
Abstract
There are emerging advancements in the strategies used for the discovery and development of food-derived bioactive peptides because of their multiple food and health applications. Bioinformatics and peptidomics are two computational and analytical techniques that have the potential to speed up the development of bioactive peptides from bench to market. Structure–activity relationships observed in peptides form the basis for bioinformatics and in silico prediction of bioactive sequences encrypted in food proteins. Peptidomics, on the other hand, relies on “hyphenated” (liquid chromatography–mass spectrometry-based) techniques for the detection, profiling, and quantitation of peptides. Together, bioinformatics and peptidomics approaches provide a low-cost and effective means of predicting, profiling, and screening bioactive protein hydrolysates and peptides from food. This article discuses the basis, strengths, and limitations of bioinformatics and peptidomics approaches currently used for the discovery and analysis of food-derived bioactive peptides.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bioactive peptides derived from food have attracted significant interest in the food and health sectors. However, the classic approach to food peptide production (involving reacting a protein with different proteases and conducting bioactivity assays on the hydrolysates) is laborious, time-consuming, and therefore not cost-effective for industrial-scale production [1,2,3]. To circumvent the limitations, the use of bioinformatics tools has emerged as a strategic approach for the production of known and novel peptide sequences from food proteins. This approach is used to indicate the occurrence of bioactive peptides encrypted in protein sequences and also to give an indication of the types and specificities of proteases that have a high probability to release the bioactive peptide sequences. With this approach, initial mining of bioactive peptides is performed with in silico tools, and this allows researchers to focus on a small number of peptide candidates that are most likely to have high potency of the desired biological activities. This approach is highly desirable as it eliminates guesswork and allows technologists to predict beforehand the kinds and potency of peptides that can be released from a food protein, and the most suitable protease to be used, before wet laboratory work is undertaken with the selected combination of protein and enzyme(s) [4]. The bioinformatics approach to the discovery of bioactive peptides also allows the peptides generated to be characterized for their theoretical physicochemical, bioactive, and sensory properties [1]. The ability of bioinformatics to predict physicochemical properties can help in designing a cascade of purification steps that will be suitable for separating peptides of interest during the wet laboratory stage [3]. Furthermore, the ability to predict the desirable biological (e.g., antimicrobial, enzyme inhibition) properties as well as the potency is important for the development of novel bioactive peptide sequences. Also, knowledge of the undesirable biological properties (e.g., allergenicity, toxicity) is useful when the peptides are intended for functional food or pharmaceutical applications [5]. This information will help the food technologist to take steps to remove or limit the production of allergenic or toxic peptides, or to change the protein–enzyme combination to one that is low in encrypted allergenic or toxic peptides. Moreover, knowledge of the taste-evoking potential of peptides (especially bitterness) is vital for the development of food and drug formulations intended for oral consumption since taste has a significant impact on consumer acceptability of a product [1, 6]. The bioinformatics approach has been used to study encrypted bioactive peptides in cereal proteins (ribulose-1,5-bisphosphate carboxylase/oxygenase, RuBisCO) [7, 8], soybean proteins [9], egg proteins [10], milk proteins [11], cheese [12] and meat proteins [13,14,15].
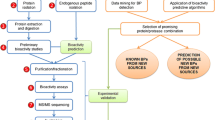
On another note, the use of “hyphenated” mass spectrometric techniques is gaining much interest for the analysis, discovery, and quantitation of the food peptidome, peptides, and food protein hydrolysates [16]. Peptidomics affords an untargeted approach for rapid detection and quantification of a wide range of peptides [17], as well as their in vivo bioactivity networks and signaling systems [18]. Considering that the human body contains several proteases, it is expected that dietary peptides will be subjected to degradation at some point in the body, leading to the release of several smaller peptides or amino acids, with concomitant loss or retention of biological properties. Because of the capability of handling a large quantity of data, peptidomics could be useful for tracking the fate of food peptide sequences and their adsorption, digestion, metabolism, and excretion profile en route in the human body. Figure 1 shows a schematic of the development of food-derived bioactive peptides by combination of the predictive power of bioinformatics and the high-throughput analytical capabilities of peptidomics. This article discusses the prospects and limitations of combining bioinformatics with peptidomics for the discovery and analysis of food-derived bioactive peptides.

Strategy for the discovery of bioactive peptides using tools in bioinformatics and peptidomics. GI gastrointestinal, LC liquid chromatography, MS mass spectrometry
Databases and in silico tools used in bioinformatics-driven discovery of bioactive food peptides
Databases for protein selection and in silico digestion
One of the outcomes of advancements in proteomics research is the large datasets of protein structures. The pioneering studies of Sanger [19], who sequenced the first protein in 1952, paved the way for several other proteins to be characterized and for their sequences to be deposited in searchable online databases [20]. It is estimated that the amino acid sequences of more than eight million proteins are available in databases [21]. This is an important storehouse of amino acid sequences that can be explored in the discovery of bioactive peptides. Bioinformatics discovery of bioactive peptides typically begins with the acquisition of the amino acid sequences of proteins (mostly food proteins) from databases such as UniProtKB (UniProt Consortium), Protein (National Center for Biotechnology Information), and the Protein Data Bank (Research Collaboratory for Structural Bioinformatics). Following the selection of proteins of known primary sequence, an in silico digestion is conducted that uses “protein/peptide cutting” functions of protein digestion databases. This allows the user to select single proteases or combinations of proteases, whose cleavage specificities are taken into account in the generation of peptides from the selected protein. Two of the most widely used bioinformatics tools for in silico protein digestion are BIOPEP “Enzyme action” and ExPASy PeptideCutter. After digestion, these databases generate a map of the protein sequence with an indication of cleavage sites (ExPASy PeptideCutter) or a list of cleaved peptides (BIOPEP) for further processing. A list of other protein databases and tools for in silico digestion is provided in Table 1. A number of computations (also called “descriptors”) are undertaken to identify the frequency and plausibility of release of bioactive peptides from a protein. For example, the BIOPEP database uses descriptors or parameters such as A, B, AE, and W as described in Eqs. 1, 2, 3, and 4 [22, 23]. Descriptor A measures the frequency of occurrence of bioactive peptide fragments in a given protein sequence, and B estimates the potential of a protein to have a particular biological activity; for example, angiotensin-converting enzyme (ACE) inhibition. Parameters A and B are calculated by the following formulae:
where a is the number of peptide fragments in the protein sequence with a specific bioactivity and N is the total number of amino acid residues in the protein, and
where a i is the number of repetitions of the ith bioactive property of interest in the protein sequence, IC50i is the concentration of the ith bioactive property of interest corresponding to its half-maximal activity in micromoles per litre, k is the number of different fragments with the bioactive property of interest, and N is the number of amino acid residues in the protein.
Moreover, there are descriptors for the frequency (AE) and relative frequency (W) with which bioactive peptide sequences are released from the protein sequence by the chosen protease(s). These parameters are calculated by the following formulae:
where d is the number of peptide fragments with a specific bioactivity releasable from the protein sequence by the protease and N is the number of amino acid residues in the protein, and
where AE is the frequency of release of peptide fragments with given activity by the selected protease(s) and A is the frequency of occurrence of bioactive peptide fragments in the protein sequence.
The theoretical degree of hydrolysis (DHt) is also used to estimate the percent degree of hydrolysis of the in silico digestion process:
where d is the number of hydrolyzed peptide bonds and D is the total number of peptide bonds in the primary sequence of the protein.
The significance of these descriptors is illustrated in the following example. Suppose a researcher is interested in identifying (1) the kinds of bioactive properties and (2) the potency of those bioactivities of peptides found in a given protein (e.g., RuBisCO large chain; UniProtKB accession number P04991). Entering the primary sequence of P04991 in the BIOPEP database, under the “Calculations” option, will give 12 different bioactivities and their corresponding A values. Among the bioactivities, dipeptidyl peptidase IV (DPP-IV) inhibition and ACE inhibition have the highest A values (0.4967 and 0.3783, respectively), meaning that peptides with DPP-IV- and ACE-inhibitory activities are the most abundant encrypted sequences in the large chain of RuBisCO. Furthermore, to identify the most suitable protease(s) for releasing the DPP-IV- and ACE-inhibitory peptides, the “Enzyme action” function is used for in silico digestion of the sequence by the trialing of several enzymes and calculation for descriptors such as AE, W, and DHt. For instance, in silico digestion of RuBisCO large chain with the enzyme subtilisin (EC 3.4.21.62) gives respective AE values of 0.0214 and 0.0165 for DPP-IV- and ACE-inhibitory activities, whereas chymotrypsin (EC 3.4.21.1) gives values of 0.0165 and 0.0146. Thus, subtilisin appears to be a better protease for producing DPP-IV- and ACE-inhibitory peptides from RuBisCO large chain. The BIOPEP descriptors are therefore useful for providing a numerical evaluation of the suitability of an enzyme–protein combination to give peptide sequences with desired biological properties.
In silico tools for peptide characterization
Following digestion, the peptides generated are characterized for their (1) physicochemical (molecular weight, theoretical pI, aliphatic index, average hydropathicity, etc.), (2) biological (bioactivities, toxicity, potential allergenicity), and (3) sensory (presence of sweet, bitter, umami, and other taste-evoking peptide sequences) properties. In silico characterization of peptides uses chemometrics and cheminformatics techniques as well as sequence homology in identifying potential bioactivities of peptides [24]. Broadly, these techniques use computational and statistical tools to collect and analyze biochemical data, and design models for understanding and predicting the behaviors of biochemical systems [25, 26]. It is possible to use in silico and computational tools to study the properties and potential biological activities of peptides because peptides (like proteins) exhibit a high degree of structure–activity behaviors. The presence and sequence arrangements of certain amino acid residues provide an indication of the properties and potential bioactivities of a peptide. For example, a peptide sequence with high amounts of cysteine as well as hydrophobic, aromatic, and/or amphiphilic amino acid residues is likely to have antioxidative properties in emulsions [27]. Most renin- and ACE-inhibitory peptides contain small and hydrophobic amino acids at the N-terminus, together with bulky or aromatic/aliphatic amino acids at the C-terminus or penultimate C-terminus [28]. Antimicrobial peptides, on the other hand, often have longer chains and are often amphipathic with a net positive charge [29]. Quantitative modeling of the relationship between physicochemical or structural and biological properties (also called “quantitative structure–activity relationship) is done by regression analysis using techniques such as partial least squares regression, principal component analysis, and artificial neural networks [4, 30]. Examples of some structure descriptors used in quantitative structure–activity relationship studies to score principal properties of amino acid residues/peptides include (1) the z score, which combines into descriptor scores the hydrophilicity/hydrophobicity (z1), molecular size/bulkiness (z2), and electronic properties/charge (z3) of molecules [31]; (2) the molecular surface–weighted holistic invariant molecular (WHIM) indices, a variant of WHIM-based indices, which provide concise, “holistic” three-dimensional molecular surface recognition information about amino acid/peptide–ligand interactions, on the basis of molecular size, shape, symmetry, and atom distribution [32, 33]; (3) Markovian chemicals in silico design (MARCH-INSIDE) descriptors, which use the Markov (stochastic or probability) chain theory to model intramolecular electron delocalization and time-based vibrational decay and codify this information as a numerical description of molecular structure [34]; (4) vectors of hydrophobic, steric, and electronic properties (VHSE) [35], which use the hydrophobicity, stericity, and electronic property of molecules to predict their activity/property; and (5) the divided physicochemical property scores (DPPS) descriptor [36], which uses the same structural properties as VHSE in addition to hydrogen bonds.
Peptide bioactivity prediction can also be achieved by molecular docking studies. Molecular docking is often used for the virtual screening of bioactive molecules and rational design of drug candidates [37, 38]. This technique has been used in a number of studies to select peptides for biological evaluation after peptidomic analyses. For instance, pharmacophore mapping of six peptides from Coix glutelin hydrolysate identified peptides VGQLGGAAGGAF and QSGDQQEF as potential ACE inhibitors [39]. Molecular docking simulation and optimization by in silico proteolysis were then used to demonstrate that GGAAGGAF might have the highest ACE inhibitory activity of all the fragments derived from the peptides. In vivo study then showed that the octapeptide effectively decreased systolic blood pressure of hypertensive rats [39]. In another study, nine peptides were identified from weaver ant protein hydrolysate fraction followed by in silico docking that predicted favorable binding for only peptides FFGT and LSRVP, which were then confirmed to display considerable in vitro ACE-inhibitory activity [40]. Furthermore, molecular docking and calculation of binding interactions and binding free energies were used to explain the mechanism and potency of ACE-inhibitory peptides derived from Mactra veneriformis [41] and Kluyveromyces marxianus [42] proteins. A list of tools and software used for molecular docking of peptides on their biological targets, which are typically proteins, is provided in Table 1.
Clearly, advancements in bioinformatics have provided important tools for efficient discovery of food peptides with biological activities, particularly for preselection of protease/protein precursor candidates, analyzing large protein and peptide datasets, and understanding interactions with biological targets and structure–activity relationships. However, there are a number of practical limitations in using the in silico tools. Considering the short chain length of the food peptides (mostly dipeptides and tripeptides), searching for the occurrence of bioactive sequences in other proteins is limited by the capability of current similarity search tools (e.g., BLAST), which require the input of longer peptide chains to return results. Furthermore, in silico platforms do not have the complete capability for comprehensive peptide analysis (e.g., the lack of multienzyme hydrolysis function), except for BIOPEP. This necessitates the use of multiple tools in peptide analysis. As the platforms are not presently connected, it is cumbersome to export data from one platform to another for further analyses.
Peptidomic analyses of food peptides
As captured in Fig. 1, prediction of bioactive peptides can be achieved with the in silico tools listed in Table 1. Thereafter, the actual wet laboratory production of the peptide is undertaken, followed by identification of peptides present in the whole hydrolysate or in its fractions, often through comprehensive analysis with high-throughput peptidomics [43]. The word “peptidomics” was first introduced in 2001 to describe the comprehensive structural characterization of peptides present in a biological sample [44]. The approach has since been applied to several areas of research, including food science, where it is used to identify and quantify peptides of nutritional or biofunctional relevance [22, 45], as well as for product authentication [46]. Peptidomic studies are conducted by liquid chromatography coupled with tandem mass spectrometry (MS). Hydrolyzed food proteins are complex mixtures, and generally contain hundreds of peptides of different chain length and relative abundance, and this makes it challenging to detect all the peptides. To improve the analysis, protein hydrolysates are often fractionated by various methods [ultrafiltration, hydrophobic high-performance liquid chromatography (HPLC), ion-exchange HPLC, capillary electrophoresis] before peptidomic analysis [47]. Two main ionization methods are used for MS analyses. The most common is electrospray ionization (ESI) because about half of known bioactive peptides in the literature contain fewer than ten amino acids [16]. The other method is matrix-assisted laser desorption/ionization (MALDI), which is used for larger sequences. Peptides of molecular mass less than 500 Da fall within the low mass range, where matrix interference is overwhelming. Consequently, the applicability of MALDI MS in peptidomic studies is limited to detection of larger peptides. However, matrix-free MALDI sample preparation, such as by nano-assisted laser desorption/ionization, is gradually being used to overcome this challenge [48].
In peptidomic analysis, MS/MS data are interpreted with bioinformatics tools. MS/MS spectra that are used for identification (i.e., sequencing) are the result of automated signal processing algorithms that transform raw spectra into generic lists of peaks. Numerous software programs developed for this purpose over the years have been reviewed elsewhere [49]. Peptidomics approaches are used to obtain profiles of peptides natively present in foods, in hydrolyzed proteins, or in fractions obtained after purification. In general, methods such membrane filtration and chromatography (ion-exchange, reversed-phase, and size-exclusion chromatography) are often used to generate concentrated fractions with enhanced activity before MS analysis. Peptidomics has been used to study the properties of several food samples, especially milk from humans, cows, and other mammals. For instance, Dallas et al. [50] identified more than 300 peptides in human milk, and concluded, on the basis of the data obtained, that proteolysis in the mammary gland is selective because most of the peptides were derived from β-casein and none was derived from other major proteins such lactoferrin, α-lactalbumin, and secretory immunoglobulin A. Moreover, about 600 peptides were identified through peptidomics in gastric aspirates from 4- to 12-day-old infants, compared with about one third of that number of peptides present in the mothers’ milk, with the gastric aspirates containing peptides from lactoferrin and α-lactalbumin [51]. This approach led to identification of peptides with known immunomodulatory and antibacterial properties, which can be of clinical relevance in infant intestinal health promotion. Also peptidomic analysis revealed that protease selectivity differs during pregnancy as the number of endogenous peptides is greater in human milk after preterm birth compared with after term birth [45].
Apart from bioactivity, peptidomics of milk was proposed as a way to detect mastitis at the clinical and subclinical levels with the observation of a 1.5-fold increase in the total number of peptides in healthy versus subclinical mastitic milk [52]. Mastitis, an inflammation of the mammary gland, is a prevalent disease in cattle that results in low milk yield and quality. Comparison of the peptidome of healthy versus mastitic milk led to the identification of 154 peptides that can be used as diagnostic biomarkers; in addition, it was possible to use 47 of those peptides to distinguish whether the cause of mastitis originated from infection by Streptococcus aureus or Escherichia coli [53]. Furthermore, peptidomic analysis resulted in the identification of two peptides that can be used to detect adulteration of goat cheese with sheep milk, and the method was capable of detecting up to 2% sheep milk in the cheese [54]. Sassi et al. [55], identified specific peptide and protein markers of fresh bovine, water buffalo, ovine, and goat milk after direct peptidomic analysis of skimmed milk samples. The method can be applied for the detection of milk adulteration or for profiling of the differences in seasonally variable milk.
Peptides derived from food proteins by enzymatic hydrolysis have been characterized by the peptidomics approach. In many cases, the outcome is the profiling of peptides in fractions with biological activities. One area of interest has been the identification of peptides in hydrolysates or fractions with antihypertensive activity, and specifically the inhibition of ACE [1]. For example, fingerprints of all possible peptide peaks in antihypertensive salmon protein hydrolysate fractions were obtained by peptidomics with an ESI MS system, which revealed that the high bioactivity of the fractions compared with the hydrolysate was due to the presence of a higher number of inhibitory peptides with molecular mass less than 500 Da [56]. In a related study, 23 peptides were identified by peptidomics in two HPLC fractions of hydrolyzed hemp seed proteins with potent inhibitory activity against ACE and renin, and five of the peptides (WVYY, PSLPA, WYT, SVYT, and IPAGV) were later found to reduce systolic blood pressure in hypertensive rats [57]. Moreover, size-exclusion chromatography, successive revered-phase HPLC separations, and ESI quadrupole time-of-flight MS/MS led to the identification of FFGT and LSRVP, among nine peptides from weaver ant proteins hydrolysate, as potent ACE inhibitors [40]. Peptidomics has also been used to identify peptides with ACE-inhibitory activity in shrimp shell discard protein hydrolysate fraction [58], buffalo milk hydrolysates [59], low molecular weight fractions of Bifidobacterium longum-fermented milk [60], fish skin gelatin hydrolysates [61], and marine bivalve Mactra veneriformis protein hydrolysates [41]. Peptidomics has also been used to generate a large database of peptides with desirable (bioactive) structural features. For instance, 181 peptides were identified in ACE-inhibiting camel milk colostrum hydrolysates [62], more than 150 potentially active peptides were identified from ACE-inhibiting soy protein hydrolysates [63], and about 39 ACE-inhibitory peptides were identified from rice bran albumin hydrolysates [64].
To combat oxidative stress, associated cellular damage, and the initiation of chronic health conditions such as inflammation, atherosclerosis, diabetes, and cancer [65,66,67], peptidomics has been used in some cases to facilitate the identification of antioxidative peptides. For instance, peptides WVYY and PSLPA were discovered to be free-radical-scavenging antioxidants after peptidomic analysis of a hydrolyzed hemp seed protein fraction [57]. Moreover, ten peptides (with up to 37 amino acid residues) were identified in two fractions of hydrolyzed buffalo milk proteins that displayed the highest radical scavenging activity [59]. A peptidomics approach was also used to identify 20 peptides with relative molecular masses between 417 and 1809 Da (5–21 residues) from an antioxidant fraction of hydrolyzed fish gelatin [61]. Furthermore, two recent studies identified 65 and 50 peptides in antioxidant oat protein hydrolysates produced with Protamex and pepsin, respectively [68, 69], although bioactivity of the individual peptides was not reported. In addition to targeting oxidative stress, peptidomics has been used to identify 16 peptides in two whey hydrolysate fractions with DPP-IV-inhibitory activities, and for the selection of important fractions for further evaluation based on desirable structural features (amino acid residues) [70]. DPP-IV degrades and inactivates incretin hormones, which are involved in the stimulation of glucose-dependent insulin secretion after food ingestion, and as such is a target for the management of type 2 diabetes. In another study, a combination of peptidomics (liquid chromatography–MS/MS analyses) and bioinformatics (using BIOPEP tools) was used to profile the peptidic composition of an albumin hydrolysate (molecular mass less than 100 Da) that possesses α-glucosidase-inhibitory activity, leading to the identification of 40 peptides with potential antidiabetic activity [64].
Challenges in the application of bioinformatics and peptidomics in bioactive food peptide discovery
Bioinformatics
The bioinformatics approach predicts bioactivity of peptides on the basis of the type and number of amino acid residues (or peptide motif) within the sequence [71]. This is simplistic as assumptions on enzymatic release and bioactivity are made on the basis of only primary structures. In vitro, the tertiary structure and three-dimensional folded state of the protein will invariably affect the accessibility of proteases to scissile peptide bonds. This implies that the number of peptides generated in silico, with use of the aforementioned bioinformatics tools, may be more than the number of peptides generated in vitro. Furthermore, in a biological system, the frequency of occurrence of bioactive motifs alone might not be adequate to predict the extent of bioactivity because of interfering conditions encountered during laboratory work. For example, the presence of other molecules, the temperature, pH, and ionic strength of the assay, or the biological matrix will invariably affect protease activity, which may result in the production of different or modified peptide structures when compared with the in silico peptides. Therefore, future research on the design of bioinformatics tools should take into account the possible influence of the protein three-dimensional structure and reaction conditions on the protease activity and release of bioactive peptide from proteins. Moreover, peptide databases are manually curated and updated by generous individual research groups, and this may lead to delays in accessing the most up-to-date information, duplication of efforts, and the development of tools that address issues only within the area of expertise of the group.
Furthermore, the bioinformatics approach currently used in food peptide discovery does not take into account the possibility of posttranslational modification (PTM), such as oxidation, methylation, deamination, and acetylation, of amino acid residues of peptides [72]. PTM of proteins and peptides is a common phenomenon when the biomolecules undergo downstream processing steps such as heating, drying, high-pressure treatment, and dehydration. To overcome this challenge, a number of user-friendly and open-access integrative bioinformatics tools have been designed to study protein and peptide PTMs, including iPTMnet (http://research.bioinformatics.udel.edu/iptmnet/) [73], STRAP PTM (http://www.bumc.bu.edu/cardiovascularproteomics/cpctools/strap-ptm/) [74], and jEcho (http://www.healthinformaticslab.org/supp/) [75]. These tools can be integrated into existing food peptide analysis methods, particularly for identifying possible PTMs in prediction studies. Lastly, selection of protein precursors by the current approach is limited to only proteins whose primary sequences are available in open-access databases, and this may exclude potential precursors, especially minor proteins and proteins in alternative sources. This calls for research in the area of protein sequencing, particularly of proteins that can be produced sustainably. Apart from dairy and animal proteins, food-grade microorganisms (single-cell proteins), marine organisms, edible insects, and fungi are alternative sources of proteins for the production of bioactive peptides. The use of these proteins is possible only if their protein sequences are available. Bioinformatics of food-derived peptides will therefore benefit from advances in protein sequencing.
Peptidomics
A number of issues can impede bioactive food peptide development, but advances in the field of peptidomics are providing strategies to overcome some of these challenges. For instance, it is challenging to separate and identify bioactive peptides present in complex food matrices, protein hydrolysates, or peptide fractions. The peptidomics approach can facilitate peptide identification because it can be performed on samples with minimal cleanup procedures. Although this approach has the advantage of not requiring complete peptide separation, there are some limitations. Co-elution of peptides is common in liquid chromatography, and this makes the detection of less concentrated peptides difficult or impossible. In addition, a number of analyses focus on multiply charged ions (2+ to 5+). Although this eliminates the detection of non-peptides and increases sensitivity, peptides smaller than 800 Da are often omitted from the analysis. Food-derived peptides of molecular mass less than 1000 Da are often the ones with the most potent biologically relevant activities and are also considered to be the most potentially bioavailable. It has been proposed that the challenge of peptide co-elution can be overcome by tandem fragmentation techniques such as collision-induced dissociation, high-energy collision-induced dissociation, and electron transfer dissociation. A combination of these three techniques has been shown to be effective for complete profiling of peptides by liquid chromatography–Fourier transform MS/MS of peptides [76]. Some other technological advances in peptidomic analysis are currently used mostly in the biomolecular sciences, but the principles can be transferred for the analysis of food peptides in complex matrices. For example, imaging MS permits the visualization of the spatial distribution of compounds, ion mobility MS provides quick (millisecond) separation and mass detection of compounds, and direct tissue/cell MALDI time-of-flight analysis allows the profiling of peptides in intact tissue samples [77].
Prospects and future directions
Bioinformatics and peptidomics approaches for the investigation and development of bioactive peptides independently facilitate the discovery and analysis of a large number of peptides of interest in a comprehensive and cost-effective manner. Although it is thought that in silico results are not always replicated in wet laboratory analysis because of the aforementioned reasons, there is a need to establish actual predictive accuracy and comparison of the effectiveness of different in silico tools and methods. Further work is also needed on adapting recent peptidomics technologies for the discovery and analysis of bioactive food peptides. In addition, a one-stop platform that integrates all in silico tools and steps (protein/protease selection, hydrolysis, bioactivity screening, bitterness, toxicity, and allergenicity prediction, structure–activity relationship analysis, etc.) can be developed to enhance the capability and efficiency of bioinformatics in food bioactive peptide discovery and analysis. Also, it would be worthwhile to establish a centralized peer-reviewed platform where researchers can directly input new peptide sequences from the literature, or as soon as they are discovered. This crowdsourcing approach will lead to a comprehensive up-to-date open-access database of functional peptides. Lastly, the predictive capability of bioinformatics and the high-throughput analytical capability of peptidomics can be combined into a new, powerful tool for the discovery and analysis of functional peptides present in food and other complex biological matrices.
References
Udenigwe CC. Bioinformatics approaches, prospects and challenges of food bioactive peptide research. Trends Food Sci Technol. 2014;36(2):137–43. https://doi.org/10.1016/j.tifs.2014.02.004.
Agyei D, Danquah MK. Industrial-scale manufacturing of pharmaceutical-grade bioactive peptides. Biotechnol Adv. 2011;29(3):272–7.
Agyei D, Ongkudon CM, Wei CY, Chan AS, Danquah MK. Bioprocess challenges to the isolation and purification of bioactive peptides. Food Bioprod Process. 2016;98:244–56. https://doi.org/10.1016/j.fbp.2016.02.003.
Carrasco-Castilla J, Hernández-Álvarez A, Jiménez-Martínez C, Gutiérrez-López G, Dávila-Ortiz G. Use of proteomics and peptidomics methods in food bioactive peptide science and engineering. Food Eng Rev. 2012;4(4):224–43. https://doi.org/10.1007/s12393-012-9058-8.
Hayes M, Rougé P, Barre A, Herouet-Guicheney C, Roggen EL. In silico tools for exploring potential human allergy to proteins. Drug Discov Today Dis Models. 2015;17-18(Suppl C):3–11. https://doi.org/10.1016/j.ddmod.2016.06.001.
Temussi PA. The good taste of peptides. J Pept Sci. 2012;18(2):73–82. https://doi.org/10.1002/psc.1428.
Udenigwe CC, Gong M, Wu S. In silico analysis of the large and small subunits of cereal RuBisCO as precursors of cryptic bioactive peptides. Process Biochem. 2013;48(11):1794–9. https://doi.org/10.1016/j.procbio.2013.08.013.
Udenigwe CC, Okolie CL, Qian H, Ohanenye IC, Agyei D, Aluko RE. Ribulose-1,5-bisphosphate carboxylase as a sustainable and promising plant source of bioactive peptides for food applications. Trends Food Sci Technol. 2017;69A:74–82. https://doi.org/10.1016/j.tifs.2017.09.001.
Gu Y, Wu J. LC–MS/MS coupled with QSAR modeling in characterising of angiotensin I-converting enzyme inhibitory peptides from soybean proteins. Food Chem. 2013;141(3):2682–90. https://doi.org/10.1016/j.foodchem.2013.04.064.
Majumder K, Wu J. A new approach for identification of novel antihypertensive peptides from egg proteins by QSAR and bioinformatics. Food Res Int. 2010;43(5):1371–8. https://doi.org/10.1016/j.foodres.2010.04.027.
Nongonierma AB, FitzGerald RJ. Structure activity relationship modelling of milk protein-derived peptides with dipeptidyl peptidase IV (DPP-IV) inhibitory activity. Peptides. 2016;79:1–7. https://doi.org/10.1016/j.peptides.2016.03.005.
Sagardia I, Iloro I, Elortza F, Bald C. Quantitative structure–activity relationship based screening of bioactive peptides identified in ripened cheese. Int Dairy J. 2013;33(2):184–90. https://doi.org/10.1016/j.idairyj.2012.12.006.
Keska P, Stadnik J. Antimicrobial peptides of meat origin - an in silico and in vitro analysis. Protein Pept Lett. 2017;24(2):165–73. https://doi.org/10.2174/0929866523666161220113230.
Lafarga T, O'Connor P, Hayes M. Identification of novel dipeptidyl peptidase-IV and angiotensin-I-converting enzyme inhibitory peptides from meat proteins using in silico analysis. Peptides. 2014;59:53–62. https://doi.org/10.1016/j.peptides.2014.07.005.
Lafarga T, O'Connor P, Hayes M. In silico methods to identify meat-derived prolyl endopeptidase inhibitors. Food Chem. 2015;175:337–43. https://doi.org/10.1016/j.foodchem.2014.11.150.
Panchaud A, Affolter M, Kussmann M. Mass spectrometry for nutritional peptidomics: how to analyze food bioactives and their health effects. J Proteomics. 2012;75(12):3546–59. https://doi.org/10.1016/j.jprot.2011.12.022.
Liu Y, Forcisi S, Lucio M, Harir M, Bahut F, Deleris-Bou M, et al. Digging into the low molecular weight peptidome with the OligoNet web server. Sci Rep. 2017;7(1):11692. https://doi.org/10.1038/s41598-017-11786-w.
Boonen K, Creemers JW, Schoofs L. Bioactive peptides, networks and systems biology. Bioessays. 2009;31(3):300–14. https://doi.org/10.1002/bies.200800055.
Sanger F. The arrangement of amino acids in proteins. Adv Protein Chem. 1952;7:1–67.
Xu D. Protein databases on the Internet. Curr Protoc Mol Biol. 2004;68:19.4.1–15. https://doi.org/10.1002/0471142727.mb1904s68.
Levitt M. Nature of the protein universe. Proc Natl Acad Sci U S A. 2009;106(27):11079–84. https://doi.org/10.1073/pnas.0905029106.
Minkiewicz P, Dziuba J, Iwaniak A, Dziuba M, Darewicz M. BIOPEP database and other programs for processing bioactive peptide sequences. J AOAC Int. 2008;91(4):965–80.
Minkiewicz P, Dziuba J, Michalska J. Bovine meat proteins as potential precursors of biologically active peptides--a computational study based on the BIOPEP database. Food Sci Technol Int. 2011;17(1):39–45. https://doi.org/10.1177/1082013210368461.
Iwaniak A, Minkiewicz P, Darewicz M, Protasiewicz M, Mogut D. Chemometrics and cheminformatics in the analysis of biologically active peptides from food sources. J Funct Foods. 2015;16:334–51. https://doi.org/10.1016/j.jff.2015.04.038.
Bertacchini L, Cocchi M, Li Vigni M, Marchetti A, Salvatore E, Sighinolfi S, et al. The impact of chemometrics on food traceability. In: Marini F, editor. Chemometrics in food chemistry Data handling in science and technology, vol. 28. Oxford: Elsevier; 2013. p. 371–410.
Wishart DS. Introduction to cheminformatics. Curr Protoc Bioinformatics. 2007;18:14.1.1–9. https://doi.org/10.1002/0471250953.bi1401s18.
Freitas AC, Andrade JC, Silva FM, Rocha-Santos TAP, Duarte AC, Gomes AM. Antioxidative peptides: trends and perspectives for future research. Curr Med Chem. 2013;20(36):4575–94.
Wu J, Aluko RE, Nakai S. Structural requirements of angiotensin I-converting enzyme inhibitory peptides: quantitative structure−activity relationship study of di- and tripeptides. J Agric Food Chem. 2006;54(3):732–8. https://doi.org/10.1021/jf051263l.
Haney EF, Hancock REW. Peptide design for antimicrobial and immunomodulatory applications. Pept Sci. 2013;100(6):572–83. https://doi.org/10.1002/bip.22250.
Li-Chan ECY. Bioactive peptides and protein hydrolysates: research trends and challenges for application as nutraceuticals and functional food ingredients. Curr Opin Food Sci. 2015;1:28–37. https://doi.org/10.1016/j.cofs.2014.09.005.
Hellberg S, Sjoestroem M, Skagerberg B, Wold S. Peptide quantitative structure-activity relationships, a multivariate approach. J Med Chem. 1987;30(7):1126–35. https://doi.org/10.1021/jm00390a003.
Zaliani A, Gancia E. MS-WHIM scores for amino Acids: a new 3D-description for peptide QSAR and QSPR studies. J Chem Inf Comput Sci. 1999;39(3):525–33. https://doi.org/10.1021/ci980211b.
Todeschini R, Lasagni M, Marengo E. New molecular descriptors for 2D and 3D structures. Theory. J Chemom. 1994;8(4):263–72. https://doi.org/10.1002/cem.1180080405.
Ramos de Armas R, González Díaz H, Molina R, Pérez González M, Uriarte E. Stochastic-based descriptors studying peptides biological properties: modeling the bitter tasting threshold of dipeptides. Bioorg Med Chem. 2004;12(18):4815–22. https://doi.org/10.1016/j.bmc.2004.07.017.
Mei H, Liao ZH, Zhou Y, Li SZ. A new set of amino acid descriptors and its application in peptide QSARs. Pept Sci. 2005;80(6):775–86. https://doi.org/10.1002/bip.20296.
Tian F, Yang L, Lv F, Yang Q, Zhou P. In silico quantitative prediction of peptides binding affinity to human MHC molecule: an intuitive quantitative structure-activity relationship approach. Amino Acids. 2009;36(3):535–54. https://doi.org/10.1007/s00726-008-0116-8.
Jimsheena VK, Gowda LR. Arachin derived peptides as selective angiotensin I-converting enzyme (ACE) inhibitors: structure–activity relationship. Peptides. 2010;31(6):1165–76. https://doi.org/10.1016/j.peptides.2010.02.022.
Meng X-Y, Zhang H-X, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7(2):146–57.
Li B, Qiao L, Li L, Zhang Y, Li K, Wang L, et al. Novel antihypertensive peptides derived from adlay (Coix larchryma-jobi L. var. ma-yuen Stapf) glutelin. Molecules. 2017;22(1):534. https://doi.org/10.3390/molecules22010123.
Pattarayingsakul W, Nilavongse A, Reamtong O, Chittavanich P, Mungsantisuk I, Mathong Y, et al. Angiotensin-converting enzyme inhibitory and antioxidant peptides from digestion of larvae and pupae of Asian weaver ant, Oecophylla smaragdina, Fabricius. J Sci Food Agric. 2017;97(10):3133–40. https://doi.org/10.1002/jsfa.8155.
Liu R, Zhu Y, Chen J, Wu H, Shi L, Wang X, et al. Characterization of ACE inhibitory peptides from Mactra veneriformis hydrolysate by nano-liquid chromatography electrospray ionization mass spectrometry (nano-LC-ESI-MS) and molecular docking. Mar Drugs. 2014;12(7):3917–28. https://doi.org/10.3390/md12073917.
Mirzaei M, Mirdamadi S, Ehsani MR, Aminlari M. Production of antioxidant and ACE-inhibitory peptides from Kluyveromyces marxianus protein hydrolysates: purification and molecular docking. J Food Drug Anal. 2017; https://doi.org/10.1016/j.jfda.2017.07.008.
Wu C, Monroe ME, Xu Z, Slysz GW, Payne SH, Rodland KD, et al. An optimized informatics pipeline for mass spectrometry-based peptidomics. J Am Soc Mass Spectrom. 2015;26(12):2002–8. https://doi.org/10.1007/s13361-015-1169-z.
Schulz-Knappe P, Hans-Dieter Z, Heine G, Jurgens M, Schrader M. Peptidomics the comprehensive analysis of peptides in complex biological mixtures. Comb Chem High Throughput Screen. 2012;4(2):207–17. https://doi.org/10.2174/1386207013331246.
Dallas DC, Guerrero A, Khaldi N, Borghese R, Bhandari A, Underwood MA, et al. A peptidomic analysis of human milk digestion in the infant stomach reveals protein-specific degradation patterns. J Nutr. 2014;144(6):815–20. https://doi.org/10.3945/jn.113.185793.
Montowska M. Using peptidomics to determine the authenticity of processed meat. In: Colgrave ML, editor. Proteomics in food science. London: Acedemic; 2017. p. 225–40.
Saavedra L, Hebert EM, Minahk C, Ferranti P. An overview of “omic” analytical methods applied in bioactive peptide studies. Food Res Int. 2013;54(1):925–34. https://doi.org/10.1016/j.foodres.2013.02.034.
Capriotti AL, Cavaliere C, Piovesana S, Samperi R, Laganà A. Recent trends in the analysis of bioactive peptides in milk and dairy products. Anal Bioanal Chem. 2016;408(11):2677–85. https://doi.org/10.1007/s00216-016-9303-8.
Hernandez P, Müller M, Appel RD. Automated protein identification by tandem mass spectrometry: Issues and strategies. Mass Spectrom Rev. 2006;25(2):235–54. https://doi.org/10.1002/mas.20068.
Dallas DC, Guerrero A, Khaldi N, Castillo PA, Martin WF, Smilowitz JT, et al. Extensive in vivo human milk peptidomics reveals specific proteolysis yielding protective antimicrobial peptides. J Proteome Res. 2013;12(5):2295–304. https://doi.org/10.1021/pr400212z.
Dallas DC, Smink CJ, Robinson RC, Tian T, Guerrero A, Parker EA, et al. Endogenous human milk peptide release is ´greater after preterm birth than term birth. J Nutr. 2014;145(3):425–33. https://doi.org/10.3945/jn.114.203646.
Guerrero A, Dallas DC, Contreras S, Bhandari A, Cánovas A, Islas-Trejo A, et al. Peptidomic analysis of healthy and subclinically mastitic bovine milk. Int Dairy J. 2015;46:46–52. https://doi.org/10.1016/j.idairyj.2014.09.006.
Mansor R, Mullen W, Albalat A, Zerefos P, Mischak H, Barrett DC, et al. A peptidomic approach to biomarker discovery for bovine mastitis. J Proteomics. 2013;85:89–98. https://doi.org/10.1016/j.jprot.2013.04.027.
Guarino C, Fuselli F, la Mantia A, Longo L, Faberi A, Marianella RM. Peptidomic approach, based on liquid chromatography/electrospray ionization tandem mass spectrometry, for detecting sheep's milk in goat's and cow's cheeses. Rapid Commun Mass Spectrom. 2010;24(6):705–13. https://doi.org/10.1002/rcm.4426.
Sassi M, Arena S, Scaloni A. MALDI-TOF-MS platform for integrated proteomic and peptidomic profiling of milk samples allows rapid detection of food adulterations. J Agric Food Chem. 2015;63(27):6157–71. https://doi.org/10.1021/acs.jafc.5b02384.
Girgih AT, Nwachukwu ID, Hasan FM, Fagbemi TN, Malomo SA, Gill TA, et al. Kinetics of in vitro enzyme inhibition and blood pressure-lowering effects of salmon (Salmo salar) protein hydrolysates in spontaneously hypertensive rats. J Funct Foods. 2016;20:43–53. https://doi.org/10.1016/j.jff.2015.10.018.
Girgih AT, He R, Malomo S, Offengenden M, Wu J, Aluko RE. Structural and functional characterization of hemp seed (Cannabis sativa L.) protein-derived antioxidant and antihypertensive peptides. J Funct Foods. 2014;6:384–94. https://doi.org/10.1016/j.jff.2013.11.005.
Ambigaipalan P, Shahidi F. Bioactive peptides from shrimp shell processing discards: antioxidant and biological activities. J Funct Foods. 2017;34:7–17. https://doi.org/10.1016/j.jff.2017.04.013.
Abdel-Hamid M, Otte J, De Gobba C, Osman A, Hamad E. Angiotensin I-converting enzyme inhibitory activity and antioxidant capacity of bioactive peptides derived from enzymatic hydrolysis of buffalo milk proteins. Int Dairy J. 2017;66:91–8. https://doi.org/10.1016/j.idairyj.2016.11.006.
Ha GE, Chang OK, Jo S-M, Han G-S, Park B-Y, Ham J-S, et al. Identification of antihypertensive peptides derived from low molecular weight casein hydrolysates generated during fermentation by Bifidobacterium longum KACC 91563. Korean J Food Sci Anim Resour. 2015;35(6):738–47. https://doi.org/10.5851/kosfa.2015.35.6.738.
Lassoued I, Mora L, Barkia A, Aristoy MC, Nasri M, Toldra F. Bioactive peptides identified in thornback ray skin's gelatin hydrolysates by proteases from Bacillus subtilis and Bacillus amyloliquefaciens. J Proteomics. 2015;128:8–17. https://doi.org/10.1016/j.jprot.2015.06.016.
Jrad Z, El Hatmi H, Adt I, Girardet J-M, Cakir-Kiefer C, Jardin J, et al. Effect of digestive enzymes on antimicrobial, radical scavenging and angiotensin I-converting enzyme inhibitory activities of camel colostrum and milk proteins. Dairy Sci Technol. 2013;94(3):205–24. https://doi.org/10.1007/s13594-013-0154-1.
Margatan W, Ruud K, Wang Q, Markowski T, Ismail B. Angiotensin converting enzyme inhibitory activity of soy protein subjected to selective hydrolysis and thermal processing. J Agric Food Chem. 2013;61(14):3460–7. https://doi.org/10.1021/jf4001555.
Uraipong C, Zhao J. Identification and functional characterisation of bioactive peptides in rice bran albumin hydrolysates. Int J Food Sci Technol. 2016;51(10):2201–8. https://doi.org/10.1111/ijfs.13204.
Chakrabarti S, Jahandideh F, Wu J. Food-derived bioactive peptides on inflammation and oxidative stress. BioMed Res Int. 2014;2014:1–11. https://doi.org/10.1155/2014/608979.
Matés JM, Segura JA, Alonso FJ, Márquez J. Intracellular redox status and oxidative stress: implications for cell proliferation, apoptosis, and carcinogenesis. Arch Toxicol. 2008;82(5):273–99. https://doi.org/10.1007/s00204-008-0304-z.
Qian Z-J, Jung W-K, Byun H-G, Kim S-K. Protective effect of an antioxidative peptide purified from gastrointestinal digests of oyster, Crassostrea gigas against free radical induced DNA damage. Bioresour Technol. 2008;99(9):3365–71. https://doi.org/10.1016/j.biortech.2007.08.018.
Baakdah MM, Tsopmo A. Identification of peptides, metal binding and lipid peroxidation activities of HPLC fractions of hydrolyzed oat bran proteins. J Food Sci Technol. 2016;53(9):3593–601. https://doi.org/10.1007/s13197-016-2341-6.
Vanvi A, Tsopmo A. Pepsin digested oat bran proteins: separation, antioxidant activity, and identification of new peptides. J Chem. 2016;2016:8. https://doi.org/10.1155/2016/8216378.
Silveira ST, Martinez-Maqueda D, Recio I, Hernandez-Ledesma B. Dipeptidyl peptidase-IV inhibitory peptides generated by tryptic hydrolysis of a whey protein concentrate rich in β-lactoglobulin. Food Chem. 2013;141(2):1072–7. https://doi.org/10.1016/j.foodchem.2013.03.056.
Huang B-B, Lin H-C, Chang Y-W. Analysis of proteins and potential bioactive peptides from tilapia (Oreochromis spp.) processing co-products using proteomic techniques coupled with BIOPEP database. J Funct Foods. 2015;19A:629–40. https://doi.org/10.1016/j.jff.2015.09.065.
Rajendran SRCK, Mason B, Udenigwe CC. Peptidomics of peptic digest of selected potato tuber proteins: post-translational modifications and limited cleavage specificity. J Agric Food Chem. 2016;64(11):2432–7. https://doi.org/10.1021/acs.jafc.6b00418.
Ross KE, Huang H, Ren J, Arighi CN, Li G, Tudor CO, et al. iPTMnet: integrative bioinformatics for studying PTM metworks. In: Wu CH, Arighi CN, Ross KE, editors. Protein bioinformatics: from Protein modifications and networks to proteomics. New York: Springer; 2017. p. 333–53.
Spencer JL, Bhatia VN, Whelan SA, Costello CE, McComb ME. STRAP PTM: software tool for rapid annotation and differential comparison of protein post-translational modifications. Curr Protoc Bioinformatics. 2013;13:13.22.1-36. https://doi.org/10.1002/0471250953.bi1322s44.
Zhao M, Zhang Z, Mai G, Luo Y, Zhou F. jEcho: an evolved weight vector to characterize the protein’s posttranslational modification motifs. Interdiscip Sci. 2015;7(2):194–9. https://doi.org/10.1007/s12539-015-0260-2.
Shen Y, Tolić N, Xie F, Zhao R, Purvine SO, Schepmoes AA, et al. Effectiveness of CID, HCD, and ETD with FT MS/MS for degradomic-peptidomic analysis: comparison of peptide identification methods. J Proteome Res. 2011;10(9):3929–43. https://doi.org/10.1021/pr200052c.
Dallas DC, Guerrero A, Parker EA, Robinson RC, Gan J, German JB, et al. Current peptidomics: applications, purification, identification, quantification, and functional analysis. Proteomics. 2015;15:1026–38. https://doi.org/10.1002/pmic.201400310.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Published in the topical collection Discovery of Bioactive Compounds with guest editors Aldo Laganà, Anna Laura Capriotti and Chiara Cavaliere.
Rights and permissions
About this article

Cite this article
Agyei, D., Tsopmo, A. & Udenigwe, C.C. Bioinformatics and peptidomics approaches to the discovery and analysis of food-derived bioactive peptides. Anal Bioanal Chem 410, 3463–3472 (2018). https://doi.org/10.1007/s00216-018-0974-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-018-0974-1