
Abstract
According to the EU and Swiss food legislation, only deregulated traits of transgenic plants are allowed to be imported and sold to the consumer. In order to control imports of soya and maize products from retailers, efficient and reliable methods for the detection and quantification are a prerequisite. The screening for specific DNA elements characteristic of transgenic plants is crucial for further analysis and has a major impact on the efficiency of the whole analysis workflow. To allow laboratories to efficiently and reliably screen food products for transgenic plant products, two novel multiplex real-time polymerase chain reaction (PCR) systems were developed and validated. One system determines DNA contents from maize, soya, cauliflower mosaic virus (CaMV) 35S promoter (P35S), NOS terminator from Agrobacterium tumefaciens and CaMV, and the second PCR system simultaneously detects DNA sequences from figwort mosaic virus promoter (PFMV), from bar gene of Streptomyces hygroscopicus, from gene coding for phosphinothricin acetyltransferase (PAT) and from a DNA construct of enolpyruvyl shikimate phosphate synthase gene (CP4-EPSPS) and Arabidopsis thaliana (CPT2). The tests exhibit good specificity and a limit of detection of at least 0.1 % for all analytes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ever since the introduction of the first transgenic plant, the Flaver SavrTM tomato, many other transgenic or genetically modified plants (GMO) have been developed and to some extent successfully commercialized. The number of commercially available transgenic plants is constantly rising. Hence, the variety as well as the number of all possible target sequences that need to be detected is steadily increasing, which represents immense analytical effort and great challenge.
According to the EU and Swiss legislation for food, only deregulated traits of transgenic plants are allowed to be imported and sold to the consumer. If the product contains more than 0.9 % of a deregulated transgenic plant material, it has to be labelled. The lion’s share of all GMO traits still remains among the crops maize and soya. In order to control imports of soya and maize products from retailers, efficient and reliable methods for the detection and quantification are a prerequisite. Most of the transgenic plants contain constructs with sequences of the cauliflower mosaic virus 35S promoter (PFMV) and/or the NOS terminator from Agrobacterium tumefaciens (Tnos). Therefore, these gm constructs are suitable as detection gene markers in screening assays and for the quantification of GMO fraction in food. It is important to estimate the overall GMO content of the tested sample in the initial screening step in order to determine whether further analysis is required. Historically, many different single PCR systems are applied for the purpose of testing raw and processed products on the presence of GMO. They are laborious and material consuming. Additionally, after the analysis, the results have to be collected and charted, which is prone to mistakes. Therefore, there was a need for a more comprehensive approach, enabling the maximum yield of information out of the same amount of extracted DNA. Multiplex PCR systems have already been developed. Some of them require visualization by gel electrophoresis, thus providing only qualitative results [1–5]. Others are composed by a multiplex PCR followed by post-PCR analyses like capillary electrophoresis [6–10] or microarray based [11–15], which offer only qualitative insights. Or they use pre-portioned single systems requiring high amount of isolated DNA and consumables like DNA polymerase [16, 17]. All these systems need post-PCR analysis which have an intrinsically contamination potential and are time-consuming and/or need expensive instrumentation.
One of the first quantitative multiplex PCR systems was designed for the determination of four transgenic maize lines [18]. Several other systems were designed for other transgenes or screening elements [19–24]. Screening methods have become increasingly important to minimize the cost of an analysis and the analytical effort and to offer an exact preselection for further analysis. Earlier publications often presented methods only for the transgenic markers without including housekeeping genes to determine the DNA species, making it impossible to calculate a relative amount of GMO. But this is a prerequisite for the decision if further analysis is required.
Here, we present the validation data of two respective systems—a pentaplex and a tetraplex quantitative real-time PCR system—for GMO screening in commodities, food and feed products.
The pentaplex system enables the simple and sensitive and quantitative estimation of the proportion of transgenic plants containing the cauliflower mosaic virus 35S promoter (P35S) and/or the NOS terminator from Agrobacterium tumefaciens (Tnos) in maize and soya samples. The tetraplex system provides a simultaneous quantitative screening for DNA sequences from figwort mosaic virus 34 S promoter (PFMV), from bar gene of streptomyces hygroscopicus, from a synthetic gene coding for phosphinothricin acetyltransferase (PAT) and from a DNA construct spanning the junction between enolpyruvyl shikimate phosphate synthase gene (CP4-EPSPS) from Agrobacterium tumefaciens ssp. strain 4 and from a chloroplast transit peptide signal sequence from Arabidopsis thaliana (CPT2).
Materials and methods
Plant material and DNA samples
Reference material was obtained from Sigma-Aldrich (Buchs, Switzerland), European Reference Material (ERM, Geel, Belgium). Further plant materials for specificity testing were collected from the market. Materials from proficiency tests organized by FAPAS (Sand Hutton, UK) and USDA Grain Inspection, Packers & Stockyards Administration (GIPSA) were used for further validation. Bt176 (ERM BF411f) served as source for the transgenic marker sequences of bar; for PAT, Bt11 (ERM 412f); for CP4-EPSPS, NK604 (ERM BF415f); and for PFMV, the transgenic potato NewLeaf plus (Russet Burbank, not certified). A concentration of 20 ng/μl was assigned as 100 %. As source for the cauliflower mosaic virus DNA, an amplicon from a single PCR was taken from a broccoli sample from the market. The PCR amplicon was confirmed by P35S-specific PCR and diluted 1 to 1E8 in PCR-grade Water. This dilution was assigned to be 10 % (or 2 ng/μl). As virus contents and transgenic content cannot be compared, this arbitrary definition seemed acceptable and practicable.
DNA extraction
DNA extraction from all sample matrices was performed using a Wizard Plus Minipreps DNA purification system (Promega, Madison, USA). In parallel, extractions were performed also using Nucleospin Food kit (Macherey–Nagel, Düren, Germany), leading to comparable results. Usually, 200 mg of grinded sample material was extracted, and DNA was eluted in 50 µl elution buffer, according to the producer’s manual. If possible, the DNA amount was determined photospectrometrically and diluted to 20 ng/μl using PCR-grade water.
Primers and probes
The primers and probes were taken either from published single PCR systems or established in “in-house” PCR systems. For all in-house designed (by Beacon Designer 5.1 software) PCR systems, specificity was first tested without probes in the single SYBR green format. This is useful to detect and avoid PCR systems amplifying unspecific sequences. After an additional first check of specificity, probes were designed according to the channels of the Rotorgene 6000. We choose FAM-, Tet-, ROX-, Cy5- and DY681-labelled probes (for details, see Tables 1 and 2).
Real-time PCR procedure
DNA extracts (5 μl) were added to 20 μl of reaction mix containing QuantiFast multiplex PCR NoROX Mastermix (Qiagen AG, Hilden, Germany), primers and probes (for final concentration, see Tables 1 and 2). All primers and probes were synthesized by Microsynth AG (Balgach, Switzerland). PCR was performed on the Rotorgene 6000 real-time system (Corbett, Australia/Qiagen), according to the following cycling protocol: for the pentaplex system “AllGVOScB”: initial step of 5 min at 95 °C, followed by 45 cycles of 5 s at 95 °C and 45 s at 60 °C, and for the tetraplex system “AllGVOScC”: initial step of 5 min at 95 °C, followed by 45 cycles of 5 s at 95 °C and 45 s at 62 °C. In parallel, the tests were conducted on Mx3005P (Stratagene, USA), showing that one thermoprofile may be used for both systems: initial step 10 min at 95 °C, followed by 45 cycles of 10 s at 95 °C, 45 s at 62 °C and 30 s at 72 °C, where single fluorescence measurement is made.
Results and discussion
Design of the multiplex real-time PCR systems
Specificity
All primers and probes were successfully checked for lack of relevant homologies by BLAST nr search within GenBank databases. DNA from a wide range of plants and other food ingredients was isolated. The success of isolation was tested by photospectrometrically determination of the DNA content (100 ng was used as template per reaction). DNA of the following organisms was isolated and tested in the multiplex PCR system AllGMOSc1: beef, trout, white beans, lentils, kidney beans, mung beans, borlotti beans, chickpeas, peas, runner beans, wheat, tomato, potato, rice, plum, apricots, peanuts, hazelnut, almonds, walnut, white lupine, blue lupine, onion, garlic, carrot, celery, parsley, nutmeg, white pepper, cinnamon, aniseed, coconut and paprika. The only cross-reactivity observed was for lupines. They showed a cross-reactivity of maximum 0.6 % in the lectin system (soya). This must be considered when examining samples containing material from lupine. To demonstrate the detectability of GMO, the following transgenic plants were tested: maize (Bt176, Bt11, MON810, GA21, T25, StarLink, MON863, TC1507, MON89034, MON88017, NK603, 59122, MIR604), rice (LL601, LL62, KMD1), soya (GTS40-3-2 (RoundUp Ready), 2704, 5547) and rapeseed (Oxy235). All of them gave the expected signals according to the screening list [29].
Sensitivity, precision and measurement uncertainty
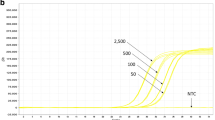
To evaluate the sensitivity, DNA extracts with a concentration of 20 ng/μl were diluted in herring sperm DNA solution (20 ng/μl) down to a transgenic concentration of 0.032 % transgenic concentration (equal to 6.4 pg/μl). Amplicons were used in dilutions as described. These single analyte dilution rows were mixed according to Tables 3 and 4 to produce multiplex standards, simulating samples containing all four and five target sequences. Each data point was analysed six times (N = 6) over a period of 1 month. All samples showed 100 % positive signals down to 0.032 % concentrations of the analytes. None of the six negative controls showed a positive signal. The quantitative data were collected, and the relative standard deviation (rSD) was calculated for the estimation of the precision. The mean deviation from the true value served for the estimation of the accuracy (Tables 5, 6).
The quantification of GMO by quantitative PCR based on screening elements is only possible for samples enclosing one single GMO trait in conjunction with the same GMO trait as calibrator, due to the different relations of marker and housekeeping genes in different GMO traits. Therefore, in practice, this quantitative approach leads only to a rough estimation of the GMO content. But this is already important to be able to divide GMO free or slightly contaminated samples from substantial mixtures of GMO.
Analysis of samples from proficiency testing programs
Table 7 shows the true values and the values measured by the system “AllGVOSc1”. Correlation between measured and assigned values was poor. This reflects mainly the problem of calibration. Samples can only be quantified using the corresponding reference material, as different insertion numbers of the measured screening elements and/or copy numbers of the housekeeping genes have a great impact on the result. As it is unknown at the moment of analysis of unknown samples, this prerequisite is impossible to be fulfilled. In addition, products made from transgenic plants often include a mixture of transgenes. In consequence, it is impossible to choose a single corresponding transgene as calibrator.
Conclusions
Herein, we showed GMO screening methods for the reliable detection of four and five target sequences, respectively, in multiplex real-time PCRs, each performed in one tube. In this way, we could accommodate the detection of up to all eight target DNA sequences in each tested sample. Furthermore, combining two multiplex systems by a common thermoprofile on one of the tested thermocyclers, we propose a way for a high throughput screening of all eight target sequences at once.
Additionally, two of these analytes in system “AllGVOSc1” were present in a roughly 100 times higher concentration. Nevertheless, it was possible to determine the minor components with high accuracy. This shows that when using optimal primers and concentrations, the parallel amplification of multiple target sequences in a balanced multiplex real-time PCR is possible without reciprocal interference. In conjunction with the here proposed two multiplex standard rows, maize and soya samples can be analysed in a very efficient manner. Cross-contaminations of maize samples with soya and inverse can easily be detected in raw and processed products, making the interpretation clearer and hence further analysis more straightforward. Furthermore, the detection of the plant-derived DNA sequences (lectin and/or mhmg) can serve as an internal control of the DNA quality, especially if DNA is derived from processed and refined sample material. The size of the amplicons is adjusted to detect the targets even in strongly fragmented DNA, common in highly processed food samples. Additionally, the maize- and soya-specific assay comprises an internal amplification control to detect inhibitions. Rare transgenic traits can be herewith detected and characterized, and they are in accordance with the compilation of the relevant GMO traits [29]. This thorough screening step is also one possible analytical strategy to detect non-approved GMOs for which no specific assays, reference materials or even the sequences are available. However, the quantification should still be done by transgene-specific PCR systems due to different insertion numbers of the here used screening elements and/or copy numbers of the housekeeping genes.
References
Permingeat H, Reggiardo M, Vallejos R (2002) Detection and quantification of transgenes in grains by multiplex and real-time PCR. J Agric Food Chem 50:4431–4436
James D, Schmidt A, Wall E, Green M, Masri S (2003) Reliable detection and identification of genetically modified maize, multiplex PCR analysis. J Agric Food Chem 51:5829–5834
Hernandez M, Rodrigez-Lazaro D, Zhang D, Esteve T, Pla M, Prat S (2005) Interlaboratory transfer of a PCR multiplex method for simultaneous detection of four genetically modified maize lines: Bt11, Mon810, T25 and GA21. J Agric Food Chem 53:3333–3337
Randhawa GJ, Chhabra R, Singh M (2009) Multiplex PCR-based simultaneous amplification of selectable marker and reporter genes for the screening of genetically modified crops. J Agric Food Chem 57:5167–5172
Randhawa GJ, Chhabra R, Singh M (2010) Decaplex and real-time PCR based detection of Mon531 and Mon15985 Bt cotton events. J Agric Food Chem 58:9875–9881
Ehlert A, Moreano F, Busch U, Engel KH (2008) Development of a modular system for the detection of genetically modified organisma in food based on ligation-dependent probe amplification. Eur Food Res Technol 227:805–812
Heide B, Dromtorp S, Rudi K, Heir E, Holck A (2008) Determination of eight genetically modified maize events by quantitative, multiplex PCR and fluorescence capillary gel electrophoresis. Eur Food Res Technol 227:1125–1137
Garcia-Canas V, Cifuentes A (2008) Simultaneous confirmatory analysis of different transgenic maize (Zea maize) lines using multiplex polymerase chain reaction-restriction analysis and capillary gel electrophoresis with laser induced fluorescence detection. J Agric Food Chem 56:8280–8286
Holck A, Pedersen B, Heir E (2010) Detection of five novel GMO maize events by qualitative, multiplex PCR and fluorescence capillary gel electrophoresis. Eur Food Res Technol 231:475–483
Luan F, Tao R, Xu YG, Wu J, Guan XJ (2012) High-throughput detection of genetically modified rice ingredients in foods using multiplex polymerase chain reaction coupled with high-performance liquid chromatography method. Eur Food Res Technol 234:649–654
Xiaodan X, Li Y, Zao H, Wen S-Y, Wang S-Q, Huang J, Huang K-L, Luo Y-B (2005) Rapid and reliable detection and identification of GM events using multiplex PCR coupled with oligonucleotide microarray. J Agric Food Chem 53:3789–3794
Bordoni R, Germini A, Mezzelani A, Marchelli R, De Bellis G (2005) A microarray platform for parallel detection of five transgenic events in foods: a combined polymerase chain reaction-ligation detection reaction-universal array method. J Agric Food Chem 53:912–918
Schmidt A, Sahota R, Pope D, Lawrence T, Belton M, Rott M (2008) Detection of genetically modified canola using multiplex PCR coupled with oligonucleotide microarray hybridization. J Agric Food Chem 56:6791–6800
Hamels S, Glouden T, Gillard K, Mazzara M, Debode F, Foti N, Sneyers M, Nuez TE, Pla M, Berben G, Moens W, Bertheau Y, Audéon C, Van den Ede G, Remacle J (2009) A PCR-microarray method for the screening of genetically modified organisms. Eur Food Res Technol 228:531–541
Mano J, Shigemitsu N, Futo S, Akyama H, Teshima R, Hino A, Furui S, Kitta K (2009) Real-time PCR array as a universal platform for the detection of genetically modified Crops and its application in identifying unapproved genetically modified crops in Japan. J Agric Food Chem 57:26–37
Gerdes L, Busch U, Pecoraro S (2011) Parallelised real-time PCR for identification of maize GMO events. Eur Food Res Technol 234:315–322
Kluga L, Folloni S, Van den Bulcke M, Van den Eede G, Quarci M (2012) (Application of the “real-time PCR-based Ready-to-use multi target analytical system for GMO detection” in processed maize matrices. Eur Food Res Technol 234:109–118
Brodmann P, Ilg E, Berthoud H, Herrmann A (2002) Real-time quantitative polymerase chain reaction methods for four genetically modified maize varieties and maize DNA content in food. J AOAC Int 85(3):646–653
Germini A, Zanetti A, Salati C, Rossi S, Forré C, Schmid S, Marchelli R (2004) Development of seven-target multiplex PCR for the simultaneous detection of transgenic soybean and maize in feeds and foods. J Agric Food Chem 52:3275–3280
Foti N, Onori R, Donnarumma E, De Santis B, Miraglia M (2006) Real-time PCR multiplex method for the quantification of roundup ready soybean in raw material and processed food. Eur Food Res Technol 222:209–216
Waiblinger HU, Ernst B, Anderson A, Pietsch K (2008) Validation and collaborative study of a P35S and T-nos duplex real-time PCR screening method to detect genetically modified organisms in food products. Eur Food Res Technol 226:1221–1228
Bahrdt C, Krech AB, Wurz A, Wulff D (2010) Validation of a newly developed hexaplex real-time PCR assay for screening for presence of GMOs in food, feed and seed. Anal Bioanal Chem 396(6):2103–2112
Dörries HH, Remus I, Grönewald C, Berghof-Jäger K (2010) Development of a qualitative, multiplex real-time PCR kit for screening of genetically modified organisms (GMOs). Anal Bioanal Chem 396(6):2043–2054
Huber I, Block A, Sebah D, Debode F, Morisset D, Grohmann L, Berben G, Stebih D, Milavec M, Zel J, Busch U (2013) Development and validation of duplex, riplex and pentaplex real-time PCR screening assays for the detection of genetically modified organisms in food and feed. J Agric Food Chem 61:10293–10301
Mazzara M, Foti N, Price S, Paoletti C, Vand den Eede G (2005) Event-specific method for the quantitation of maize line TC1507 using real-time PCR European commission. Joint Res Center (in preparation)
Swiss Book of Foodstuffs (SLMB), Chapter 52B point 2.1.7 (in preparation)
Chaouachi M, Fortabat MN, Gerldreich A, Yot P, Kerlan C, Kebdani N, Audeaon C, Pomaniuk M, Bertheau Y (2008) An accurate real-time PCR test for the detection and quantification of cauliflower mosaic virus (CaMV): applicable in GMO screening. Eur Food Res Technol 227(3):789–798
Köppel R, Zimmerli F (2010) Proposal for a performance factor to characterize real-time PCR systems, evaluate premixed DNA-polymerases and assign minimal performance criteria for individual quantitative PCR runs. Eur Food Res Technol 231:727–732
Waiblinger H, Grohmann L, Mankertz J, Engelbert D, Pietsch K (2010) A practical approach to screen for authorised an unauthorised genetically modified plants. Eur Food Res Technol 396:2065–2072
Official Collection of Test Methods (2014) Detection of a DNA sequence from the figwort mosaic virus promoter (PFMV) by real-time PCR in materials derived from genetically modified organisms (GMO) in foodstuffs—element specific method. German food and feed law—food analysis, article 64, L 00.00-148. Beuth, Berlin
German Official Collection of Test Methods (2008) Specific detection of a frequently used DNA sequence from genetically modified organisms (GMO) derived from the bar-gene of Streptomyces hygroscopicus in foodstuffs—screening method. German food and feed law—food analysis, article 64, L 00.00-124. Beuth, Berlin
Official Collection of Test Methods (2008) Detection of the CTP2-CP4-EPSPS gene sequence for screening of materials derived from genetically modified organisms (GMO) in foodstuffs—construct specific method. German food and feed law—food analysis, article 64, L 00.00-125. Beuth, Berlin
Waiblinger H-U, Huber I, Palisch A, Köppel R (2014) Collaborative trial validation of a single and a multiplex real-time PCR assay for screening of genetically modified plants (in preparation)
Acknowledgments
We thank the cantonal laboratory of Zürich for providing the resources for this work.
Conflict of interest
None.
Compliance with Ethics Requirements
This article does not contain any studies with human or animal subjects.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Köppel, R., Sendic, A. & Waiblinger, HU. Two quantitative multiplex real-time PCR systems for the efficient GMO screening of food products. Eur Food Res Technol 239, 653–659 (2014). https://doi.org/10.1007/s00217-014-2261-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-014-2261-5