, Jendrike T, Sirianant L, Wanitchakool P, Zimmermann S, Reif CM, Horling F, Schrewe H, Strünker T, Alvarez L, Schreiber R, Kunzelmann K, Wetzel CH, Weber BHF (2015) Bestrophin1 is the volume-regulated anion channel in mouse sperm and human retinal pigment epithelium. Proc Natl Acad Sci USA 112:E2630–E2639
Clapham DE (1998) The list of potential volume-sensitive chloride currents continues to swell (and shrink). J Gen Physiol 111:623–624
Jentsch TJ, Stein V, Weinreich F, Zdebik AA (2002) Molecular structure and physiological function of chloride channels. Physiol Rev 82:503–568
Nilius B, Droogmans G (2003) Amazing chloride channels: an overview. Acta Physiol Scand 177:119–147
Grunder S, Thiemann A, Pusch M, Jentsch TJ (1992) Regions involved in the opening of CIC-2 chloride channel by voltage and cell volume. Nature 360:759–762
Duan D, Winter C, Cowley S, Hume JR, Horowitz B (1997) Molecular identification of a volume-regulated chloride channel. Nature 390:417–421
Hermoso M, Satterwhite CM, Andrade YN, Hidalgo J, Wilson SM, Horowitz B, Hume JR (2002) ClC-3 is a fundamental molecular component of volume-sensitive outwardly rectifying Cl− channels and volume regulation in HeLa cells and Xenopus laevis oocytes. J Biol Chem 277:40066–40074
Arreola J, Begenisich T, Nehrke K, Nguyen HV, Park K, Richardson L, Yang B, Schutte BC, Lamb FS, Melvin JE (2002) Secretion and cell volume regulation by salivary acinar cells from mice lacking expression of the Clcn3 Cl− channel gene. J Physiol 545:207–216
Jin NG, Kim JK, Yang DK, Cho SJ, Kim JM, Koh EJ, Jung HC, So I, Kim KW (2003) Fundamental role of ClC-3 in volume-sensitive Cl− channel function and cell volume regulation in AGS cells. Am J Physiol Gastrointest Liver Physiol 285:G938–G948
Liu J, Zhang FF, Li L, Yang J, Liu J, Guan YY, Du YH (2013) ClC-3 deficiency prevents apoptosis induced by angiotensin II in endothelial progenitor cells via inhibition of NADPH oxidase. Apoptosis 18:1262–1273
Matsuda JJ, Filali MS, Moreland JG, Miller FJ, Lamb FS (2010) Activation of swelling-activated chloride current by tumor necrosis factor-alpha requires ClC-3-dependent endosomal reactive oxygen production. J Biol Chem 285:22864–22873
Dermietzel R, Hwang TK, Buettner R, Hofer A, Dotzler E, Kremer M, Deutzmann R, Thinnes FP, Fishman GI, Spray DC (1994) Cloning and in situ localization of a brain-derived porin that constitutes a large-conductance anion channel in astrocytic plasma membranes. Proc Natl Acad Sci USA 91:499–503
Okada SF, O’Neal WK, Huang P, Nicholas RA, Ostrowski LE, Craigen WJ, Lazarowski ER, Boucher RC (2004) Voltage-dependent anion channel-1 (VDAC-1) contributes to ATP release and cell volume regulation in murine cells. J Gen Physiol 124:513–526
Oeggerli M, Tian Y, Ruiz C, Wijker B, Sauter G, Obermann E, Guth U, Zlobec I, Sausbier M, Kunzelmann K, Bubendorf L (2012) Role of KCNMA1 in breast cancer. PLoS One 7:e41664
Koehl GE, Spitzner M, Ousingsawat J, Schreiber R, Geissler EK, Kunzelmann K (2010) Rapamycin inhibits oncogenic intestinal ion channels and neoplasia in APCMin/+Mice. Oncogene 29:1553–1560
Ousingsawat J, Spitzner M, Puntheeranurak S, Terracciano L, Tornillo L, Bubendorf L, Kunzelmann K, Schreiber R (2007) Expression of voltage gated potassium channels in human and mouse colonic carcinoma. Clin Cancer Res 13:824–831
Urrego D, Tomczak AP, Zahed F, Stuhmer W, Pardo LA (2014) Potassium channels in cell cycle and cell proliferation. Philos Trans R Soc Lond B Biol Sci 369:20130094
Pardo LA, Stuhmer W (2014) The roles of K+ channels in cancer. Nat Rev Cancer 14:39–48
Kunzelmann K (2005) Ion channels and cancer. J Membr Biol 205:159–173
Turner KL, Sontheimer H (2014) Cl− and K+ channels and their role in primary brain tumour biology. Philos Trans R Soc Lond B Biol Sci 369:20130095
Shukla A, Edwards R, Yang Y, Hahn A, Folkers K, Ding J, Padmakumar VC, Cataisson C, Suh KS, Yuspa SH (2014) CLIC4 regulates TGF-β-dependent myofibroblast differentiation to produce a cancer stroma. Oncogene 33:842–850
Suh KS, Mutoh M, Nagashima K, Fernandez-Salas E, Edwards LE, Hayes DD, Crutchley JM, Marin KG, Dumont RA, Levy JM, Cheng C, Garfield S, Yuspa SH (2004) The organellular chloride channel protein CLIC4/mtCLIC translocates to the nucleus in response to cellular stress and accelerates apoptosis. J Biol Chem 279:4632–4641
Leanza L, Biasutto L, Manago A, Gulbins E, Zoratti M, Szabo I (2013) Intracellular ion channels and cancer. Front Physiol. 4:227
Stavrovskaya AA (2000) Cellular mechanisms of multidrug resistance of tumor cells. Biochemistry (Mosc) 65:95–106
Lee EL, Shimizu T, Ise T, Numata T, Kohno K, Okada Y (2007) Impaired activity of volume-sensitive Cl− channel is involved in cisplatin resistance of cancer cells. J Cell Physiol 211:513–521
Ise T, Shimizu T, Lee EL, Inoue H, Kohno K, Okada Y (2005) Roles of volume-sensitive Cl− channel in cisplatin-induced apoptosis in human epidermoid cancer cells. J Membr Biol 205:139–145
Shimizu T, Lee EL, Ise T, Okada Y (2008) Volume-sensitive Cl− channel as a regulator of acquired cisplatin resistance. Anticancer Res 28:75–83
West RB, Corless CL, Chen X, Rubin BP, Subramanian S, Montgomery K, Zhu S, Ball CA, Nielsen TO, Patel R, Goldblum JR, Brown PO, Heinrich MC, van de RM (2004) The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol 165:107–113
Duvvuri U, Shiwarski DJ, Xiao D, Bertrand C, Huang X, Edinger RS, Rock JR, Harfe BD, Henson BJ, Kunzelmann K, Schreiber R, Seethala RR, Egloff AM, Chen X, Lui VW, Grandis JR, Gollin SM (2012) TMEM16A, induces MAPK and contributes directly to tumorigenesis and cancer progression. Cancer Res 72:3270–3281
Wanitchakool P, Wolf L, Koehl G, Sirianant L, Gaumann A, Schreiber R, Duvvuri U, Kunzelmann K (2014) Role of anoctamins in cancer and apoptosis. Philos Trans R Soc Lond B Biol Sci 369:20130096
Shiwarski DJ, Shao C, Bill A, Kim J, Xiao D, Bertrand C, Seethala RR, Sano D, Myers JN, Ha PK, Grandis JR, Gaither LA, Puthenveedu MA, Duvvuri U (2014) To “Grow” or “Go”: TMEM16A expression as a switch between tumor growth and metastasis in SCCHN. Clin Cancer Res 20:4673–4688
Qu Z, Yao W, Yao R, Liu X, Yu K, Hartzell HC (2014) The Ca-activated Cl channel, ANO1 (TMEM16A), is a double-edged sword in cell proliferation and tumorigenesis. Cancer Med 3:453–461
Britschgi A, Bill A, Brinkhaus H, Rothwell C, Clay I, Duss S, Rebhan M, Raman P, Guy CT, Wetzel K, George E, Popa MO, Lilley S, Choudhury H, Gosling M, Wang L, Fitzgerald S, Borawski J, Baffoe J, Labow M, Gaither LA, Bentires-Alj M (2013) Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc Natl Acad Sci USA 110:E1026–E1034
Matsuba S, Niwa S, Muraki K, Kanatsuka S, Nakazono Y, Hatano N, Fujii M, Zhan P, Suzuki T, Ohya S (2014) Downregulation of Ca2+-activated Cl− channel TMEM16A by the inhibition of histone deacetylase in TMEM16A-expressing cancer cells. J Pharmacol Exp Ther JPET
He Q, Halm ST, Zhang J, Halm DR (2011) Activation of the basolateral membrane Cl conductance essential for electrogenic K secretion suppresses electrogenic Cl secretion. Exp Physiol 96:305–316
Ruiz C, Martins JR, Rudin F, Schneider S, Dietsche T, Fischer CA, Tornillo L, Terracciano LM, Schreiber R, Bubendorf L, Kunzelmann K (2012) Enhanced expression of ANO1 in head and neck squamous cell carcinoma causes cell migration and correlates with poor prognosis. PLoS One 7:e43265
Jacobsen KS, Zeeberg K, Poulsen KA, Hoffmann EK, Schwab A (2013) The role of TMEM16A (ANO1) and TMEM16F (ANO6) in cell migration. Pflugers Arch 465:1753–1762
Berglund E, Akcakaya P, Berglund D, Karlsson F, Vukojevic V, Lee L, Bogdanovic D, Lui WO, Larsson C, Zedenius J, Frobom R, Branstrom R (2014) Functional role of the Ca-activated Cl channel DOG1/TMEM16A in gastrointestinal stromal tumor cells. Exp Cell Res 326:315–325
Sui Y, Sun M, Wu F, Yang L, Di W, Zhang G, Zhong L, Ma Z, Zheng J, Fang X, Ma T (2014) Inhibition of TMEM16A expression suppresses growth and invasion in human colorectal cancer cells. PLoS One 9:e115443
Bill A, Hall ML, Borawski J, Hodgson C, Jenkins J, Piechon P, Popa O, Rothwell C, Tranter P, Tria S, Wagner T, Whitehead L, Gaither LA (2014) Small molecule-facilitated degradation of ANO1 protein: a new targeting approach for anticancer therapeutics. J Biol Chem 289:11029–11041
Cha JY, Wee J, Jung J, Jang Y, Lee B, Hong GS, Chang BC, Choi YL, Shin YK, Min HY, Lee HY, Na TY, Lee MO, Oh U (2015) Anoctamin 1 (TMEM16A) is essential for testosterone-induced prostate hyperplasia. Proc, Natl Acad Sci USA 201423827
Prevarskaya N, Ouadid-Ahidouch H, Skryma R, Shuba Y (2014) Remodelling of Ca2+ transport in cancer: how it contributes to cancer hallmarks? Philos Trans R Soc Lond B Biol Sci 369:20130097
Schreiber R (2005) Ca2+ signaling, intracellular pH and cell volume in cell proliferation. J Membr Biol 205:129–137
Fink SL, Cookson BT (2006) Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol 8:1812–1825
Hersh D, Monack DM, Smith MR, Ghori N, Falkow S, Zychlinsky A (1999) The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc Natl Acad Sci USA 96:2396–2401
Fischer H, Clauss W (1990) Regulation of Na+ channels in frog lung epithelium: a target tissue for aldosteron action. Pflügers Arch 416:62–67
Silveira TN, Zamboni DS (2010) Pore formation triggered by Legionella spp. is an Nlrc4 inflammasome-dependent host cell response that precedes pyroptosis. Infect Immun 78:1403–1413
Kahlenberg JM, Dubyak GR (2004) Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am J Physiol Cell Physiol 286:C1100–C1108
Franchi L, Kanneganti TD, Dubyak GR, Nunez G (2007) Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J Biol Chem 282:18810–18818
Eisenhut M, Wallace H (2011) Ion channels in inflammation. Pflugers Arch 461:401–421
Casson CN, Shin S (2013) Inflammasome-mediated cell death in response to bacterial pathogens that access the host cell cytosol: lessons from legionella pneumophila. Front Cell Infect Microbiol 3:111
Chen Q, Jin Y, Zhang K, Li H, Chen W, Meng G, Fang X (2014) Alarmin HNP-1 promotes pyroptosis and IL-1beta release through different roles of NLRP3 inflammasome via P2X7 in LPS-primed macrophages. Innate Immun 20:290–300
Keyel PA, Roth R, Yokoyama WM, Heuser JE, Salter RD (2013) Reduction of streptolysin O (SLO) pore-forming activity enhances inflammasome activation. Toxins (Basel) 5:1105–1118
Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P (2014) Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 15:135–147
Barros LF, Hermosilla T, Castro J (2001) Necrotic volume increase and the early physiology of necrosis. Comp Biochem Physiol A Mol Integr Physiol 130:401–409
Linkermann A, Green DR (2014) Necroptosis. N Engl J Med 370:455–465
Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, Ouellette M, King BW, Wisnoski D, Lakdawala AS, DeMartino MP, Casillas LN, Haile PA, Sehon CA, Marquis RW, Upton J, Daley-Bauer LP, Roback L, Ramia N, Dovey CM, Carette JE, Chan FK, Bertin J, Gough PJ, Mocarski ES, Kaiser WJ (2014) RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell 56:481–495
Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148:213–227
Kaczmarek A, Vandenabeele P, Krysko DV (2013) Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38:209–223
Linkermann A, Stockwell BR, Krautwald S, Anders HJ (2014) Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol 14:759–767
Kitur K, Parker D, Nieto P, Ahn DS, Cohen TS, Chung S, Wachtel S, Bueno S, Prince A (2015) Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog 11:e1004820
Kozai D, Ogawa N, Mori Y (2014) Redox regulation of transient receptor potential channels. Antioxid Redox Signal 21:971–986
Bogeski I, Kappl R, Kummerow C, Gulaboski R, Hoth M, Niemeyer BA (2011) Redox regulation of calcium ion channels: chemical and physiological aspects. Cell Calcium 50:407–423
Takahashi N, Kozai D, Kobayashi R, Ebert M, Mori Y (2011) Roles of TRPM2 in oxidative stress. Cell Calcium 50:279–287
Simon F, Varela D, Eguiguren AL, Diaz LF, Sala F, Stutzin A (2004) Hydroxyl radical activation of a Ca2+-sensitive nonselective cation channel involved in epithelial cell necrosis. Am J Physiol Cell Physiol. 287:C963–C970
Barros LF, Stutzin A, Calixto A, Catalan M, Castro J, Hetz C, Hermosilla T (2001) Nonselective cation channels as effectors of free radical-induced rat liver cell necrosis. Hepatology 33:114–122
Miller BA (2006) The role of TRP channels in oxidative stress-induced cell death. J Membr Biol 209:31–41
Patel AJ, Lauritzen I, Lazdunski M, Honore E (1998) Disruption of mitochondrial respiration inhibits volume-regulated anion channels and provokes neuronal cell swelling. J Neurosci 18:3117–3123
Okada Y, Maeno E, Shimizu T, Manabe K, Mori S, Nabekura T (2004) Dual roles of plasmalemmal chloride channels in induction of cell death. Pflugers Arch 448:287–295
McConkey DJ, Orrenius S (1996) The role of calcium in the regulation of apoptosis. J Leukoc Biol 59:775–783
Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG (2014) Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 16:55–65
Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Li W, Zheng X, Chen P, Han J (2014) Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res 24:105–121
Diaz RJ, Harvey K, Boloorchi A, Hossain T, Hinek A, Backx PH, Wilson GJ (2014) Enhanced cell volume regulation: a key mechanism in local and remote ischemic preconditioning. Am J Physiol Cell Physiol 306:C1191–C1199
Sciacca MF, Milardi D, Messina GM, Marletta G, Brender JR, Ramamoorthy A, La Rosa C (2013) Cations as switches of amyloid-mediated membrane disruption mechanisms: calcium and IAPP. Biophys J 104:173–184
Su L, Quade B, Wang H, Sun L, Wang X, Rizo J (2014) A plug release mechanism for membrane permeation by MLKL. Structure 22:1489–1500
Karch J, Kanisicak O, Brody MJ, Sargent MA, Michael DM, Molkentin JD (2015) Necroptosis interfaces with MOMP and the MPTP in mediating cell death. PLoS One 10:e0130520
Tait SW, Oberst A, Quarato G, Milasta S, Haller M, Wang R, Karvela M, Ichim G, Yatim N, Albert ML, Kidd G, Wakefield R, Frase S, Krautwald S, Linkermann A, Green DR (2013) Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep 5:878–885
Schulze-Lohoff E, Hugo C, Rost S, Arnold S, Gruber A, Brune B, Sterzel RB (1998) Extracellular ATP causes apoptosis and necrosis of cultured mesangial cells via P2Z/P2X7 receptors. Am J Physiol 275:F962–F971
Adinolfi E, Pizzirani C, Idzko M, Panther E, Norgauer J, Di Virgilio F, Ferrari D (2005) P2X(7) receptor: death or life? Purinergic Signal 1:219–227
Schreiber R, Faria D, Skryabin BV, Rock JR, Kunzelmann K (2014) Anoctamins support calcium-dependent chloride secretion by facilitating calcium signaling in adult mouse intestine. Pflügers Arch 467:1203–1213
Sirianant L, Ousingsawat J, Tian Y, Schreiber R, Kunzelmann K (2014) TMC8 (EVER2) attenuates intracellular signaling by Zn2+ and Ca2+ and suppresses activation of Cl− currents. Cell Signal 26:2826–2833
Kunzelmann K, Schreiber R (2014) Chloride secretion, anoctamin 1 and Ca2+ signaling. Channels (Austin) 8:387–388
Kunzelmann K, Schreiber R, Kmit A, Jantarajit W, Martins JR, Faria D, Kongsuphol P, Ousingsawat J, Tian Y (2012) Expression and function of epithelial anoctamins. Exp Physiol 97:184–192
Dixon SJ, Stockwell BR (2014) The role of iron and reactive oxygen species in cell death. Nat Chem Biol 10:9–17
Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz PS, Weinlich R, Vanden Berghe T, Vandenabeele P, Pasparakis M, Bleich M, Weinberg JM, Reichel CA, Brasen JH, Kunzendorf U, Anders HJ, Stockwell BR, Green DR, Krautwald S (2014) Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci USA 111:16836–16841
Vorobjeva NV, Pinegin BV (2014) Neutrophil extracellular traps: mechanisms of formation and role in health and disease. Biochemistry (Mosc) 79:1286–1296
Yipp BG, Kubes P (2013) NETosis: how vital is it? Blood 122:2784–2794
Waring P (2005) Redox active calcium ion channels and cell death. Arch Biochem Biophys 434:33–42
Andrabi SA, Dawson TM, Dawson VL (2008) Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann N Y Acad Sci 1147:233–241
Linkermann A, Brasen JH, Darding M, Jin MK, Sanz AB, Heller JO, De Zen F, Weinlich R, Ortiz A, Walczak H, Weinberg JM, Green DR, Kunzendorf U, Krautwald S (2013) Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci USA 110:12024–12029
Laster SM, Wood JG, Gooding LR (1988) Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol 141:2629–2634
Golstein P, Kroemer G (2007) Cell death by necrosis: towards a molecular definition. Trends Biochem Sci 32:37–43
Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Metivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G (2007) Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 13:54–61
Hetz CA, Hunn M, Rojas P, Torres V, Leyton L, Quest AF (2002) Caspase-dependent initiation of apoptosis and necrosis by the Fas receptor in lymphoid cells: onset of necrosis is associated with delayed ceramide increase. J Cell Sci 115:4671–4683
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11:700–714
Acknowledgments
This work was supported by DFG SFB699-A7/A12, DFG KU756/12-1 and Volkswagenstiftung AZ 87 499. We thank Prof. Dr. Stefan Krautwald and PD Dr. Andreas Linkermann (Division of Nephrology and Hypertension, Christian-Albrechts-University Kiel) for providing the NIH-3T3 cell line and critical discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kunzelmann, K. Ion channels in regulated cell death. Cell. Mol. Life Sci. 73, 2387–2403 (2016). https://doi.org/10.1007/s00018-016-2208-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2208-z