
Abstract
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The controversy surrounding agribiotech crops since the 1990s has been particularly important in the European Union. It also led to the release of several European directive and regulations (2001/18/EEC, 1829/03/EC and 1830/03/EC) able to keep the consumers their freedom of choice. Therefore, processed food and feed containing or derived from genetically modified organisms (GMOs) have to be labelled above a threshold of fortuitous presence. For approved GMO, this threshold is 0.9% per ingredient in EU [1], 5% in Japan [2] and 3% in South Korea (http://www.doh.gov.tw/).
The development and the application of reliable and cost-effective methods based on molecular tools to detect GMOs are thus essential in order to precisely label and control food and feed for offering free choice to consumer and for ensuring the traceability from “farm to fork” and vice-versa [3] This traceability will allow the tracing of GMO materials and derived products along the supply chains [4]. All these aspects are studied in a recent European project called Co-Extra (Co-Existence and Traceability; 2005–2009), which should provide practical tools and methodologies for practically implementing co-existence.
The reference technique for gene quantification is real-time PCR. It is proved to be more suitable for the diagnostic laboratory than conventional PCR, due its quantitative performance, its sensitivity and the use of closed tube assays [5]. The real time PCR have been used in most of the methods developed in quantitative analysis of GMO [6]. Many authors looked for more sensitivity and reviewed the application of alternative techniques such as the ligation-dependent probe amplification (LPA) technique by Moreano et al. [7], a quartz crystal microbalance (QCM) DNA-sensor by Passamano et al. [8] or, recently, microarrays by Leimanis et al. [9].
To use the developed methods in routine analysis, its validation have to be performed through inter-laboratory studies which is the case for example of the P35S test validated in 15 French laboratories and based on the strategy promoted by the European Network of GMO Laboratories (ENGL, http://engl.jrc.it/) [10]. Also, in Europe, the Community Reference Laboratory for GM Food and Feed (CRL) has as a core task, the assessment and the validation of the GMO detection methods as part of the European Commission authorization procedure (http://gmo-crl.jrc.it/).
To maintain cost-effectiveness of detection methods, the routine detection labs use generally an initial step of “screening” which target elements present in the inserts of numerous GMOs [11]. In case of positive results, a second step of specific PCR attempts identifies and quantifies the GMO [12] relatively to the plant taxon [13]. The accuracy of the first step (screening) by, e.g., avoiding false positive or false negative results is a crucial requirement.
Screening tests are of major importance for routine analysis in food and feed but also in other kind of materials such as seeds and grains. According to the biotech crop databases BATS (Biosafety Assessment, Technology and Sustainability, http://www.bats.ch/gmo-watch/) and Agbios (http://www.agbios.com), promoters and terminators coming from the donor organisms CaMV and Agrobacterium tumefaciens are the most commonly used in the currently approved GMO. Some tests have to be developed to distinguish the presence of donor organism, i.e., the taxon from which the sequences of the GMO insert originated. While some of these controls are qualitative, quantitative tests are required to clarify the situation when both GMO and donor organism are present and for use, e.g., in quantitative differential PCR for assessing the presence of unknown GMOs (Ancel et al. unpublished data).
Cauliflower mosaic virus (CaMV) is a 8 kbp member of caulimovirus whose genome is composed of a double stranded circular DNA [14]. It is made of six open reading frames (ORF) and encodes for six proteins [15–17]. Our PCR targeted region is located in the ORFIII [10] and encodes a small basic protein organized into two functional domains located towards the N and C terminal regions. Both domains are required for CaMV infectivity [18] and are thus always present in CaMV strains, in particular the few strains of CaMV (W260, D4 and B29) known to infect solanaceous or both solanaceous and brassicacae (D/H, Bari and CabbS) [19]. As it is known, many genetically modified plants, such as potatoes or papaya, have been created to confer resistance to viruses (e.g., NewLeaf Y Russet Burbank potato) [20]. Since the ORFIV used by Cankar et al. codes for a 56 kDa viral capsid protein (P4), this target might be used later in the construction of GMOs. Thus, probable discordance in specificity results might be observed in the future causing false positives. To avoid such problems, we have chosen the ORFIII as target for designing our CaMV specific test. It codes for a 15 kDa (kilodalton) protein (P3) of 129 AA involved in the CaMV infectivity. Our work describes a quantitative assay that can distinguish the presence of CaMV in presence or not of GMO, and its advantages when compared to other previously developed tests. It demonstrates also the ability of this test to be used in a combination with our previously validated P35S method [10] used worldwide.
Materials and methods
Plant material
Samples from Solanaceae family such as pepper (Capsicum annuum, Capsicum baccatum, Capsicum cardenasii, Capsicum chacoense and Capsicum chinense) and tomato varieties (Lycopersicon esculentum, Lycopersicon cerasiforme, Lycopersicon pimpinellifolium) and eggplant (Solanum melongena), Mock tomato (Solanum aethiopicum subsp. Kumba, Solanum aethiopicum subsp. aculeatum, Solanum aethiopicum subsp. Shum and Solanum aethiopicum subsp. Gilo) were provided by the laboratoire de Génétique et d’Amélioration des Fruits et Légumes, (INRA, Domaine St Maurice, France). Brassicaceae species of the triangle of U [21] were also tested. Cauliflower (Brassica oleraceae), Chinese cabbage (Brassica rapa), mustard (Brassica juncea), black mustard (Brassica nigra) were provided by the Laboratoire d’Amélioration des Plantes et Biotechnologies Végétales (INRA; Rennes, France). The other species used for the specificity test were Arabidopsis thaliana, sugar beet (Beta vulgaris subsp. vulgaris), cotton (Gossypium barbadense and Gossypium hirsutum), Barley (Hordeum vulgare), Soybean (Glycine max), rice (Oryza sativa), Line (Linum usitatissimum), alfalfa (Medicago truncatula), white bean (Phaseolus aureus), pea (Pisum sativum), cereal rye (Secale cereale), wheat (Triticum durum and Triticum aestivum), maize and teosinte (Zea mays and Z. diploperennis). Transgenic material used was maize T25, MON810, Bt176, Bt11, Bt10, GA21 and CBH351, soybean RR, sugarbeet GTSB77, rapeseed (GT73, RF1, RF2, RF3, MS1, MS8) and rice (LL62, provided for research only from JRC; Ispra, Italy). As previously reported, plants were grown in greenhouses and than collected and stored at −20°C until DNA extraction [22].
Donor organism material
The origin of CaMV, FMV and CERV strains used in this paper are provided in Table 1. Several types of DNA were then used for the tests: DNA extracted from infected leaves, pure CaMV virus and DNA, and plasmids either containing the targeted amplicon, or a wider overlapping fragment already published by our laboratory [22]. Agrobacterium tumefaciens (strain EHA105) was also used for specificity assessment in order to confirm the reliability even in the presence of a contamination with the promoter and terminator NOS donor organism.
Extraction of the CaMV virus and its DNA
Plants were infected using the classical method with mechanical transmission a of the virus inoculums on the whole surface of the leaves. The method used to extract the virus genome from the infected plants was carried out according to Hull et al. (1976) [23]. The leaves were grinded in a phosphate buffer (0.5 M, pH 7.5), 2.5% Triton 100× and urea 1 M and placed at 4 °C overnight. After a 20 min centrifugation at 4°C, 7,000g, the supernatant was filtered and centrifuged again for 90 min at 70,000g between 10 and 25 °C. Then, the supernatant was discarded and the pellet was homogenized in 0.5 mL of ultra pure water. A chloroform extraction was then performed. After a 15 min centrifugation at 15 °C, 1,200g, the supernatant was loaded on a 25% saccharose gradient (kept at −20 °C overnight). A centrifugation was performed during 2 h at 10 °C, 90,000g and the virus particles were collected from one of the saccharose phases (white color). This extract was diluted twice in ultra pure water and centrifuged during 1 h 30 min at 200,000g at 10 °C. The pellet containing the virus particles was gently resuspended in 100–200 μL of ultra pure water. This pellet was treated with DNAseI (10 μg/mL) during 10 min at 37 °C to eliminate exogenous cellular DNA. EDTA (1 mM) was finally added to stop the reaction. As an average, 1 kg of infected leaves gives approximately 10 mg of virus DNA (unpublished data).
To extract the DNA from the virus particles, SDS buffer (0.5%) was added in a 1 mL final volume with proteinase K (50 μg/mL) and Tris 0.1 M. The mixture was incubated overnight at 37 °C. The day after, a phenol–chloroform extraction was performed with a 10 min centrifugation at 90,000g, 15 °C. The supernatant was collected and mixed with two volumes of cold absolute ethanol, 1 μL of glycogen (2 mg/mL), NaCl (1 M). A precipitation was carried on at −80 °C during 15 min minimum. Then, a centrifugation was performed at 4 °C during 30 min, 90,000g. After the supernatant was eliminated, the pellet was dried and resuspended in 50 μL of ultra pure water.
The CaMV particles and the resulting DNA were both quantified by a spectrophotometer at 260 nm wavelength: 7 OD units correspond to 1 mg of virus particles and 1 OD unit corresponds to 50 μg/mL of double-strand DNA. The quality of virus DNA was checked on a 0.8% agarose electrophoresis gel.
Extraction of plant material
The total DNA was extracted from plant leaves with a CTAB method [24]. In order to check and quantify the DNA extractions, the samples were loaded on 0.8% agarose gels stained with Ethidium Bromide and quantified by Bio1D software (Vilbert Lourmat, Marne la Vallée, France) in comparison with a calibration curve of commercial λDNA (MBI Fermentas, Vilnius, Lituania) with a concentration range (10, 20 and 30 ng). The DNA amplifiability was assessed by the universal test [25].
Sequencing of the ORFIII 260 bp-fragment
The sequencing of the 260 bp amplicons [22] was performed in a 20 μL reaction containing 3.2 pmol of primer (forward or reverse), 4 μL of BigDye Terminator Cycle Sequencing reaction mixture (Perkin–Elmer), 1.6 μL of 5× sequencing buffer, 3.2 μL of H2O and 8 μL of the purified PCR product. The products of this reaction were purified using a G50 gel filtration [Sephadex® G-50 superfine (Amersham Biosciences, Hercules, CA, USA)] and loaded to an ABI 310 sequencer. Sequence alignments and detection of polymorphism were performed using the software Chromas version 2.32.
Primers and probe design and PCR conditions
The primers and probe (Table 2) were designed with the Primer Express software (version 2.1, Applied Biosystems, Foster, California) inside the 260 bp fragment previously described [10, 22], in to amplify a 152 bp amplicon. The DNA plant quality was checked with universal plant primers. All the simplex experiments were made with a TaqMan® probe labelled with FAM and black Hold quencher (BHQ).
The real-time amplifications were performed with the Real-time Master MIX provided by Applied Biosystems, 200 nM of the probe, 300 nM of the primers, and 2.5 mM of Mg2+.
Cloning of the 152 bp target
The 152 bp amplicon was cloned from the PCR amplicon obtained from infected leaves of the PV148 CaMV strain in a TA-cloning pGEM-T vector (Promega, Charbonnières, France). This plasmid is available after its deposit in the BCCM / LMBP Ghent (Belgium; http://bccm.belspo.be/) plasmid bank by Institut Scientifique de Santé publique, (ISP; Section de Biosecurité et Biotechnologie, SBB; Brussels, Belgium). It was used in this work for the sensitivity studies.
Results and discussion
Qualitative test with conventional PCR: specificity of the 152 bp test
The specificity is one of the PCR test performance criteria (EN ISO 24276) [26]. The Procedural Manual of the Codex Alimentarius and EN ISO 24276 defines this criterion as the ability of a method to respond exclusively to the characteristic or analyte. In other terms, it describes the absence of false positives. Theoretical and experimental specificity were assessed.
Theoretical specificity
Sequence specificity of the CaMV primers and probes was examinated by the Basic Local Alignment Search Tool (Blast) on the website of the National Centre for Biotechnology Information (NCBI) (http://www.ncbi.nih.gov/BLAST/) using DDBJ (DNA Database of Japan; release 5.0 December 2002), EMBL (European Molecular Biology Laboratory) and GenBank (Accession number X79465). The results of Blast confirmed the theoretical specificity of the targeted region. It was almost conserved among the CaMV strains while no obvious homology was observed with other virus groups such as Figwort Mosaic virus (FMV) and Carnation Etched Ring virus (CERV) (data not shown).
Experimental specificity
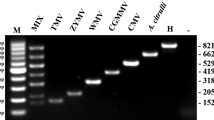
Experimental specificity of the primers targeting the ORF III of the CaMV was assessed against several targets: Agrobacterium tumefaciens, CaMV, CERV and FMV strains, GMOs, and non-transgenic plants including especially the CaMV and the CERV hosts. Results are presented in the Table 3. Only samples containing the CaMV genome or the cloned 152 bp fragment were amplified (Fig. 1). Parallel tests with the primers SF/SR targeting P35S, were performed with all the templates containing this promoter. We concluded that the two tests are independent and no interferences can occur between the two targets. According to these results, our qualitative PCR test is highly specific of the ORFIII of CaMV strains and does target neither CERV nor FMV viruses.

Migration of the PCR products in 4% agarose gel. Experimental specificity with GMO samples 1 low marker, 2 Ji virus, 3 no template control, 4 cabbage, 5 event T25 maize, 6 Mon810 maize, 7 Bt11 maize, 8 GTS40-3-2 “RRS” soybean, 9 Starlink (CBH351) maize and 10 event 176 maize
Confirmation of the specificity by sequencing
To further assess the specificity of our PCR test and to confirm the specificity of the internal probe, the PCR amplicons of seven CaMV strains (Fig. 2) namely, PV148, PV303, PV0227, PV0229, D4, Ji and CabbS were sequenced. The alignments show a conserved 152 bp sequence for the PV0227, PV0229, D4 strains, for the CabbS and Ji virus DNAs, while a 154 bp amplicon was observed for the PV148 and PV303 strains (data not shown). The main differences between viruses in the collection tested are the genome size and the infectivity spectra.

Migration of the PCR products in 4% agarose gels. Single PCR amplification of the fragment ORFIII. Gel amplification products of PCR corresponding to: 1 biomarker low ladder, 2 virus Ji, 3 virus CabSS, 4 clone 148 bp, 5 clone CabbS, 6 PV148, 7 PV0229, 8 PV303, 9 PV0227, 10 C.K1, 11 C.K2
Cloning of the 152 bp amplicon
According to Mattarucchi et al. [27] the use of a linearised or not plasmid DNA as a standard in GMO detection and quantification could provide a cheaper and more flexible alternative to conventional reference materials (pure CaMV DNA or DNA of infected leaves) for quantifying the GMOs and donor organisms. It can thus ensure the continuity and stability, necessary for reference material, provided commutability is demonstrated. The target amplicon of CaMV (152 bp) was thus cloned in a p-GEMT plasmid. This clone (pPV148) is available after its deposit to the international Ghent collection.
Quantitative real time PCR
Estimation of the DNA copy number in the samples
The copy number of a virus DNA solution is calculated as follow:
(N = Avogadro number = 6.023 × 1023 molecules; Ji: CaMV virus strain). The virus genome size is assumed to be 8,030 bp [17]. As an example a DNA solution of 200 ng/μL contains:
Experiments were carried out to determine the relationship between DNA plasmid and leaves infected virus genome copies. Serial dilutions were performed from a 109 copies of either plasmid or infected leaves DNA for assessing the absolute limit of detection (LODa) of the test.
The ΔRn (fluorescence value obtained at the end of the PCR amplification curve) of the infected leaves, cloned 152 bp and CabbS and pure virus DNA ranged from 1.6 to 1.8, 1.5 to 2, 1.2 to 1.4 and 1 to 1.2, respectively. These values demonstrates the good amplification efficiency of the PCR reaction whose differences were not statistically significant.
Amplification efficiency of the TaqMan® assay
The PCR efficiency was evaluated with all the CaMV available materials: virus DNA, plasmids and DNA extracted from infected leaves. The mean efficiency was determined at 90 ± 5%. The squared coefficient of correlation (R 2) ranged from 0.993 to 0.998 (Fig. 3). This demonstrates that there is a high correlation between the original amount of genomic DNA in the template and the real-time PCR Ct values. As the experiment was performed at least three times with different batches of primers and probes, the test seems to be highly reproducible. Statistically speaking, no significant differences were observed in the efficiencies between plasmid and virus DNA (ANOVA, t test at 1%).

Example of amplification plot and standard curve using as sample the DNA of the strain Ji. Tenfold dilution series ranged from 106 copies to 0.1 copies. Each point was analysed in triplicate and each real-time experiment was repeated three times. The absolute limit of detection was better than ten copies
Determination of the dynamic range and the absolute limit of detection (LODa)
Generally, the sensitivity of a given test is determined using the limit of detection (LOD) performance criterium. There are two types of LOD, the absolute LOD calculated in our study using one target. Besides, relative LOD is calculated in presence of a template other than the targeted called ballast DNA [28] Here, the LODa was determined with qualitative and quantitative real time PCR on six dilutions in triplicates and done three times independently (9 data per dilution), using ranges of the targets varying from 106 copies to one copy per dilution. The LODa results are similar for the Ji and CabbS virus, as for the clone PV148: between one and ten copie(s) (Fig. 3). According to the amplification efficiency and LODa data, the correlation factor between plasmid and virus DNA copy numbers contents is therefore equal to one.
Determination of the absolute limit of quantification (LOQa)
The LOQ is defined as the lowest amount of the target to be reliably detected and quantified under ideal conditions with a probability ≥95% with a RSDr (relative standard deviation of the repeatability) ≤25% (EN ISO 21570) [28]. In order to validate the LOQ, we used two purified viruses Ji and CabbS DNA. The same templates for the determination of the limit of detection were used with tenfold dilution series from 106 to one copy of CaMV genome. Each dilution was tested in triplicate. The results are summarized in the Table 4. As expected, the SD values increased in the case of low amounts of CaMV genome copies. We were able to detect CaMV in all three parallels down to ten copies, whereas only one of the parallels was positive when we used an approximate average of one copy. From the results shown in the Table 4, we concluded that under our conditions, approximate 100 initial CaMV genome copies were required for reliable quantification.
Development of a qualitative and quantitative duplex CaMV/P35S
In addition to the simplex test described in this paper, we developed a new qualitative and quantitative test for the simultaneous detection of P35S (having either the virus or the GMO or both as origin) and CaMV. This test is of a great importance especially in quantitative trials, because it will allow the segregation between the quantity of P35S present in the construction of the GMOs and the P35S of the CaMV using a differential equation. The Fig. 4 shows the result of the qualitative duplex test with two infected leaves using PV148 strain. We have also developed a quantitative duplex test using the two specific sets of primers and probes, P35S and CaMV, for the detection of CaMV virus (strains CK1 and CK2, respectively) in rapeseed (Brassica napus var. westar) leaves and cauliflower (Brassica oleracea). Three dilutions of the total extracted DNA (genomic plant DNA and virus DNA all together) were tested. The LODa and the PCR efficiencies of the duplexes were similar to the simplexes in presence of low content of both targets (data not shown). Thus, the combination of both targets, P35S and CaMV is feasible since the performance criteria do not look affected.

Migration of the PCR products in 4% agarose gels. Example of end point amplification with the duplex assay combining the P35S (amplicon 79 bp) and the CaMV control [amplicon 152 bp. 1 water, 2 low marker, 3 CaMV positive control (Clone 152 bp PV148), 4 inoculated rapeseed (Brassica napus) with strain PV148, 5 inoculated cabbage (Brassica rapa) with the strain C.K1 (see Table of virus material)]
Conclusions
The P35S screening test is the most used one because of its sequence high frequency in the currently approved GMOs. Thus, the CaMV identification test is needed to discard false positive, a requirement in the French accreditation system currently reached by the change, laboratory by laboratory, of our previously published qualitative test. Although there is no published data about the frequency of false positive in P35S screening, numerous requests over the last years of enforcement laboratories to use the qualitative test let suspect that the presence of CaMV can occur more frequently than supposed even in plant samples without known GMO like sunflower seeds, probably contaminated by dusts.
After our previous development of a qualitative CaMV test already in use in numerous detection laboratories, we developed qualitative and real-time PCR systems suitable for detection, identification and, most of all, quantification of the CaMV virus. In comparison with CaMV tests already published which target the ORFIV [29] or ORFV and ORFVI [30] our primers and probes were assessed against a larger set of samples and showed a very good specificity altogether with a similar sensitivity. Moreover, as the capsid (ORFIV) protein sequence is often used in GMOs for plant protection, our ORFIII based test might be a better choice for long-term uses, since the ORFIV might be used in future virus resistant GM plants.
The absolute limit of detection (LODa) was estimated below 10 genome or plasmid copies and the absolute limit of quantification (LOQa) below 100 copies using TaqMan® chemistry which is less expensive than MGB probe while the two chemistries look to perform equally [31, 32]. The measured uncertainty, an important factor in quantitative analysis, is in good compliance with the current thresholds set in regulations [33]. This test looks quite robust as observed with the similar PCR efficiencies on DNA extracted by several methods. Moreover, plasmids prepared in this study can be used as alternative standards for the quantification of the CaMV virus (correlation factor of 1) as it is the case of many plasmids constructed for precise GMO quantification [34, 35].
Finally, a duplex qualitative and quantitative test (P35S and CaMV) is routinely working, thus providing a time- and cost-effective detection method. The average cost of 1 quantitative real-time PCR reaction is about 0.75 $ per tube and thus the cost is not the most important factor to be taken into account. However, the ability of directly detecting CaMV in a P35S positive sample could is time-effective and should provide rapid indications of cross-contamination even for plants, such as solanaceous, whose infection by the CaMV is generally unknown from the enforcement laboratories or in samples whose P35S positive character may result of dusts contamination as observed for sunflower seeds. The CaMV P35S tests quantitative duplex tests can thus be used for detecting unknown GMO by one the strategy under study in the European research program Co-Extra, namely the “quantitative differential PCR” (Ancel et al., unpublished data).
Finally, an inter-laboratory validation should be now started for the routine uses by the detection laboratories of our tests.
References
Regulation (EC) No 1829/2003 of the European Parliament and of the Council of 22 September 2003 concerning the traceability and labelling of genetically modified organisms and the traceability of food and feed products produced from genetically modified organisms and amending directive 2001/18/EC. 2003. Official journal of the European Commnities. No L268:24–28
Hino A (2002) Toxicol Pathol 30:126–128
Anklam E, Gadani F, heinze P, Pijnenburg H, Van Den Eede G (2002) Eur Food Res Technol 214:3–26
Gryson N, Messens K, Van Laere D, Eeckhout M (2006) Eur Food Res Technol. doi:10.1007/s00217-006-0511-x
Kubista M, Andrade JM, Bengtsson M, Forooton A, Jiri J, Lind K, Sindelka R, Sjöback R, Björn S, Stömbom L, Stahlberg A, Zoric N (2006) Mol Aspects Med 27:95–125
Gachon C, Mingam A, Charrier B (2004) J Exp Bot 402:1445–1454
Moreano F, Ehlert A, Busch U, Engel K-H (2006) Eur Food Res Technol 222:479–485
Passamano M, Pighini M (2006) Sens Actuators B 118:177–181
Leimanis S, Hernandez M, Fernandez S, Boyer F, Burns M, Bruderer S, Glouden T, Harris N, Kaeppeli O, Philipp P, Pla M, Puidomenech P, Vaitilingom M, Bertheau Y, Remacle J (2006) Plant Mol Biol 61:123–139
Feinberg M, Fernandez S, Cassard S, Bertheau Y (2005) J AOAC Intl 88(2):558–573
Yang L, Xu S, Pan A, Yin C, Zhang K, Wang Z, Zhou Z, Zhang D (2006) J Sci Food Agric 53:9312–9318
Ronning SB, Berdal KG, Andersen CB, Holst-Jensen A (2006) J Agric Food Chem 54:68–71
Hernandez M, Esteve T, Pla M (2005) J Agric Food Chem 53:7003–7009
Richard C, Howarth GAJ, Hahn P, Brown-Luedi M, Shepherd RJ, Messing J (1981) Nucleic Acid Res 9:2871–2881
Balazs E, Guilley H, Jonard G, Richards K (1982) Gene 19:239–249
Chenault KD, Melcher U (1993) Gene 123:255–257
Franck A, Guilley H, Jonard G, Richards K, Hirth L (1980) Cell 1:285–94
Leh V, Jaquot E, Geldreich A, Hass M, Blanc S, Keller M, Yot P (2001) J Virol 100–106
Piqué M, Mougeot JL, Geldreich A, Guidasci T, Mesnard JM, Leubeurier G, Yot P (1995) Gene 155:305–306
Jaccaud E, Hohne M, Meyer R (2003) J Agric Food Chem 51:550–557
U, N (1935) Jap J Bot 7:389–452
Fernandez S, Charles-Delobel C, Geldreich A, Berthier G, Boyer F, Collonnier C, Coue-Philippe G, Diolez A, Duplan MN, Kebdani N, Romaniuk M, Feinberg M, Bertheau Y (2005) J AOAC Int 88(2):547–557
Hull R, Shepherd RJ (1976) Virology 70:217–220
Côté MJ, Allison JM, Raymond P, Dollar C (2005) J Agric Food Chem 53:6691–6696
Taberlet P, Gielly L, Pautou G, Bouvet J (1991) Plant Mol Biol 17:1105–1109
European Committee for Standardization/International Organization for standardization (2006) EN ISO 24276, Brussels, Belgium
Mattarucchi E, Weighardt F, Barbati C, Querci M, Van den Eeden G (2005) Eur Food Res Technol 221:511–519
European Committee for Standardization/International Organization for standardization (2006) EN ISO 21570, Brussels, Belgium
Cankar K, Ravinkar M, Zel J, Gruden C, Toplak N (2005) J AOAC Int 88:814–822
Wolf C, Scherzinger M, Wurz A, Pauli U, Hübner P, Lüthy J (2000) Eur Food Res Technol 210:367–372
Andersen CB, Holst-Jensen A, Berdal KG, Thorstensen T, Tengs T (2006) J Agri Food Chem 54:9658–63
Yao Y, Nellåker C, Karlsson H (2006) Mol Cell Probes 20:311–316
Zel J, Gruden K, Cankar K, Stebih D, Blejec D (2007) J AOAC Int 2:582–586
Kuribara H, Shindo Y, Matsuoka T, Takubo K, Futo S, Aoki N, Hirao T, Akiyama H, Goda Y, Toyoda M, Hino A (2002) J AOAC Int 85:1077–1189
Block A, Schwarz G (2003) Eur Food Res Technol 216:421–427
Acknowledgments
We thank Dr. Katarina Cankar (National Institute of Biology, Department of Plant Physiology and Biotechnology, Ljubljana, Slovenia) and Dr. Camille Kerlan (Bio3P, INRA, Rennes, France) for providing us the CERV and CK samples, respectively.
Author information
Authors and Affiliations
Corresponding author
Additional information
Maher Chaouachi and Marie Noelle Fortabat are contributed equally to this work.
Rights and permissions
About this article
Cite this article
Chaouachi, M., Fortabat, M.N., Geldreich, A. et al. An accurate real-time PCR test for the detection and quantification of cauliflower mosaïc virus (CaMV): applicable in GMO screening. Eur Food Res Technol 227, 789–798 (2008). https://doi.org/10.1007/s00217-007-0787-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-007-0787-5