
Abstract
N ε-(carboxymethyl) lysine (CML) and N ε-(carboxyethyl) lysine (CEL) are two advanced glycation end products. Few studies have focused on the simultaneous determination of CML and CEL content in foods, especially in Eastern foods. In this study, a stable isotope dilution LC–MS/MS method was developed for the simultaneous determination of CML and CEL in foods. The CML and CEL contents in three cereal foods consumed in China were determined by the developed method. Sample preparation consisted of lyophilization, defatting, grinding, reduction, protein precipitation, acid hydrolysis, and solid-phase extraction. The limit of quantification for CML and CEL was 4 and 3 ng/g, respectively. CML and CEL content in fried dough stick was determined for the first time. CML and CEL contents in fried dough sticks were 28.06–66.69 and 10.67–30.22 μg/g of fried dough sticks, respectively. The highest CML and CEL contents in biscuits were 117.53 and 46.09 μg/g of biscuits, respectively. CML and CEL contents in bread crusts were higher than those in bread crumbs.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Advanced glycation end products (AGEs) constitute a group of heterogeneous compounds formed by Maillard reaction (MR), a non-enzymatic reaction between reducing sugars and proteins, amino acids, or nucleic acids [1, 2]. At the beginning of MR, reducing sugars react with amino groups of proteins, forming Schiff bases, which are unstable N-substituted glycosylamine molecules. Subsequently, Schiff bases change to more stable Amadori products by isomerization reactions called Amadori rearrangements. Amadori products produce highly reactive dicarbonyl compounds such as glyoxal (GO) [3], methylglyoxal (MGO) [4], and 3-deoxyglucosone (3-DG) [5] via dehydration and rearrangement reactions. The reactions between these dicarbonyl compounds and amino or guanidine groups of proteins result in the formation of stable and irreversible AGEs. AGEs can be formed in vivo and in vitro. Additionally, diet is a significant source of exogenous AGEs [6]. Studies have reported that a typical diet provides 25–75 mg of AGEs [2] per day and approximately 10 % of ingested AGEs are absorbed [7]. Whether formed in vivo or obtained from the diet, AGEs have the same detrimental effects [6]. Many studies have suggested that AGEs play potential roles in the pathogenesis of diabetes [8–10], Alzheimer’s disease (AD) [11], atherosclerosis [12–14], renal diseases [9, 12], and aging [15, 16].
N ε-(carboxymethyl) lysine (CML) is an AGE formed in vivo and during food processing. CML is formed by the reaction between GO and the epsilon-amino group of lysine [17]. N ε-(carboxyethyl) lysine (CEL), which is formed by the reaction between MGO and lysine, is a homolog of CML [18]. CML and CEL can be used as markers of MR, oxidative stress, and thermal damage in foods. When CML and CEL are formed through lipid peroxidation, they are called advanced lipoxidation end products (ALEs) [19, 20]. The content of AGEs in foods is affected by the food matrix and food processing conditions. Foods rich in proteins and fats can contribute to more AGEs than foods rich in carbohydrates [21]. Cooking with dry heating (e.g., grilling, roasting, and broiling) can produce more AGEs than cooking with moisture heating (e.g., boiling, poaching, and stewing) [6]. The content of CML in several foods has been determined [6, 21–23]; however, few studies have focused on CEL content in foods. In addition, the AGE content in several Eastern foods, such as frying sticks, has not been reported. Therefore, more information on the AGE content in foods is needed to improve the assessment of dietary intakes of AGE.
Several methods have been developed to determine AGE content in foods: fluorescence measurements [24, 25], enzyme-linked immunosorbent assay (ELISA) [6, 26, 27], high-performance liquid chromatography (HPLC) [28–30], liquid chromatography with tandem mass spectrometry detection (LC–MS/MS) [22, 23, 31–35], and gas chromatography–mass spectrometry (GC–MS) [36, 37]. Fluorescence measurements are easy to manipulate; however, they can only detect AGEs with fluorescent properties (e.g., pentosidine and crossline) and not AGEs without fluorescent properties (e.g., CML and CEL) [38]. Even though ELISA is rapid, the results are expressed in arbitrary units instead of actual concentrations. Furthermore, the specificity of the assay is affected by the food matrix. Additionally, ELISA requires the use of specific antibodies for each compound. Instrumental methods such as LC–MS/MS and GC–MS can provide more accurate results; however, they require more time compared to other methods and highly trained personnel. Nevertheless, LC–MS/MS has been used by several researchers. Few studies have focused on the simultaneous determination of CML and CEL by LC–MS/MS in foods [32], especially in Eastern foods.
The objective of this study was to develop a stable isotope dilution LC–MS/MS method for the simultaneous determination of CML and CEL in foods. CML and CEL contents in breads, biscuits, and fried dough sticks were determined by the validated method, which may contribute to the assessment of AGEs in Chinese foods.
Materials and methods
Chemicals
N ε-(carboxymethyl) lysine, CEL, N ε-(carboxymethyl) lysine-d4 (CML-d4), and N ε-(carboxyethyl) lysine-d4 (CEL-d4) were purchased from Santa Cruz Co. (CA, USA). The purity of these standards was >98 % (CML), >98 % (CEL), >98 % (CML-d4), and >98 % (CEL-d4). HPLC grade methanol was purchased from Merk Co. (Darmstadt, Germany). Ultrapure water was obtained using a Superpure Water System (Shanghai, China). All other reagents were of analytical grade and purchased from Sinopharm Chemical Reagent Co. Ltd.
Food samples
Breads and biscuits were purchased from local supermarkets in Wuxi, Jiangsu Province, China. Breads from six different companies consisted of wholemeal breads and white breads; biscuits from eight different companies consisted of soda crackers, crisp biscuits, tough biscuits, and cookies. Fried dough sticks were obtained from local markets in Wuxi and at cafeterias from Jiangnan University and Wuxi Institute of Technology.
Protein determination
Bread crust and bread crumbs were assessed separately. Protein content of bread crust, bread crumbs, biscuits, and fried dough sticks was determined by the Kjeldahl method using the conversion factor 6.25 for cereal foods [39].
Moisture determination
Moisture content of bread crust, bread crumbs, biscuits, and fried dough sticks was determined by the oven-drying method [40].
Sample preparation
Bread crust, bread crumbs, and fried dough sticks were first lyophilized; the weight change was recorded. The lyophilized food samples were ground in a mill; the biscuits were ground without lyophilization. A quantity of ground food samples, equivalent to 5 mg of protein, were defatted twice using hexane (2 ml) before reduction with sodium borohydride. The hexane extract was removed with a Pasteur pipet, and the residue was completely dried under a stream of nitrogen gas. Sodium borate (0.2 M, pH 9.2, 1 ml) was initially added to the defatted foods, and sodium borohydride (1 M in 0.1 M NaOH) was added resulting in a final concentration of 0.1 M sodium borohydride. The samples were incubated at 4 °C for 0, 2, 4, 6, 8, and 10 h to obtain an optimal reduction time. To precipitate protein, trichloroacetic acid (TCA, 60 %, w/v) was added to the reduced food samples resulting in a final concentration of 20 % (w/v). The food samples were centrifuged at 10,000 rpm for 15 min. The resulting supernatants were removed with a Pasteur pipet, and the precipitates were used for acid hydrolysis. The precipitates were mixed with 5 ml of 6 M hydrochloric acid (HCl) and incubated at 110 °C for 24 h. Following the 24-h incubation, the hydrolysates were filtered and diluted to 10 ml. Approximately 400 μl of the diluted acid hydrolysate (equivalent to 200 μg of protein) was dried under a stream of nitrogen gas at 60 °C using a pressured gas-blowing concentrator (Jintan, China). To the dried acid hydrolysates, 2 ml of 0.1 M HCl, 100 μl of 0.4 μg/ml CML-d4, and 100 μl of 0.4 μg/ml CEL-d4 were added. The reconstituted acid hydrolysates were subjected to solid-phase extraction (SPE). In SPE, a STYRE-SCREEN SSDBX® (Sepax technology, USA) cartridge (200 mg, 3 ml) was first preconditioned with 3 ml of methanol and 3 ml of 0.1 M HCl at a flow rate of 1 ml/min. The reconstituted acid hydrolysates were loaded into the preconditioned cartridge at a flow rate of 0.5 ml/min, and the cartridge was washed with 3 ml of 0.1 M HCl and 3 ml of ultrapure water. Finally, the sample was eluted with 3 ml of 5 % ammonia/methanol (v/v) at a flow rate of 0.5 ml/min. The eluent was dried under a stream of nitrogen gas at 40 °C using a pressured gas-blowing concentrator, redissolved in 200 μl of ultrapure water, filtered through a 0.22-μm membrane, and stored at −20 °C prior to LC–MS/MS.
LC–MS/MS analysis
The samples were analyzed using a Waters Acquity UPLC (Waters Corporation, MA, USA) coupled to an Acquity TQD mass spectrometer (Waters Corporation, MA, USA). Separations were performed in a Waters XSELECT™ HSS T3 column (4.6 × 150 mm, 5 μm) equipped with a column oven at 35 °C. Solutions of 0.1 % (v/v) formic acid in water (solvent A) and 0.1 % (v/v) formic acid in methanol (solvent B) were used as eluents. A linear gradient was performed: 0–0.5 min, 20 % B; 0.5–6.5 min, 20–100 % B; 6.5–7.5 min, 100 % B; and 7.5–8 min, 100–20 % B. The flow rate was 0.5 ml/min, and the injection volume was 2 μl. The detection was carried out by MS in an electrospray ionization (ESI) positive mode with multiple reaction monitoring (MRM). MS had a source temperature of 120 °C, desolvation temperature of 380 °C, capillary voltage of 3.5 kV, cone voltage of 30 V, cone gas flow rate of 50 l/h, desolvation gas flow rate of 800 l/h, and collision gas flow rate of 0.13 ml/min. The optimized MRM conditions are shown in Table 1.
The determination of CML and CEL was based on calibration curves obtained by linear regressions of analyte/internal standard peak area ratio against analyte concentration. Seven calibration points were prepared for each calibration curve. Each sample was analyzed in triplicate; the results were expressed as mean ± standard deviation (SD). CML and CEL contents in samples were expressed as μg/g protein and μg/g sample. The method was validated by assessing selectivity, linearity, precision, and accuracy of the method. Limit of detection (LOD) and limit of quantification (LOQ) were defined as the concentration (ng/g) for which the signal-to-noise ratio of the quantifier was 3 and 10, respectively.
Results and discussion
Optimization of sample preparation
The sample preparation consisted of lyophilization, grinding, defatting, reduction, hydrolysis, and SPE. Due to their high moisture content, bread and fried dough sticks had to be freeze-dried prior to grinding. Biscuits could be easily ground without lyophilization. Ground samples can provide more accurate results.
Both CML and CEL can be formed by lipid peroxidation; therefore, it is important to remove fat from the food samples. Otherwise, there may be an overestimation in the results. In this study, we compared the CML and CEL content in bread crusts with and without delipidation. In defatted bread crusts, CML and CEL contents were 73.67 ± 3.72 and 58.46 ± 3.25 μg/g of bread crust, respectively. In non-defatted bread crusts, CML and CEL contents were 86.16 ± 4.86 and 70.15 ± 4.18 μg/g of bread crust, respectively.
Prior to hydrolysis, a reduction step was performed to reduce Amadori products (e.g., fructoselysine) and lipid oxidation products, which may prevent the formation of CML and CEL during acid hydrolysis [22, 28]. In this study, we used 0.1 M sodium borohydride. The effect of reduction time on CML and CEL contents was assessed. As shown in Fig. 1, there were no significant differences in CML and CEL contents when the reduction time was 6 h. Therefore, the reduction conditions in this study involved the use of 0.1 M sodium borohydride with a reduction time of 6 h. Acid hydrolysis, which can lead to a complete release of CML and CEL from protein, was also performed in this study. Samples were subjected to SPE using a STYRE-SCREEN SSDBX® (Sepax technology, USA) cartridge (200 mg, 3 ml).

Influence of reduction time on CML and CEL content. a Influence of reduction time on CML content. b Influence of reduction time on CEL content
Optimization of chromatography conditions
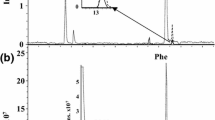
Due to their high polarity, CML and CEL are difficult to retain in most reversed-phase columns. However, CML was successfully analyzed by a C18 column using an ion-pairing reagent called nonafluoropentanoic acid (NFPA) [22, 23, 31, 32, 34, 41]. NFPA can contribute to increased retention times of CML and has no effect on the sensitivity of CML. Nevertheless, NFPA may result in the deterioration of the reversed-phase column because it can lead to a mobile phase of low pH (~2) [33]. To avoid the formation of NFPA residues, the chromatographic column and mass detector must be thoroughly cleaned. Therefore, to avoid the use of NFPA, a LC–MS/MS method has been developed to assess CML and other AGEs using Hypercarb™ columns; the mobile phases consist of 26 mM ammonium formate (pH 3.8) and acetonitrile [35]. However, the method takes a very long period of time (35 min). Alternatively, a hydrophilic ion liquid chromatography (HILIC) column has been used to determine CML; the eluents consist of 5 mmol/l ammonium acetate buffer (pH 4) and 0.025 % (v/v) formic acid in acetonitrile [33]. However, this method has certain disadvantages including long chromatographic runs (29 min). In this study, a LC–MS/MS method was developed to simultaneously determine CML and CEL using a Waters XSELECT™ HSS T3 column, which is able to retain polar compound such as CML and CEL. The eluents consisted of 0.1 % (v/v) formic acid in water and 0.1 % (v/v) formic acid in methanol; the total run time was only 13 min. Mass chromatograms of standard solutions and bread crust are shown in Figs. 2 and 3, respectively. The retention time of CML and CEL in bread crust was consistent with that in the standard solution, and there was no peak interference for CML or CEL in the sample. The column used was very stable and robust, with no obvious shifts in retention times.

Typical mass chromatograms of standard solutions

Typical mass chromatograms of bread crust
Validation of the method
The developed method was validated by assessing CML and CEL content in bread crusts. Mass conditions were optimized, and the results for MRM are shown in Table 1. Both CML and CEL had two product ions. One product ion was at m/z 84, and the other was at m/z 130; the most intense product ion was at m/z 84. The product ion at m/z 84 was used for quantification, and the product ion at m/z 130 was used for confirmation. The corresponding m/z 88 and m/z 134 product ions of the internal standards (CML-d4 and CEL-d4) were used for quantification and confirmation, respectively. As shown in Table 2, the correlation coefficients (R 2) were >0.99. The linear range (20–2,500 ng/ml) was wide enough to assess CML and CEL content in cereal foods. LOQ of CML and CEL was 4.0 and 3.0 ng/ml, respectively (Table 2).
The recoveries of exogenous CML and CEL added to bread crusts were determined at three concentrations (Table 2). The low, middle, and high exogenous concentrations for both CML and CEL were 30, 300, and 1,000 ng/ml, respectively. The recovery experiments were conducted five times for each concentration. The recoveries for CML and CEL were 88.76–93.21 and 87.64–95.15 %, respectively.
Precision was expressed as relative standard deviation (RSD). For intra-day precision (i.e., repeatability), each sample was analyzed three times a day; for inter-day precision (i.e., reproducibility), each sample was analyzed once a day for five consecutive days. RSD of intra-day precision for CML and CEL was 6.84 and 5.68 %, respectively. RSD of inter-day precision for CML and CEL was 8.62 and 9.36 %, respectively.
Matrix effects were assessed by recovery analyses. In CML and CEL, there were no differences in recoveries whether the standards were added to bread crust extracts or aqueous solutions. Stable isotope-labeled CML-d4 and CEL-d4 can decrease matrix effects.
CML and CEL content in cereal foods
The CML content in foods has been determined by instrumental methods [22, 23, 37, 41]; however, there are few reports on the CEL content in foods [32, 42]. In this study, CML and CEL content in three types of cereal foods was determined by the validated LC–MS/MS method. Bread crust and bread crumb were analyzed separately. As shown in Tables 3 and 4, CML and CEL contents in bread crust were much higher than those in bread crumbs. This may be attributed to a low moisture content and high processing temperatures in bread crust [22]. In our study, the content of CML in bread crust (58.12–94.29 μg/g) was higher than that of bread crusts reported by Assar et al. [22] (36.1 μg/g of white bread crust and 46.1 μg/g of wholemeal bread crust), which may be attributed to differences in sample preparation and bread ingredients. In this study, CEL content in bread crusts was 22.65–71.49 μg/g. To the best of our knowledge, no study has reported CEL content in bread.
As shown in Table 5, the highest content of CML and CEL was present in biscuits (117.53 and 46.09 μg/g of biscuits, respectively). The lowest content of CML and CEL was also present in biscuits (50.73 and 12.16 μg/g of biscuits, respectively). The CML and CEL content differences may be due to differences in biscuit ingredients and processing temperatures.
Fried dough sticks constitute a traditional Chinese food. The CML and CEL contents in fried dough sticks were simultaneously determined for the first time (Table 6). The highest content of CML and CEL in fried dough sticks was approximately three times the lowest content of CML and CEL. Differences in the ingredients, frying temperatures, and processing times may result in the differences in CML and CEL contents in fried dough sticks.
Conclusion
We developed a stable isotope dilution LC–MS/MS method for the simultaneous determination of CML and CEL content in cereal foods. The developed method was highly sensitive and rapid (total run time of 13 min). The recovery, precision, and linearity data were satisfactory. The method was successfully applied to determine CML and CEL content in three types of cereal foods consumed in China. The CML and CEL content in fried dough sticks was assessed for the first time. The results of this study can be used to inform consumers about AGE intakes.
References
Singh R, Barden A, Mori T, Beilin L (2001) Advanced glycation end-products: a review. Diabetologia 44:129–146
Henle T (2003) AGEs in foods: do they play a role in uremia? Kidney Int 63:S145–S147
Arribas-Lorenzo G, Morales FJ (2010) Analysis, distribution, and dietary exposure of glyoxal and methylglyoxal in cookies and their relationship with other heat-induced contaminants. J Agric Food Chem 58:2966–2972
Nemet I, Varga-Defterdarovic L, Turk Z (2006) Methylglyoxal in food and living organisms. Mol Nutr Food Res 50:1105–1117
Thornalley PJ, Langborg A, Minhas HS (1999) Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J 344:109–116
Uribarri J, Woodruff S, Goodman S, Cai W, Chen X, Pyzik R, Yong A, Striker GE, Vlassara H (2010) Advanced glycation end products in foods and a practical guide to their reduction in the diet. J Am Diet Assoc 110:911–916
Vlassara H, Palace MR (2003) Glycoxidation: the menace of diabetes and aging. Mt Sinai J Med 70:232–241
Yamagishi S, Matsui T, Nakamura K (2008) Possible link of food-derived advanced glycation end products (AGEs) to the development of diabetes. Med Hypotheses 71:876–878
Sebekova K, Somoza V (2007) Dietary advanced glycation end products (AGEs) and their health effects—PRO. Mol Nutr Food Res 51:1079–1084
Ahmed N (2005) Advanced glycation end products—role in pathology of diabetic complications. Diabetes Res Clin Pract 67:3–21
Li J, Liu D, Sun L, Lu Y, Zhang Z (2012) Advanced glycation end products and neurodegenerative diseases: mechanisms and perspective. J Neurol Sci 317:1–5
Lieuw-A-Fa MLM, van Hinsbergh VWM, Teerlink T, Barto R, Twisk J, Stehouwer CDA, Schalkwijk CG (2004) Increased levels of Nε-(carboxymethyl)lysine and Nε-(carboxyethyl)lysine in type 1 diabetic patients with impaired renal function: correlation with markers of endothelial dysfunction. Nephrol Dial Transplant 19:631–636
Semba RD, Bandinelli S, Sun K, Guralnik JM, Ferrucci L (2009) Plasma carboxymethyl-lysine, an advanced glycation end product, and all-cause and cardiovascular disease mortality in older community-dwelling adults. J Am Geriatr Soc 57:1874–1880
Wang Z, Jiang Y, Liu N, Ren L, Zhu Y, An Y, Chen D (2012) Advanced glycation end-product N(varepsilon)-carboxymethyl-lysine accelerates progression of atherosclerotic calcification in diabetes. Atherosclerosis 221:387–396
Grillo MA, Colombatto S (2007) Advanced glycation end-products (AGEs): involvement in aging and in neurodegenerative diseases. Amino Acids 35:29–36
Baynes JW (2001) The role of AGEs in aging: causation or correlation. Exp Gerontol 36:1527–1537
Van Nguyen C (2006) Toxicity of the AGEs generated from the Maillard reaction: on the relationship of food-AGEs and biological-AGEs. Mol Nutr Food Res 50:1140–1149
Ahmed MU, Frye EB, Degenhardt TP, Thorpe SR, Baynes JW (1997) N-epsilon-(carboxyethyl) lysine, a product of the chemical modification of proteins by methylglyoxal, increases with age in human lens proteins. Biochem J 324:565–570
Miyata T, Kurokawa K, De Strihou CVY (2000) Advanced glycation and lipoxidation end products role of reactive carbonyl compounds generated during carbohydrate and lipid metabolism. J Am Soc Nephrol 11:1744–1752
Fu M-X, Requena JR, Jenkins AJ, Lyons TJ, Baynes JW, Thorpe SR (1996) The advanced glycation end product, N-(carboxymethyl) lysine, is a product of both lipid peroxidation and glycoxidation reactions. J Biol Chem 271:9982–9986
Goldberg T, Cai W, Peppa M, Dardaine V, Baliga BS, Uribarri J, Vlassara H (2004) Advanced glycoxidation end products in commonly consumed foods. J Am Diet Assoc 104:1287–1291
Assar SH, Moloney C, Lima M, Magee R, Ames JM (2009) Determination of Nɛ-(carboxymethyl)lysine in food systems by ultra performance liquid chromatography-mass spectrometry. Amino Acids 36:317–326
Niquet-Léridon C, Tessier FJ (2011) Quantification of Nε-carboxymethyl-lysine in selected chocolate-flavoured drink mixes using high-performance liquid chromatography–linear ion trap tandem mass spectrometry. Food Chem 126:655–663
Yanagisawa K, Makita Z, Shiroshita K, Ueda T, Fusegawa T, Kuwajima S, Takeuchi M, Koike T (1998) Specific fluorescence assay for advanced glycation end products in blood and urine of diabetic patients. Metabolism 47:1348–1353
Yanagisawa K, Makita Z, Shiroshita K, Ueda T, Fusegawa T, Kuwajima S, Takeuchi M, Koike T (1998) Specific fluorescence assay for advanced glycation end products in blood and urine of diabetic patients. Metabolism 47:1348–1353
Dittrich R, Hoffmann I, Stahl P, Muller A, Beckmann MW, Pischetsrieder M (2006) Concentrations of N-epsilon-carboxymethyllysine in human breast milk, infant formulas, and urine of infants. J Agric Food Chem 54:6924–6928
Vay D, Vidali M, Allochis G, Cusaro C, Rolla R, Mottaran E, Bellomo G, Albano E (2000) Antibodies against advanced glycation end product Nɛ-(carboxymethyl) lysine in healthy controls and diabetic patients. Diabetologia 43:1385–1388
Hartkopf J, Pahlke C, Lüdemann G, Erbersdobler HF (1994) Determination of Nε-carboxymethyllysine by a reversed-phase high-performance liquid chromatography method. J Chromatogr A 672:242–246
Drusch S, Faist V, Erbersdobler HF (1999) Determination of N-carboxymethyllysine in milk products by a modified reversed-phase HPLC method. Food Chem 65:547–553
Ahmed N, Argirov OK, Minhas HS, Cordeiro CAA, Thornalley PJ (2002) Assay of advanced glycation end products (AGEs): surveying AGEs by chromatographic assay with derivatization by 6-aminoquinolyl-N-hydroxysuccinimidyl-carbamate and application to N-epsilon-carboxymethyl-lysine- and N-epsilon-(1-carboxyethyl)lysine-modified albumin. Biochem J 364:1–14
Teerlink T, Barto R, Ten Brink HJ, Schalkwijk CG (2004) Measurement of Nepsilon-(carboxymethyl)lysine and Nepsilon-(carboxyethyl)lysine in human plasma protein by stable-isotope-dilution tandem mass spectrometry. Clin Chem 50:1222–1228
Zhang G, Huang G, Xiao L, Mitchell AE (2011) Determination of advanced glycation end products by LC–MS/MS in raw and roasted almonds (Prunus dulcis). J Agric Food Chem 59:12037–12046
Schettgen T, Tings A, Brodowsky C, Müller-Lux A, Musiol A, Kraus T (2007) Simultaneous determination of the advanced glycation end product Nɛ-carboxymethyllysine and its precursor, lysine, in exhaled breath condensate using isotope-dilution–hydrophilic-interaction liquid chromatography coupled to tandem mass spectrometry. Anal Bioanal Chem 387:2783–2791
Delatour T, Hegele J, Parisod V, Richoz J, Maurer S, Steven M, Buetler T (2009) Analysis of advanced glycation endproducts in dairy products by isotope dilution liquid chromatography–electrospray tandem mass spectrometry. The particular case of carboxymethyllysine. J Chromatogr A 1216:2371–2381
Thornalley PJ, Battah S, Ahmed N, Karachalias N, Agalou S, Dawnay A (2003) Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem J 375:581–592
Petrovic R, Futas J, Chandoga J, Jakus V (2005) Rapid and simple method for determination of Nepsilon-(carboxymethyl)lysine and Nepsilon-(carboxyethyl)lysine in urine using gas chromatography/mass spectrometry. Biomed Chromatogr 19:649–654
Charissou A, Aitameur L, Birlouezaragon I (2007) Evaluation of a gas chromatography/mass spectrometry method for the quantification of carboxymethyllysine in food samples. J Chromatogr A 1140:189–194
Trégoat V, Brohée M, Cordeiro F, van Hengel AJ (2009) Immunofluorescence detection of advanced glycation end products (AGEs) in cookies and its correlation with acrylamide content and antioxidant activity. Food Agric Immunol 20:253–268
Chang SKC (2010) Protein analysis. In: Nielsen SS (ed) Food analysis. Springer Science+Business Media, LLC, Boston, pp 133–146
Bradley RL (2010) Moisture and total solids analysis. In: Nielsen SS (ed) Food analysis. Springer Science+Business Media, LLC, Boston, pp 85–104
Hull GL, Woodside JV, Ames JM, Cuskelly GJ (2012) Nε-(carboxymethyl) lysine content of foods commonly consumed in a Western style diet. Food Chem 131:170–174
Ahmed N, Mirshekar-Syahkal B, Kennish L, Karachalias N, Babaei-Jadidi R, Thornalley PJ (2005) Assay of advanced glycation end products in selected beverages and food by liquid chromatography with tandem mass spectrometric detection. Mol Nutr Food Res 49:691–699
Acknowledgments
This work was financially supported by the National Basic Research Program of China (973 Program) (2012CB720801), the National Natural Science Foundation of China (Grant No. 31000751), and the National Key Scientific Instrument, and Equipment Development Special Fund (2011YQ170067). The authors are grateful to Guanjun Tao, Fang Qin, Shuang Zhang, and Yahui Gao for their assistance in LC–MS/MS.
Conflict of interest
None.
Compliance with Ethics Requirements
This article does not contain any studies with human or animal subjects.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
He, J., Zeng, M., Zheng, Z. et al. Simultaneous determination of N ε-(carboxymethyl) lysine and N ε-(carboxyethyl) lysine in cereal foods by LC–MS/MS. Eur Food Res Technol 238, 367–374 (2014). https://doi.org/10.1007/s00217-013-2085-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-013-2085-8