
Abstract
Furosine is a well-know marker of the extent of the thermal treatment applied to foodstuffs. A fast, reproducible and accurate capillary zone electrophoresis (CZE) method to analyse furosine focused on complex matrices, such as cocoa-added breakfast cereals, is presented. A representative group of 17 commercial cocoa-added breakfast cereals were determined by the reference method (HPLC) and by CZE. In addition, eight ready-to-eat breakfast cereals were tested to point out the application of the procedure to a simpler matrix. Data from both methodologies are compared by F-test and unpaired Student's t-test, showing no significant differences among them and an adequate correlation between methods was found (r 2=0.9852; p=0.000). The CZE method provides a valuable analytical tool for determination of furosine in cocoa-added breakfast cereals and is suitable for routine analysis where presence of highly retained material could limit the resolution and shelf-life of the analytical columns applied for HPLC. The analysis is carried out in 3 min. The stability of the migration time and the good correlation between HPLC–CZE results support the effectiveness of the method.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cereal flours pass through many manufacturing processes to finally obtain the usually consumed breakfast cereals. After drying and toasting, extrusion is the most applied procedure by the cereal industry to get appropriate tasty and texture of raw flours [1]. The use of high temperature (>180°C) during the steps of manufacturing in combination with low moisture (<15%) favoured the development of the Maillard reaction (MR) [2].
The MR, known as the non-enzymatic reaction between reducing sugars and primary amino acids, is a complex set of reactions that take place during the processing, domestic cooking and storage of most foods [3]. MR may compromise the nutritional value of foods through the blocking and destruction of essential amino acids and subsequently by limiting their bioavailability, such as lysine [4]. Maillard reaction products (MRPs) are responsible of brown colour and many organoleptic properties of food [5].
Furosine (ɛ-N-2-furoylmethyl-l-lysine) is an amino acid generated after acid hydrolysis of the Amadori compounds, early MRPs, formed from the interaction of the ɛ-amino groups of lysine with glucose, lactose and maltose [6], inducing the loss of amino groups [7]. For this reason, the estimation of the extent of protein damage caused by heating in the first stages of that reaction are often based on determinations of the amount of furosine that is formed during the acid hydrolysis of foods [8].
Furosine is a highly valuable heat-induced index in cereal products, since lysine is usually their limiting amino acid. Knowing the furosine content in these foods would help to set adequate manufacturing conditions to ensure the highest nutritional value [3].
Although gas chromatography after derivatisation to heptafluorobutyryl isobutyl esters and amino acids analysis have also been used, furosine determination has been traditionally performed by ion-pairing reverse-phase high performance liquid chromatography (HPLC) in many type of products. It has been successfully applied to cereal products such as pasta [9], dairy products [10], commercial baby cereal [11, 12] and bread [13, 14]. Recently, capillary zone electrophoresis (CZE) is being used for this purpose [15] because of the speed, the efficient separation of peaks and the low cost of the analysis, which is due to saving of solvents [16]. On the other hand, depending on the type of samples, the shelf-life of the analytical columns used in the ion-pairing reverse-phase HPLC technique could be affected. In addition, retention times will change on the basis of the retention of hydrophobic material by the column. This fact is especially noteworthy for cocoa-added foodstuffs and will limit the use of chromatographic techniques in a routine analysis, besides increasing the solvent cost. However, CZE always offers a clean and conditioned capillary for each sample analysis.
Some researchers have found the method to be suitable in terms of repeatability, efficiency, and sensibility but inadequate in terms of accuracy when low levels of furosine were determined [15]. Subsequently, Vallejo-Córdoba et al. [16] have developed a new method to improve the accuracy in furosine determination on dairy products by CZE applying reverse polarity and using the additive hexadecyl trimethylammonium bromide (HDTAB) in the running buffer.
This study presents a fast, reproducible and accurate CZE method to analyse furosine content in complex samples, such as cocoa-added breakfast cereals. Results are compared with the reference method by ion-pairing reverse-phase HPLC. Moreover, the separation of furosine peak under reproducible conditions is obtained without the addition of additives to the running buffer or the use of reverse polarity conditions.
Material and methods
Chemicals
All chemical used were of analytical grade and were obtained from Merck (Darmstadt, Germany), unless mentioned otherwise. Furosine was obtained from Neosystem Laboratories (Strasbourg, France).
Samples
A total number of seventeen commercial cocoa-added breakfast cereals from seven different manufactures were collected from supermarkets. Another eight ready-to-eat cereals non cocoa added were also included to check the utility of the method in a simpler matrix. Breakfast cereals containing dried fruit were excluded of this study to avoid bias during data interpretation. The whole product contained in each package was powdered, homogenised and stored in polyethylene containers under vacuum at 4°C until analyses were performed.
Analytical techniques
Total protein content
Samples (0.800–1.000 g) were heated to 1,050°C following AOAC 992.15 [17] in a LECO model FP-2000 (Leco Instruments, Madrid, Spain) protein/nitrogen analyser calibrated with EDTA (Dumas method). The nitrogen-to-protein conversion factor considered was 5.70, 5.95 or 6.25% for wheat, rice and the rest of cereals, respectively. Results were expressed as grams of protein/100 g of products.
Ion-pairing HPLC determination of furosine
Furosine determination was performed following the methods described by Delgado et al. [18] with some modifications. Briefly, 30 mg of the sample were hydrolysed with 4 ml of 7.95 M HCl at 110°C for 23 h in a Pyrex screw-cap vial with PTFE-faced septa. Hydrolysis tubes were sealed under nitrogen. The hydrolysates were aerated and cooled at room temperature and subsequently centrifuged at 14,000×g for 10 min. A 0.5-ml portion of the supernant was applied to a Sep-pak C18 cartridge (Millipore) pre-wetted with 5 ml of methanol and 10 ml of deionized water and was then eluted with 3 ml of 3 M HCl. The dried sample was dissolved in 1 ml of a mixture of water, acetonitrile and formic acid (95:5:0.2). A degassed mobile phase was prepared with 5 mM sodium heptane sulphonate including 20% of acetonitrile and 0.2% of formic acid. An Extrasyl-ODS2 analytical column (25×0.40 cm, 5-μm particle size, Tecknokroma, Barcelona, Spain) was used at 32°C. The elution was isocratic and flow rate was 1.0 ml/min. The injection volume was 20 μl and detection at 280 nm. Furosine was quantified by the external standard method. Calibration curves were built from a stock solution (1.2 mg/ml of furosine) in the ranges 1.25–10.00 mg/l and 10.00–75.00 mg/l of furosine in distilled deionized water. The HPLC system consisted of a MD-420 pump, a MD-465 autosampler, a MD-432 ultraviolet-visible detector and a DT-450/MT v.3.90 computing integrator connected to a PC, all from Kontron Instruments (Milan, Italy).
Capillary zone electrophoresis analysis
Electromigration was carried out with a HP3D system equipped with a built-in diode-array detector and a HP ChemStation for system control, data collection, and data analysis from Hewlett-Packard (Madrid, Spain). Capillary zone electrophoresis was performed on an uncoated fused silica capillary with 48.5 cm total length (40 cm effective length), 50 μm internal diameter, and an extended light path (bubble factor×3) supplied by Hewlett-Packard (Madrid, Spain). Capillary was conditioned by filling (highflush 5.0 bar for 1 min) with 1 M NaOH and maintained so for 25 min. Afterward, it was washed with water for 1 min and then buffer was passed for 1 min. All the samples tested in the HPLC technique, prepared as described before, were also determined by CZE. They were hydrodynamically injected by applying 50 mbar pressure for 4 s at the anionic end of the capillary. After every run, the capillary was hiflushed (5 bar) with 0.1 M NaOH (0.2 min), and 50 mM sodium phosphate pH 7.0 for 0.4 min. Electrolyte solutions were renewed every five runs with 0.5 ml per pot. Temperature of the cartridge was maintained at 25°C, and total running time was set at 3 min. A standard electrophoretic run was performed at a constant voltage of 25 kV with the anode at the inlet side. Electropherograms were monitored at 280 nm wavelength, with raw spectral data collection between 190 and 700 nm. Benzyl alcohol (3.2 min) was used as marker of the electroosmotic flow (EOF) and a benzoic acid solution (1 mg/ml, 9.8 min) as internal standard. Peak identification was confirmed by migration time, standard addition and PDA spectrum, and quantified by an external standard. Same calibration curves used for HPLC determinations were also run for CZE analysis.
Statistical analysis of data
Statistical analysis of data was performed by F-test (variance comparison) and unpaired Student's t-test (means comparison) and the level of significance was set at 95%. Statgraphic v.2.0 software (Statistical Graphics Corp., Rockville, MD, USA) was used. Samples were analysed by duplicate at least.
Results and discussion
The repeatability of the CZE method was investigated by performing eight determinations at two different levels of the standards (10 and 75 mg/l) as well as in a sample. Residual standard deviations were found to be 0.79% (10 mg/l), 0.85% (75 mg/l) and 3.68% (sample). Furosine migration time was highly reproducible in all measurements (Table 1). Then it was not necessary to apply corrected area values for quantitative purposes. Therefore, adsorption of analyte on the capillary wall did not take place and it was not necessary to operate the instrument with reverse (sample-side negative) polarity. This is a procedure used in previous works aimed to apply CZE to determine the furosine content in dairy products [16]. Main cause of adsorption to the fused silica wall is ionic interactions between cationic solutes and the negatively charged wall. But furosine is positively charged at the analytical conditions. In this way, migration time for furosine remained limited to 2.60–2.65 s and it was not observed tailing peaks. Current was about 84 mV at the analytical conditions described. Additionally, linearity of the volume of sample loaded was evaluated in the range of 50–500 mbar·s (n=8) at a fixed pressure of 50 mbar. A good correlation coefficient was obtained (r 2=0.9964) for cocoa-added breakfast cereals. Injection time up to 10 s could be applied for low levels of furosine content. However, all samples analysed were carried out at 4 s (50 mbar).
Detection limit (DL) and quantification limit (QL) for furosine were determined by the standard deviation approach in the estimated limit of quantification, both in HPLC and CZE techniques (Table 2). It was confirmed a higher sensibility in the HPLC method than in the CZE one, as expected. Usually, linear detection range in the CZE equipment is significantly lower than that observed in liquid chromatography because of the small size and curvature of the capillary, but this is partially overcome by using extended path-light capillaries.
Two levels of calibration curves for each method, corresponding to high and low levels of furosine, were necessary to meet high accuracy and precision in the measurement (Table 2). By application of a unique calibration curve up to 75 mg/l, furosine levels close to the low range were underestimate as compared with the HPLC method. In this sense, some authors have reported the inadequate accuracy of CZE when samples contain low furosine levels [15], probably the use of an exclusive calibration curve for those type of samples would be indicated.
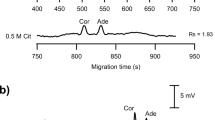
Figure 1 shows a typical electropherogram of a cocoa-added breakfast cereal, current curve, which was stable during the analysis, and the UV-scan for furosine, with a maximum value of absorbance at 280 nm, are also depicted. Figure 1 shows the symmetrical peak shape and the high resolution of the method, demonstrating that it was not necessary to add a surfactant in the running buffer. Thus, CZE gave higher resolution compared to reverse-phase HPLC, which is in agreement with observations of Tomlinson et al. [19] and Ames et al. [20] who analysed MRPs by both methodologies.
Some authors have described the appearance of tail in the furosine peak, what they associated with the difference, into the capillary, between initial pH values the running buffer zone and the sample zone [21]. In our study, there was no evidence of tailing peaks which indicates that the hydrolysis, clean-up step, dried-vacuum and reconstitution in a water/acetonitrile/formic acid (95:5:0.2) solution bear an adequate final pH of sample. It is known that acid-hydrolysed samples cannot be directly injected into the CZE system due to the high ionic strength [14].
Data from both methodologies were consistent, as manifested the mean furosine content in all samples by HPLC and CZE (315.27±239.31 vs. 306.52±234.04 mg/100 g protein, respectively) (Fig. 2). Samples analysed cover the usual distribution of furosine in breakfast cereals, including low-range sample (1.40 mg/l, corresponding to 38.79 mg/100 g protein) and high-range sample (17.3 mg/l, corresponding to 822.35 mg/100 g protein). The variation among the methods ranged from 12.52 to 0.22%, with a mean value of 6.99±4.41%. F-test (p=0.9294) and t-test (p=0.9156) showed no significant differences between both methods. A high correlation was obtained between the HPLC and the CZE method (r 2=0.9852; p<0.001).

Typical electropherogram of a cocoa-added breakfast cereal (617.17 mg furosine/100 g of protein) corresponding to 124.76 μg of furosine injected (4 nl). Current profile and the furosine UV-scan (λmax 280 nm) are included. Conditions: buffer 50 mM phosphate (pH 7.0), voltage 25 kV, capillary 48.5 cm length and 50 μm i.d., 25°C, injection 200 mbar×s

Relationship between furosine determination by HPLC and by CZE (mg/100 g of protein). Error bars (y, x axes) lies inside symbols and denote standard deviation from two independent analyses
On the other hand, this fast method was previously applied in several commercial breakfast cereals without added cocoa and with different fibre content. Data obtained by the present procedure showed adequate correlation with those obtained from HPLC analysis, independently of fibre content (Table 3).
In summary, the fast, reproducible and accurate CZE method presented in this study provides a quantitative determination of furosine in complex breakfast cereals such as those to which cocoa was added. The analysis is carried out in 3 min and, therefore there is a significant saving of solvents as compared with classical HPLC analysis. On the other hand, the presence of hydrophobic material in the extract did not affect to the resolution of the analysis or the shelf-life of the capillary. The effectiveness of the method is also supported by the stability of the furosine migration time and the adequate correlation between data obtained by HPLC and CZE. Finally, addition of HTDAB in order to improve the resolution or operating the instrument with reverse (sample-side negative) polarity is not needed.
References
Rada-Mendoza M, García-Baños JL, Villamiel M, Olano A (2004) J Cereal Sci 39:167–173
Cheftel JC (1986) Food Chem 20:263–283
Friedman M (1996). J Agric Food Chem 44:631–653
Finot PA (1990). Metabolism and physiological effects of Maillard reaction products. In: Finot PA, Aeselbacher H, Hurrell RF, Liardon R (eds) The Maillard reaction in food processing, human nutrition and physiology. Birkhauser Verlag, Basel, pp 259–271
Borrelli RC, Mennella C, Barba F, Russo M, Russo GL, Krome K, Erbersdobler HF, Faist V, Fogliano V. (2003) Food Chem Toxicol 41:1367–1374
Erbersdobler HF, Hupe A (1991) Zeitschrift für Ernährugswissenschaft 30:46–49
Morales FJ, Romero C, Jiménez-Pérez S (1996) Int J Food Sci Tech 31:411–418
Nursten LE (1981). Food Chem 6:263–277
Resmini P, Pagani AM, Pellegrino L (1991) Tecnica Molitoria 41:821–826
Corzo, N., Villamiel, N., Arias M., Jiménez-Pérez S., Morales FJ. (2000) Food Chem 71:255–258
Guerra-Hernández E, Corzo N (1996) Cereal Chem 73:729–731
Guerra-Hernández E, Corzo N, García-Villanova B (1999) J Cereal Sci 29:171–176
Ramírez-Jiménez A, Guerra-Hernández E, García-Villanova B (2000) J Agric Food Chem 48:4176–4181
Ramírez-Jiménez A, García-Villanova B, Guerra-Hernández E (2001) J Sci Food Agric 81:513–518
Tirelli A (1998). J Food Prot 61:1400–1404
Vallejo-Córdoba B, Mazorra-Manzano MA, González-Córdova AF (2004). J Agric Food Chem 52:5787–5790
Association of Official Analytical Chemists (1995). Official Methods of the Association of Official Analytical Chemists, 16th edn. AOAC, Washington, DC, pp 3–6
Delgado T, Corzo N, Santa-María G, Jimeno ML, Olano A (1992) Chromatographia 33:374–376
Tomlinson AJ, Mlotkiewicz JA, Lewis IASA (1994) Food Chem 49:219–223
Ames JM, Arnoldi A, Bates L, Negroni M (1997) J Agric Food Chem 45:1256–1263
Tirelli A, Pellegrino L (1995) Ital J Food Sci 7:379–385
Acknowledgement
We are indebted to D. Gomez for technical assistance. This research was supported by two postdoctoral grants from Consejeria de Educacion y Ciencia (Junta de Andalucia) and by the Spanish Ministry of Science and Technology under project AGL20001452
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Delgado-Andrade, C., Rufián-Henares, J.A. & Morales, F.J. Fast method to determine furosine in breakfast cereals by capillary zone electrophoresis. Eur Food Res Technol 221, 707–711 (2005). https://doi.org/10.1007/s00217-005-0030-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-005-0030-1