
Abstract
Milk proteins have been extensively studied for their ability to yield a range of bioactive peptides following enzymatic hydrolysis/digestion. However, many hurdles still exist regarding the widespread utilization of milk protein-derived bioactive peptides as health enhancing agents for humans. These mostly arise from the fact that most milk protein-derived bioactive peptides are not highly potent. In addition, they may be degraded during gastrointestinal digestion and/or have a low intestinal permeability. The targeted release of bioactive peptides during the enzymatic hydrolysis of milk proteins may allow the generation of particularly potent bioactive hydrolysates and peptides. Therefore, the development of milk protein hydrolysates capable of improving human health requires, in the first instance, optimized targeted release of specific bioactive peptides. The targeted hydrolysis of milk proteins has been aided by a range of in silico tools. These include peptide cutters and predictive modeling linking bioactivity to peptide structure [i.e., molecular docking, quantitative structure activity relationship (QSAR)], or hydrolysis parameters [design of experiments (DOE)]. Different targeted enzymatic release strategies employed during the generation of milk protein hydrolysates are reviewed herein and their limitations are outlined. In addition, specific examples are provided to demonstrate how in silico tools may help in the identification and discovery of potent milk protein-derived peptides. It is anticipated that the development of novel strategies employing a range of in silico tools may help in the generation of milk protein hydrolysates containing potent and bioavailable peptides, which in turn may be used to validate their health promoting effects in humans.

The targeted enzymatic hydrolysis of milk proteins may allow the generation of highly potent and bioavailable bioactive peptides.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Milk proteins contain peptide motifs known as bioactive peptides, which can positively impact on a range of health biomarkers [1]. Bioactive peptides may be released by several means involving the digestion of food proteins, i.e., by microbial fermentation along with physical, chemical, or enzymatic breakdown. Enzymatic hydrolysis is one of the most frequently used approached employed during the generation of bioactive peptides.
The bioactive properties of intact or hydrolyzed milk proteins have been extensively studied over the past 40 y. In particular, their mineral binding, opioid, antihypertensive, antimicrobial, anti-inflammatory, antidiabetic, and anticancer properties have been reported [2, 3]. Most of these properties have been determined using in vitro assays and animal studies. However, human intervention studies with milk protein hydrolysates have also been carried out, most particularly in the areas of anticarcinogenic, antihypertensive, and antidiabetic activities [4, 5]. Furthermore, the release of bioactive peptides during milk protein digestion in humans has also been demonstrated in numerous studies [for reviews, see:6, 7, 8]. Some of the milk peptides may be bioavailable and reach the circulation of humans, as previously demonstrated [9,10,11]. Similarly, a recent study has reported on the identification of caseinophosphopeptides (CPPs) in human plasma following 7 d intake of 100 g/d parmigiano reggiano cheese [11]. While bioactive peptides may be released in the gastrointestinal tract of humans following ingestion of intact milk proteins [12], increased bioavailability and potency have been used as arguments to justify the need to hydrolyze milk proteins before their ingestion. However, milk protein hydrolysis does not always allow achievement of superior bioavailability and potency compared with unhydrolyzed milk proteins. The reason for this may be linked to the fact that the peptides are not optimally released during hydrolysis due to food matrix effects. In addition, the peptides may not be able to survive gastrointestinal conditions (digestive enzymes, degradation by the microbiota, pH modifications, etc.). Finally, they may not be able to cross the intestinal barrier (low bioavailability). In this context, it has been proposed that specific/targeted proteolysis of food proteins may allow the release of peptides that are more likely to be bioactive in vivo [13].
To date, the generation of milk protein hydrolysates has been based on rather empirical approaches. These approaches, also termed conventional/classic approaches, have been described in different reviews [14,15,16]. In the empirical approach, the choice of the starting substrate, enzyme preparation(s), and hydrolysis parameters is based on previous studies published in the literature or in-house know-how based on preliminary studies. The conventional approach, however, has a number of drawbacks, mainly that it limits the discovery process, since it provides very little scope for innovation. In fact the same enzyme preparation(s) and rather similar hydrolytic conditions have been used in a range of studies based on the assumption that parameters that previously yielded bioactive hydrolysates would also allow the generation of bioactive samples. The main issue with this approach is that justification for the selection of the initial conditions has not always been provided in the literature. Furthermore, the hydrolysis parameters may not always have been selected solely to optimally generate bioactive samples. Other parameters, which are not always considered, such as cost, availability of reagents (i.e., enzyme and substrate), and equipment as well as technological limitations (e.g., requirement for a certain extent of hydrolysis, pH, total solid content, solubility, etc.), may also play a role in the final choice of hydrolytic reagents and parameters. In addition, certain hydrolytic conditions may not apply to every situation as it is well known that interactive effects between hydrolysis parameters (e.g., hydrolysis temperature, pH, time, enzyme, and substrate concentration, etc.) can affect peptide release [17, 18]. For this reason, it may be helpful to employ a more targeted approach during the generation of food protein hydrolysates. Targeted approaches help to build stronger scientific hypotheses to better understand how protein and enzyme combinations together with various hydrolysis parameters may affect peptide release during hydrolysis [14]. In silico methods are the basis for the targeted enzymatic release of bioactive peptides [14,15,16, 19]. In silico methods have been applied to a wide range of food proteins and, in several instances, have led to the generation of potent enzymatic hydrolysates and in the identification of novel bioactive peptides.
Several studies have described the utilization of in silico methodologies for the release of bioactive peptides from a range of food proteins from animal and plant sources [20,21,22,23,24,25,26,27,28]. Recent review articles have discussed how the application of in silico methods may help in the discovery of bioactive peptides originating from a range of dietary proteins [16, 19, 29]. More specifically, two recent review articles have described the application of in silico methodologies in the analysis of milk bioactive peptides [15, 30]. However, the link between in silico methodologies and improvement in the bioactive properties of milk protein hydrolysates and in the detection of bioactive peptides has not been specifically addressed in earlier reviews. Therefore, the aim of this review was to collate and critique the targeted strategies currently employed during the generation and identification of bioactive milk protein hydrolysates. The main in silico tools [i.e., peptide cutters, molecular docking, quantitative structure activity relationship (QSAR), artificial neural networks (ANN), and design of experiments (DOEs)] specifically applied to predict the bioactive properties of milk protein-derived peptides are outlined. Furthermore, the commercial enzyme preparations commonly used during the production of milk protein hydrolysates are described. Finally, the usefulness of targeted enzymatic release in the subsequent identification of bioactive peptides within milk protein hydrolysates is discussed.
Milk protein substrates and commercial food-grade enzyme preparations used in the generation of milk protein hydrolysates
Bovine milk proteins are the most studied protein substrates for the generation of bioactive food protein hydrolysates. However, there is an increased interest in milk proteins from other species as a source of bioactive peptides. These include milks originating from mare, goat, camel, buffalo, yak, and sheep [31,32,33]. Protein hydrolysates may be prepared using milk proteins or fractionated milk proteins [e.g., casein (CN) or whey protein (WP) concentrates and isolates]. Fractionated milk proteins are obtained using different unit operations involving selective precipitation of proteins and membrane filtration systems such as ultra- and diafiltration [34, 35]. Milk protein fractions (30%–90% protein) have also been employed during enzymatic hydrolysis with a view to increase the yield of specific bioactive peptides. Furthermore, preparations enriched in individual milk proteins (e.g., individual CNs or WPs) have been used during the generation of bioactive peptides [36,37,38,39,40,41].
Together with the milk protein substrate, selection of the enzyme preparation plays a key role in determining the peptides released and therefore in the generation of bioactive peptides. Protein hydrolysates intended for human nutrition must be prepared using food-grade protocols. This therefore restricts the range of enzymes that may be used during the manufacture of food protein hydrolysates. Numerous food-grade enzyme preparations are available on the market. These preparations originate from mammalian (porcine, bovine, etc.), microbial (bacterial and fungal), or plant sources [42, 43]. These preparations generally consist of a crude extract and, as a consequence, they contain several enzyme activities. The low purity of commercial enzyme preparations has been linked to the high costs associated with enzyme purification [44]. In addition, processes involved in extensive purification steps may require the use of solvents that may not be generally recognized as safe (GRAS).
Gastrointestinal enzymes (pepsin, trypsin, chymotrypsin, and pancreatic elastase) have commonly been employed for the generation of bioactive peptides. There are a number of reasons for this. First, several studies have attempted to understand the release of bioactive peptides during in vivo digestion of milk proteins [for reviews, see: 6, 7]. Recently, a correlation between peptide release from milk proteins during their digestion in humans and in vitro simulated gastrointestinal digestion has been established [45]. Another reason to use gastrointestinal enzymes is linked to the assumption that peptides released by digestive enzymes may be relatively stable during transit in the gastrointestinal tract of humans. This is a clear rationale to design milk protein hydrolysates containing peptides that are bioavailable in vivo. Finally, gastrointestinal digestive enzymes such as pepsin, trypsin, chymotrypsin, and pancreatic elastase have been studied for many years. Therefore, their cleavage specificity is relatively well known, which can be relevant for studies linking predicted peptide release to bioactive properties.
The globalization of food production and trade as well as the development of products for specific diets and food product certification has led to a move to enzymes that are compliant with Hallal, Kosher, and vegetarian/vegan status. Enzyme preparations that are compliant with the above classifications correspond generally to microbial and plant-derived preparations. Several of these enzyme preparations have been designed for specific food applications, e.g., meat, brewery, bakery applications, etc. However, an enzyme preparation may be used for milk protein hydrolysis provided that its optimum operating conditions (pH, temperature, ionic strength, etc.) are compatible with the milk protein sample being hydrolyzed. The commercial proteinase/peptidase preparations that have typically been used during the manufacture of food protein hydrolysates have recently been summarized in two review papers [46, 47].
The utilization of crude enzyme preparations has a number of drawbacks. One of these is the lack of characterization of enzyme activities within the preparations, i.e., lack of knowledge regarding the side activities present within the preparation. A limited number of studies have attempted to characterize the enzyme activities present within commercial food-grade enzyme preparations. Some enzyme activities that have been identified in commercial food-grade enzyme preparations used for the manufacture of milk protein hydrolysates are summarized in Table 1. As can be seen, besides the main activity reported to be present in the enzyme preparation, a wide range of other activities, some of which are not proteinases and peptidases, were present. The occurrence of proteinase and peptidase side-activities is therefore likely to affect peptide release during milk protein hydrolysis.
Batch-to-batch variability has also been highlighted within commercial enzyme preparations. The stability of the enzyme activities and the batch-to-batch variability of Flavourzyme, an Aspergillus oryzae-derived enzyme preparation containing both endo- and exopeptidases, has been studied [52]. Leu aminopeptidase activity, the main peptidase activity of Flavourzyme, was similar in the 12 batches of Flavourzyme tested and this activity was relatively stable over 22 mo storage at 7 °C. However, significant batch-to-batch variability was observed for the seven other enzyme activities assessed (various aminopeptidases, dipeptidyl peptidase, proteinase, and amylase). This was particularly the case for the general endoproteinase activity as measured by the azocasein assay, which varied between 9% and 100% between batches. These differences in activities, and more particularly those of the endoproteinase activity, were shown to affect the release of peptides during bovine CN hydrolysis. This resulted in a variation in the degree of hydrolysis (DH) obtained, which ranged between 55.5% and 64.1% when Flavourzyme was added on a weight basis, at the same enzyme to substrate ratio (E:S), in the CN solution during hydrolysis [52]. These modifications in peptide release may ultimately affect the bioactivity of milk protein hydrolysates.
To date, most enzyme activities within proteolytic preparations have been identified using synthetic substrates. However, liquid chromatography tandem mass spectrometry (LC-MS/MS) may be employed to characterize the cleavage specificity of enzyme preparations. Peptide sequences released from milk proteins can be used to identify cleavage sites on milk protein sequences [36, 41, 53, 54]. This is particularly relevant when individual milk proteins are digested with enzyme preparations. For example, the C-terminal amino acids of peptides released during enzymatic hydrolysis of β-lactoglobulin (β-Lg) with a pancreatic elastase preparation revealed the presence of enzyme (i.e., Glu-specific exopeptidase or glutamyl endopeptidase-, trypsin-, and chymotrypsin-like) activities other than pancreatic elastase [36].
In silico methods to assist in the selection of milk protein substrates for the generation of bioactive peptides
Several in silico strategies have been described in the literature [for reviews, see: 15, 19] for a wide range of food proteins. The in silico methods that have been employed for the targeted release of milk protein-derived bioactive peptides are summarized in Fig. 1. Utilization of these approaches requires knowledge of individual milk protein sequences. The sequences of the major milk proteins from different mammals (e.g., human, cow, donkey, pig, sheep, goat, camel, etc.) have been well characterized. These are publicly available through online repositories such as the National Center for Biotechnology Information (NCBI, [55]) and UniProt [56].

Summary of in silico strategies employed to study the release of bioactive peptides from milk proteins. ANN: artificial neural network; DOE: design of experiments; RSM: response surface methodology; QSAR: quantitative structure activity relationship
In silico strategies have been used to investigate the occurrence of bioactive peptides within individual milk proteins. This has consisted of identifying known bioactive peptide sequences or peptide sequences with known features within milk proteins. A number of databases of bioactive peptides, comprising milk protein-derived sequences, exist, e.g., BIOPEP [57, 58] and the Milk Bioactive Peptide Database (MBPDB) [59, 60]. In silico searches for bioactive peptides having antimicrobial, antioxidant, immunomodulatory, angiotensin converting enzyme (ACE) inhibitory, and dipeptidyl peptidase IV (DPP-IV) inhibitory activities within milk proteins have been reported in the literature [20, 23, 61,62,63]. The frequency of occurrence of bioactive peptide sequences within individual milk protein molecules (A) may be determined using Equation 1 [64];
with a, the number of bioactive peptide sequences found within the individual protein, and N, the number of amino acid residues within the protein.
Other means to estimate the potential of individual milk proteins to act as sources of bioactive peptides have been proposed, which take into account the bioactive potency of the peptides within individual proteins [for review, see: 29]. The potential biological activity of a protein, B, may be calculated using Equation 2 [64],
with ai, number of repetitions of peptide i within the protein sequence; EC50,i, the half maximal effective concentration of peptide i, and N, the number of amino acid residues within the protein.
Other indexes have also been described in the literature. For instance, a potency score incorporating the half maximal inhibitory potency of peptides (IC50) and the molecular weight (MW) of the protein was described [61]. A potency index (PI) combining the IC50, MW of the protein, and the fact that each amino acid from the protein can only contribute to a single peptide was also proposed [63]. The outcomes of the in silico analysis may vary depending on the model used. For example, it was predicted, using the A value approach, that bovine β-CN would contain the highest abundance of DPP-IV inhibitory peptides [14, 23]. In contrast, calculation of the PI for bovine milk proteins indicated that κ-CN was the milk protein containing the most potent DPP-IV inhibitory peptide sequences [63].
Prediction of peptide release using peptide cutter programmes
In silico analysis of individual protein sequences for bioactive peptides do not take into account the fact that these peptides may or may not be released during enzymatic digestion. Therefore, methods consisting of predicting the release of bioactive peptides during the digestion of individual milk proteins have been developed. Peptide cutters have been used to predict the release of bioactive peptides during in silico digestion of individual milk proteins [13, 37, 65,66,67,68]. Peptide cutters represent software used to perform in silico digestion of proteins based on the cleavage specificity of selected enzyme activities [14]. Several peptide cutter programs are freely accessible online [for reviews, see: 15, 19]. Peptide cutters commonly employed for in silico digestion of milk proteins can be found on the ExPASy [69] and BIOPEP [58] (enzyme action tool) portals. A frequency of occurrence of bioactive peptide release (AE) has been developed by Minkiewicz et al. [24] (Equation 3),
with AE, the frequency of occurrence of bioactive peptides predicted to be released in silico following digestion of a protein by a specific enzyme activity; d, the number of bioactive peptide sequences released following in silico digestion of a protein, and N, the number of amino acid residues within the protein.
Peptide cutters are useful in predicting the structure of peptides that may be released during enzymatic digestion of milk proteins. However, several limitations exist. Most peptide cutters appear to only take into account the main cleavage sites of enzymes. Nevertheless, most enzyme activities are able to cleave additional peptide bonds. For example, pepsin has been reported to specifically cleave at the C-terminus of Phe, Tyr, Trp, and Leu residues [69]; however, cleavages may also be observed post Ala, Asp, and Glu residues [70]. Even though these sites are not preferentially cleaved, this may affect bioactive peptide release. In addition, peptide cutter cleavage rules are based on incorrect assumptions regarding protein structure. Owing to disulphide bonds, several food proteins, including milk proteins, are present as globular structures in their native form and they can form aggregates during processing, notably following pH and temperature modifications [for review, see: 29]. As a consequence, protein aggregation can affect peptide release during enzymatic digestion [71]. The other assumption of peptide cutters is that all potentially scissible peptide bonds have the same probability to be cleaved during in silico digestion, leading to complete digestion of the protein. However, it is well known that peptide bond selectivity may be affected by various parameters such as pH, total solid concentration, temperature, etc. [43, 53, 54]. In addition, it has been demonstrated that the kinetics of peptide bond cleavage during bovine WP hydrolysis with a purified Bacillus licheniformis protease differed depending on the cleavage site [72]. Of the peptide bonds expected to be cleaved (i.e., at the C-terminal side of Glu and Asp residues), some bonds were hydrolyzed following first order kinetics, whereas others followed a demasking kinetic model (i.e., they could only be hydrolyzed following prior hydrolysis of other peptide bonds) and, furthermore, some peptide bonds were not hydrolyzed (i.e., missed cleavage).
Another limitation of in silico digestion of food proteins lies in the fact that peptide cutter software assumes a binary system consisting of a single protein substrate and single enzyme activity, whereas during in vitro digestion of food proteins, the protein substrate is never pure and therefore contains a mixture of proteins together with other components (e.g., carbohydrates, lipids, salts, etc.). The same situation is found for enzyme preparations, which generally contain several enzyme activities (Table 1). It is possible with certain peptide cutters to conduct protein hydrolysis using sequential digestion with several enzyme activities. Nevertheless, peptide cutters are not able to predict the release of peptides when several enzyme activities and proteins are concomitantly present. Finally, the range of availability of enzyme activities, which are relevant for food applications, are limited in peptide cutters. Peptide cutters only account for pepsin, trypsin, chymotrypsin, pancreatic elastase, thermolysin, glutamyl endopeptidase, and prolyl endopeptidase activities. However, commercial food-grade enzyme preparations contain a wide range of other activities (Table 1) that are not accessible through conventional peptide cutters. Owing to all these limitations, the predicted in silico digestion of proteins has been difficult to replicate in vitro.
The wider application of MS and the increased accuracy of MS analyzers have allowed identification of a wide range of bioactive peptides within food protein hydrolysates [73, 74]. Peptide identification within milk protein hydrolysates generated following targeted enzymatic release has been mainly achieved using LC-MS/MS analysis [75]. Milk protein hydrolysates are composed of complex mixtures of peptides as they can typically contain hundreds to thousands of different constituents. The precise identification of peptides within milk protein hydrolysates can therefore be a challenging and time-consuming task. The information generated during in silico prediction may be incorporated in a so-called “include list”, which comprises the m/z of the predicted peptide sequences. The targeted release of bioactive peptides may therefore be used as a means to enhance peptide identification within complex milk protein hydrolysates. However, in studies conducted with milk proteins, large differences between peptides predicted to be released and actual peptide release, as determined by LC-MS/MS, have been reported (Table 2). For example, during hydrolysis of bovine β-CN [76] and α-CN [38] with glutamyl endopeptidase, it was observed that 70% and 57%, respectively, of the peptide bonds predicted to be cleaved (C-terminal side of Glu and Asp residues) were actually hydrolyzed. Similarly, there was a relatively low match, i.e., 30% and 50%, between the in silico prediction and actual peptide release following hydrolysis of bovine β-CN [77] and α-CN [78], respectively, with Brewer’s Clarex (DSM, Heerlen, The Netherlands), an Aspergillus niger-derived prolyl endoproteinase. In another in silico study conducted on the generation of bioactive peptides during bovine α-lactalbumin (α-La) hydrolysis with pancreatic elastase, 60% of the peptides predicted to be released were detected by LC-MS/MS [75]. In the case of the digestion of bovine β-Lg with pancreatic elastase, only 2.5% of the peptides predicted to be released were actually identified by LC-MS/MS [36]. This was attributed to the globular structure of bovine β-Lg and to the reduced accessibility of certain peptide bonds within the protein. However, other studies have reported the release of target peptides from milk proteins based on peptide cutter predictions. This was the case for Ile-Lys-His-Gln-Gly-Leu-Pro-Gln-Glu [caseicin A, αs1-CN (f6-17)], an antimicrobial peptide from bovine milk, for which precursor peptides were predicted to be released by the action of thermolysin [80]. Caseicin A could be identified by LC-MS/MS in a sodium caseinate hydrolysate generated with Thermoase PC10F (Amano Enzymes, Nagoya, Japan), a commercial enzyme preparation containing thermolysin, at an E:S of 0.5% (w/v) when incubated at 37 °C for 6 h.
Most studies describing the link between in silico and in vitro hydrolysis have been conducted with intact milk proteins as starting substrates. There are a limited number of studies that have also analyzed the predicted and actual cleavage of milk protein-derived peptides as starting substrates. Such studies are interesting as milk protein hydrolysates are made of peptides that are prone to further degradation during the hydrolytic reaction. Five synthetic peptides, which were predicted to be released in silico following digestion of bovine β-CN by glutamyl endopeptidase (Fig. 2A), were incubated with this enzyme [76]. It was shown that certain peptide bonds that were predicted to be cleaved within the selected β-CN-derived peptides remained intact following incubation with glutamyl endopeptidase. This was the case for the Glu-Glu bond of β-CN (f16-26) and the Glu-Met bond of β-CN (f105-119), which were not observed to be cleaved. Interestingly, the cleavage of β-CN (f16-26) was not affected by phosphorylation of the three Ser residues present within this peptide. The peptide bond Glu(117)-Pro(118) was hydrolyzed in β-CN (f115-125) (Fig. 2A), in contrast with β-CN, where no cleavage was observed for this bond, suggesting that accessibility of peptide bonds depended on peptide sequence up- and downstream. A similar study was conducted with bovine CN-derived peptides incubated with DPP-IV (Fig. 2B) at two different E:S, i.e., 1 and 10 U: 1 g peptide [83]. As expected, peptide bonds were cleaved to a lower extent at the lowest E:S. All peptide bonds predicted to be cleaved by DPP-IV were actually hydrolyzed in vitro when the highest E:S was used. In peptides possessing two possible cleavage sites (i.e., Leu-Pro-Tyr-Pro-Tyr and Leu-Pro-Leu-Pro-Leu), peptide bonds located closer to the N-terminus of the peptide were cleaved to a greater extent. This was due to the fact that DPP-IV is an aminopeptidase and, therefore, cleavage of the second peptide bond will be required to allow further digestion of the product released (i.e., Tyr-Pro-Tyr and Leu-Pro-Leu, respectively). The requirement for this sequence of events was evidenced by the fact that at the lowest E:S the cleavable peptide bond located furthest from the N-terminus of the peptide did not appear to be hydrolyzed. This study also demonstrated the importance of enzyme dosage to hydrolysis of peptide bonds. Therefore, adequate enzyme dosages need to be determined in order to increase the agreement in the match between predicted in silico and actual in vitro release of peptides.

Hydrolysis of peptides corresponding to specific sequences from (A) bovine β-casein (CN) by glutamyl endopeptidase (incubation at 37 °C for 2 h [76]), and (B) specific synthetic CN-derived peptides by dipeptidyl peptidase IV (DPP-IV, incubation at 37 °C for 18 h [83]). Full arrow peptide bond predicted to be cleaved and observed to be cleaved; broken arrow: peptide bond predicted to be cleaved but not observed to be cleaved
Several studies have revealed nonspecific (or aspecific) cleavage of milk proteins during enzymatic hydrolysis [36, 38, 76, 78, 84]. For example, during bovine β-CN hydrolysis with glutamyl endopeptidase, cleavage occurred at the C-terminal side of Thr(128) and Leu(192) residues [76]. In this study, the role of temperature on peptide bond cleavage was also evaluated. Differences in peptide bond cleavage were observed when hydrolysis was conducted at 37 °C and 50 °C. Cleavage of the Glu(20)-Glu(21) peptide bond was observed at 37 °C and not at 50 °C, whereas for the Glu(36)-Glu(37) bond, cleavage occurred at 50 °C but not at 37 °C. Similarly, cleavage of Asp(43)-Glu(44) and Asp(129)-Val(130) only occurred at 37 °C and 50 °C, respectively. Similarly, aspecific cleavages of bovine α-CN following incubation with glutamyl endopeptidase were reported, which differed at 37 °C and 50 °C [38]. Nonspecific cleavage of bovine β-CN with Brewer’s Clarex was also reported. For instance, two peptides which were not predicted to be released, Lys-Val-Leu-Pro [β-CN (f169-172)] and Arg-Asp-Met-Pro [β-CN (f183-186)], were detected in the hydrolysate [77]. These peptides, however, were not very potent inhibitors of ACE as both displayed IC50 values >1000 μM. Atypical cleavage at the C-terminal side of Glu and Lys residues was also shown during the digestion of bovine β-Lg with pancreatic elastase, which has been reported to cleave at the C-terminal side of Ala, Ile, Leu, Val, Ser, Tyr, Thr [36]. This atypical cleavage was attributed to possible side activities (i.e., trypsin- and glutamyl endo/exopeptidase-like) within the elastase preparation used in the study. Similarly, the formation of aspecific peptides has been demonstrated during hydrolysis of bovine WPs with a purified B. licheniformis protease [84]. It was demonstrated using synthetic peptides, matching the sequences of bovine β-Lg (f56-62), (f75-85), (f135-157), and (f138-157), which were incubated at 40 °C, pH 8.0 up to 3 or 18 h in the absence of the purified B. licheniformis protease, that some of these aspecific peptides were formed as a result of a spontaneous hydrolysis. The extent of cleavage of these aspecific peptides was increased in the presence of purified B. licheniformis protease. This suggested that some “unstable” peptide bonds were prone to enzyme-mediated hydrolysis, even though they should not have been cleaved based on the enzyme specificity.
Virtual screening of peptides using molecular docking
In silico tools have also been used to carry out virtual screening of peptides. Virtual screening of peptides derived from milk proteins has typically been carried out with molecular docking approaches. Molecular docking is suitable for the analysis of bioactive properties, for example involving peptide binding to receptors or active sites of enzymes [85]. For this reason, molecular docking has mostly been employed to predict the potential of milk protein-derived peptides to bind to different metabolic enzymes such as ACE, DPP-IV, and xanthine oxidase (XO). To date, molecular docking has been applied to large sets of peptides regardless of the peptide origin [86, 87]. However, a limited number of studies are available where molecular docking has solely focused on milk protein-derived peptides for their opioid agonistic as well as their ACE, DPP-IV, and XO inhibitory potential [88,89,90,91]. Molecular docking has been used to determine specific interactions (i.e., hydrogen bonding, electrostatic and hydrophobic) involved in the binding at the active site of specific bioactive milk protein-derived peptides having opioid agonistic [91], ACE [86, 89], xanthine oxidase [88], and DPP-IV [88, 90] inhibitory activities. Molecular docking allows the study of large numbers of peptides. Nevertheless, molecular docking needs to be applied with caution. One of the assumptions of molecular docking of enzyme inhibitory/receptor binding peptides implies peptide binding at the active site of enzymes/receptors. However, several peptides are able to bind to enzymes and receptors outside the active site. Such peptides, for example, can act through a noncompetitive mode of inhibition of enzymes. It has been shown that the bioactivity of noncompetitive inhibitory milk protein-derived peptides of DPP-IV and XO could not be predicted using molecular docking approaches [88, 90]. Furthermore, limitations of molecular docking also exist for competitive peptides. This is particularly the case for peptides that act through substrate-type inhibition mechanisms, i.e., peptides that can be degraded by the enzyme [83]. The possibilities for peptide instability are generally not taken into account during molecular docking, which represents further limitations for its widespread use as a predictive tool.
QSAR modeling and ANN approaches applied to the study of milk bioactive peptides
QSAR models have been developed for a wide range of bioactive properties such as antioxidant, antimicrobial, ACE, renin, and DPP-IV inhibition [92]. Similar to molecular docking, most of the QSAR models have been employed for large peptide datasets (regardless of peptide origin), or these models have targeted peptides originating from protein substrates other than milk [93,94,95,96]. The application of QSAR to milk peptides has been described in a number of publications. More specifically, QSAR models were developed to predict the ACE [97] or DPP-IV [98] inhibitory activity of milk peptides. A QSAR model was developed with 36 milk protein-derived peptides by Pripp et al. [97]. The role of the last two C-terminal residues in peptides ≤6 amino acids on ACE inhibition was demonstrated. The hydrophobicity of the C-terminal amino acid (C1) as well as the positive charge and molecular volume of the next to the C-terminal amino acid (C2) were positively correlated with high ACE inhibition. In this study, however, no obvious correlation was found between amino acids present at the N terminal side of peptides and their ACE inhibitory potency.
The combination of several in silico strategies has been employed for the identification of milk peptides with potential to act as ACE inhibitory peptides. Indexes determined with in silico models (e.g., AE) solely rely on the availability of data of the peptide’s potency (i.e., IC50 values and EC50 values), which may be accessed from the literature or in-house by the experimenter. Because peptide potencies are not always available for particular sequences, Pripp [13] proposed prediction of the potency of peptides using QSAR models. This strategy was applied to bovine milk proteins to predict ACE IC50 values of individual peptides. Peptides from the major bovine milk proteins (i.e., αs1-, αs2- β-, κ-CN, β-Lg, and α-La) were independently digested 20 times in silico in order to release peptides possessing one of each of the 20 amino acid residues at the C-terminal side. The peptides released were then subjected to sequential in silico digestion with pepsin, trypsin, and chymotrypsin. Predicted IC50 values for (a) all peptides or (b) di- and tripeptides released in silico in the peptide mixture were then calculated. Di- and tripeptides were selected because they are more likely to be bioavailable than larger peptide sequences. For each of the six individual major milk proteins studied, the predicted ACE IC50 value of the peptide mixture was based on the QSAR predicted IC50 of each peptide [13]. This analysis allowed the finding that hydrolysis at the C-terminal side of Ile and Pro residues prior to gastrointestinal digestion should allow production of particularly potent ACE inhibitory samples.
3D-QSAR models with high correlation coefficients (R2 > 0.9) have been specifically developed for milk protein derived di- and tripeptides with ACE inhibitory properties [89]. A combination of several in silico tools comprising 3-D QSAR, molecular docking, and in silico digestion of milk proteins by peptide cutter, was described. This study predicted that selected dipeptides originating from milk proteins would be good candidates for future human studies due to their high ACE inhibitory potency. One of these peptides, Lys-Pro, was predicted to have a relatively low ACE IC50 of 11 μM.
The utilization of QSAR combined with knowledge of the cleavage specificity of enzymes has been described to generate a particularly potent ACE inhibitory bovine β-CN hydrolysate. Peptides containing C-terminal Pro residues have been associated with high levels of ACE inhibition. Therefore, Pripp [13] proposed that hydrolysis of milk proteins with a prolyl endoproteinase activity would enhance the generation of ACE inhibitory peptides. Thus, the release of peptides with a C-terminal Pro during the generation of food protein hydrolysates may lead to samples with high ACE inhibitory properties. Based on this assumption, bovine β-CN, a substrate rich in Pro residues, was hydrolyzed with Brewer’s Clarex, an A. niger prolyl endoprotease preparation [77]. A hydrolysate displaying a particularly low ACE IC50 value of 16.4 μg mL-1 was obtained after 24 h incubation at 55 °C, pH 6.0. Of the peptides identified within the hydrolysate and tested for their ACE inhibitory properties, the most potent was Leu-Pro-Pro, which had previously been reported to have an IC50 value of 9.6 μM [99]. The most potent novel ACE inhibitory peptide identified within the β-CN hydrolysate was Phe-Leu-Gln-Pro, having an IC50 value of 68.7 μM [77].
Similar to QSAR, a software such as PeptideRanker [100], which is based on an ANN approach, has also been employed as a selection tool to predict the bioactivity of milk protein-derived peptides [101, 102]. PeptideRanker predicts the probability of a peptide to be bioactive based on its structure. A few studies have employed this software to predict the bioactive properties of milk protein-derived peptides. However, only a limited number of studies have conducted confirmatory studies with synthetic peptides. For example, the identification of antioxidant and ACE inhibitory peptides within donkey milk was conducted using PeptideRanker and synthetic peptides [101].
Optimization of bioactive peptide release from milk proteins using DOE and response surface methodology (RSM)
Due to the compositional complexity of milk proteins and the enzyme preparations used for their hydrolysis, limitations exist to accurately predict peptide release during enzymatic hydrolysis. Therefore, other predictive tools that do not require detailed knowledge of the substrate composition and enzyme activities present in the enzyme preparations may be employed [16]. DOEs followed by RSM allow characterization of hydrolysates at a macromolecular level as they take into account the whole hydrolysate for, e.g., the optimization of hydrolysate generation. Optimization through DOE and RSM are classified as in silico methods as they utilize specific software to carry out the modeling of experimental data and the prediction of optimum hydrolysis parameters.
Components that are generally incorporated into the DOE include hydrolysis parameters such as protein substrate, enzyme preparation, concentration of reactants, pH, ionic strength, temperature, time, etc. Selected parameters that may impact on the release of bioactive peptides are studied and their limits are determined generally as a function of the optimal operating conditions for the enzyme preparation (pH, temperature, E:S, cofactors, etc.) and industrially relevant conditions (i.e., reagents, substrate concentration, etc.). When the parameters are selected and the boundaries of the experimental domain (low and high limits for each parameter) are chosen, a DOE may be employed to study concomitant variation of all parameters (Fig. 3). A mathematical model consisting of a multilinear regression (MLR) is usually generated to link the experimentally determined biological activity of hydrolysates to hydrolysis conditions. Optimum hydrolysis parameters are subsequently determined mathematically by minimising or maximising the MLR model. A confirmatory step consists in the generation of the hydrolysate(s) using the optimum parameters and assessment of the biological activity of the resultant sample(s) in order to validate the MLR model (Fig. 3).

General workflow for the elucidation of optimum hydrolysis parameters during the generation of bioactive milk protein hydrolysates using design of experiments (DOE) and response surface methodology (RSM) approaches (adapted from van der Ven et al. [18], Contreras et al. [17], and Nongonierma et al. [103])
One of the first DOE to be used during the optimum generation of food protein hydrolysates was applied to the release of ACE inhibitory peptides following bovine WP hydrolysis with a porcine gastrointestinal preparation, Corolase PP (AB Enzymes, Darmstadt, Germany) [18]. Five parameters, i.e., pretreatment temperature of the protein suspension, E:S, pH, temperature, and hydrolysis time were included in the DOE. The optimum conditions for the generation of a potent ACE inhibitory hydrolysate were determined to be 45 °C (pretreatment temperature), pH 8.0, E:S 2.19% (w/w) for durations between 1.0 to 5.5 h. These conditions yielded the generation of relatively potent WP hydrolysates with ACE IC50 values ranging from 0.148 (5 h) to 0.202 mg mL-1 (1 h).
DOE approaches have since been applied for the optimization of milk protein hydrolysates with a wide range of bioactive properties. These include antioxidant [17, 103,104,105], reduced antigenicity [106, 107], ACE inhibitory [18, 104, 108,109,110,111,112,113,114], and DPP-IV inhibitory [82, 103, 115, 116] properties. Generally, a good correlation has been found between the predicted and experimentally determined bioactive properties of milk protein hydrolysates. This was illustrated for the DPP-IV inhibitory potency of tryptic bovine milk protein isolate (MPI) hydrolysates [116] and camel milk protein hydrolysates [82], where R2 values of 0.93 and 0.98, respectively, were found between predicted and experimental DPP-IV IC50 values (Fig. 4).

Correlation between the experimental and predicted dipeptidyl peptidase IV (DPP-IV) half maximal inhibitory concentration (IC50) value from tryptic (A) bovine [116] and (B) camel milk protein hydrolysates [82]. Values were obtained using the same design of experiments (DOE). H16 corresponds to the predicted optimized sample
In most studies, the optimization of bioactive peptide release yielded the generation of relatively potent bioactive milk protein hydrolysates. In addition, the predicted potency of the optimum hydrolysate (H16 in Fig. 4A) was generally of the same order as the experimentally determined value. In other cases, the MLR model slightly underestimated the IC50 value of the optimum hydrolysate (Fig. 4B). This has been explained by limiting parameters (substrate availability, enzyme autolysis and retro-inhibition by peptides, etc.), which may have pertained during the enzymatic hydrolysis reactions [116].
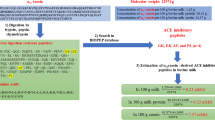
Optimization of the yield of specific peptides has been conducted during milk protein hydrolysis using DOE and RSM approaches. The optimum release of His-Leu-Pro-Leu-Pro (β-CN (f134-138), an ACE inhibitory peptide, was studied using a DOE [114]. Bovine sodium caseinate was hydrolyzed, using three parameters, concentration of Corolase PP, concentration of Peptidase 433P (Biocatalysts, Cardiff, UK), and hydrolysis duration. The optimum release of His-Leu-Pro-Leu-Pro was obtained after 24 h hydrolysis with a 6% (w/w) E:S of Corolase PP and without Peptidase 433P.
Peptide identification following targeted release of bioactive peptides
During LC-MS/MS identification of peptides within milk protein hydrolysates, sequences originating from major milk proteins are generally taken into account. This is done on the basis that peptides must be present in sufficiently high concentrations to display a bioactive effect. However, minor milk proteins may contain relatively potent peptides, which are likely to play an important role in the overall bioactive properties of milk protein hydrolysates. In addition, various genetic variants or post-translational modifications (PTM) induced as a result of milk processing may be found within the starting substrates. It was demonstrated using LC-MS/MS that β-casomorphin 7, an opioid peptide, was released in larger amounts from β-CN variant A1 than from β-CN variants A2 and I following simulated gastrointestinal digestion [117]. Variations in the protein sequence need to be sufficiently characterized to determine the composition of starting milk substrates. Various LC-MS/MS-based approaches that have been used to determine PTM and genetic polymorphism have been reviewed [118, 119]. Most approaches are based on a protein fractionation (sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), reverse-phase high performance liquid chromatography (RP-HPLC), ion exchange chromatography (IEC), or immobilized metal ion affinity chromatography (IMAC)) followed by specific digestion with an enzyme presenting a known cleavage specificity (e.g., L-1-tosylamide-2-phenylethyl chloromethyl ketone (TPCK)-treated trypsin) and peptide mapping, which is used to assign sequences to known proteins. For example, shot-gun proteomic approaches combining SDS-PAGE and LC-MS/MS may be employed to determine the protein composition of the milk protein substrate [118, 119]. The exact composition of the protein substrate may be employed to build an “in-house” database that can be used to confirm peptide identification within specific hydrolysate samples. In addition, there is a requirement to take into account the secondary and tertiary structure of proteins in LC-MS/MS peptide identification strategies. To date, most protein databases rely solely on the primary structure of food proteins. Peptides identified by LC-MS/MS are generally mapped to the primary structure of proteins to validate their belonging to individual milk proteins. Therefore, peptides released from regions of the protein where disulphide bridges occur are less likely to be identified as these particular sequences do not show in the primary structure of the protein. In addition, the resultant peptide fragmentation may be difficult to interpret due to the high noise/signal ratio and the presence of multicharged ions [119]. It is therefore likely that some peptides originating from cross-linked regions of milk proteins, which may be bioactive, are not taken into account for further studies. There is a need to build protein databases or software that can assign peptides to protein regions where cross-linkages occur.
Food protein hydrolysates generally contain very large numbers of peptide sequences and free amino acids. For this reason, enrichment and/or fractionation protocols are often applied to milk protein hydrolysates [for reviews, see: 3, 30, 120]. While the compositional complexity is generally decreased following fractionation, other challenges exist. It is common to obtain several bioactive fractions, due to the fact that active peptides may partition between different fractions. Therefore, instead of simplifying the system, researchers are faced with a larger number of samples for analysis in an effort to try to understand the origin of the bioactivity. Alternatively, it is possible to re-fractionate the hydrolysates or selected fractions using other fractionation protocols. In addition, overall peptide recovery during fractionation is never 100%, implying that losses/degradation of peptides may occur during the fractionation steps. Furthermore, losses of activity may occur during fractionation due to possible synergistic peptide effects in the hydrolysates [121]. As a consequence, part of the information on hydrolysate composition may be lost during fractionation.
Targeted hydrolysis strategies may have some benefits over conventional approaches. For instance, the predictive nature of several in silico tools (i.e., peptide cutters, molecular docking, and QSAR modeling) may allow assessment of the exact structure of those peptide sequences that are predicted to be released and/or bioactive. Therefore, a first step for peptide identification is to carry out a mining exercise whereby predicted peptide sequences are specifically searched for within milk protein hydrolysates [38, 75,76,77,78, 82, 84].
Preliminary knowledge of peptide sequence may help to improve analysis of hydrolysates during MS detection. Multiple reaction monitoring (MRM) may be applied to improve fragmentation of selected peptides and increase the likelihood to detect these peptides if they are present within milk protein hydrolysates. The identification of di- and tripeptides containing branched-chain amino acid (BCAA) within WP hydrolysates was carried out using MRM [122, 123]. In addition, knowledge of the target sequences may be used to specifically determine parameters such as optimum collision energy and transfer time to improve their detection during LC-MS/MS [9, 122, 123]. Synthetic peptides may also be employed to spike samples and confirm their presence based on their specific retention times during LC separation and their fragmentation spectra following MS [122].
In silico predictive tools have been described for specific analysis of short peptides (2–4 amino acids) within milk protein hydrolysates. It has been suggested that short peptides have a potential to be bioavailable. The identification of short peptides within complex mixtures is challenging due to their lack of structural diversity (i.e., isobaric peptides and low amount of information in their fragmentation spectra) and the fact that they can over- or underfragment during MS analysis [122, 124]. However, the use of multi-stage methodologies, combining information such as accurate mass, peptide occurrence within target proteins, retention time, and fragmentation spectra can help in the identification of short peptide sequences within food protein hydrolysates [for review, see: 29]. Short peptides may be separated using hydrophilic interaction liquid chromatography (HILIC) columns, which may be more suitable for their chromatographic separation. Front end separation with HILIC columns has been employed to develop retention time prediction models [125, 126]. In order to improve short peptide separation and gain more confidence in their subsequent identification, analysis with different LC matrices has been used prior to MS detection [126]. For instance, LC separation with two different columns, e.g., HILIC and RP [127], has allowed identification of short peptide sequences within fractions of WP hydrolysates. Interestingly, some of these peptides (i.e., Ile-Pro-Ile, Ile-Pro-Ala, Val-Leu, and Trp-Leu), were shown to possess particularly potent DPP-IV inhibitory properties (IC50 < 100 μM) in vitro.
The information obtained during QSAR outcomes also plays an important role in assisting the identification of novel bioactive peptides. QSAR models can be used to predict the potency of peptides. This strategy has been applied to assist in the identification of novel ACE inhibitory peptides within water soluble extracts (WSE) from Idiazabal cheese [128]. A total of 273 peptides were identified in the WSE from the cheese. Their ACE inhibitory activity was predicted with a QSAR model. The predicted ACE IC50 values of selected peptides were then compared with the experimental values determined in vitro. This study allowed peptide identification without further fractionation of the cheese WSE. Novel milk protein-derived peptides (10) with ACE inhibitory properties were identified within the WSE of Idiazabal cheese. Within these peptides, Gln-Asn-Ala-Leu-Ile-Val-Arg-Tyr-Thr-Arg [bovine serum albumin (BSA) f(403-412)] was the most potent ACE inhibitory peptide with an IC50 value of 4.6 μM [128]. Taking a similar approach, peptides identified within tryptic camel milk protein hydrolysates were analyzed based on the features of known DPP-IV inhibitory peptides and their QSAR predicted DPP-IV IC50 value. This allowed the discovery of nine novel DPP-IV inhibitory peptides derived from camel milk proteins. Two of these peptides, Leu-Pro-Val-Pro [β-CN (f172-175)] and Met-Pro-Val-Gln-Ala [β-CN (f186-190)], displayed a particularly low DPP-IV IC50 values (<100 μM) and were competitive inhibitors of DPP-IV [129]. Other examples of the application of QSAR to large milk protein-derived peptide datasets are found in the literature. The DPP-IV inhibitory potential (IC50 value) has been predicted by QSAR for bovine milk protein-derived peptides identified in the gastrointestinal tract of humans following the consumption of milk or other dairy products. Ten peptides, which were previously shown to be bioaccessible in humans following milk ingestion [12], were identified as novel DPP-IV inhibitory peptides [98]. Within these novel sequences, the most potent peptide was Leu-Pro-Val-Pro-Gln [β-CN (f171-175)], which had a DPP-IV IC50 value of 43.8 μM.
The utilization of QSAR to predict the biological activity of peptides, however, also has a few limitations. These include inaccurate prediction of peptide potency. It is not unusual to find differences between QSAR-predicted and experimental IC50 values of peptides [98, 128]. These differences arise from the robustness of the QSAR model. Furthermore, the data incorporated in QSAR models may not be obtained under the same experimental conditions, i.e., arise from different laboratories. This may present some issues since the assay conditions employed have been shown to have a significant impact on the potency values obtained for bioactive peptides [130,131,132]. In addition, the mode of action of peptides involved in the inhibition of metabolic enzymes (i.e., competitive versus non-competitive, mixed type, etc.) should be taken into account when building QSAR models as relationships between biological activity and structure should only be built for peptides acting through the same mechanism of action [98].
To date, peptide identification has generally been conducted with database-driven approaches where sequences are matched to the primary sequence of milk proteins. However, peptide mapping to the primary sequence of milk proteins may present difficulties as several milk proteins are folded in their native state as outlined earlier. This may lead to unsuccessful assignment of peptide sequences to milk proteins and therefore non-identification of such sequences. Other approaches such as de novo analysis have been proposed during peptide identification to overcome this issue [30, 120]. De novo analysis is based on sequencing algorithms and it does not rely on knowledge of the parent protein sequence [133]. The utilization of de novo sequencing may be employed to complement classic database-driven peptide identification of milk peptides [30, 119]. De novo approaches also present some advantages when the predictive ability of the in silico approach employed is low. For this reason, several studies have also incorporated de novo sequencing of milk peptides to generate additional information on the peptides detected within milk protein hydrolysates [36, 115, 134].
Conclusions and perspectives
In silico studies conducted at a molecular level with milk protein-derived peptides, although very worthwhile, have several limitations. In fact several in silico methods are not able to accurately predict peptide release or bioactivity of peptides. This is mainly based on inappropriate assumptions being made during in silico analysis of peptides. These assumptions arise using models that (1) do not represent the manner in which proteins/peptides are actually cleaved during enzymatic digestion, or (2) incorrectly assume specific interactions between peptides and metabolic enzymes (e.g., that all biologically active enzyme inhibitory peptides bind to the active site). There is a need to develop models that better describe the behavior of proteins and peptides during enzymatic hydrolysis and that take into account mechanistic approaches in relation to their bioactivity. In this context, the development of peptide cutters, which take in account the multidimensional (i.e., globular) structure of food proteins, may help to achieve predictions that are more representative of actual peptide release. The role of PTM during enzymatic digestion has not been extensively studied and is poorly understood; therefore, more studies in this area are also needed to better predict peptide release during enzymatic digestion of dietary proteins. In addition, methods such as DOE and RSM have proven to be very useful to identify optimum hydrolysis conditions for the generation of bioactive milk protein hydrolysates. These methods do not necessitate prior knowledge of the protein composition or the range of activities within the enzyme preparation. Overall, they can inform the generation of milk protein hydrolysates and have been quite successful in allowing the production of potent bioactive milk protein hydrolysates. Because the optimization is actually based on outputs obtained from the actual biological activity of milk protein hydrolysates, these methods take into account the complexity of hydrolytic reactions. Therefore, future targeted release of bioactive peptides may be based initially on DOE and RSM approaches. Subsequently, other in silico tools may be informed by the data obtained during this process to identify more potent and bioavailable peptides that are more relevant to human health.
Although a large range of bioactive peptide sequences have been reported in the scientific literature, there is still significant scope for the discovery of more potent and bioavailable sequences. This may be supported to a great extent by the array of in silico tools described in this review. For instance, the application of QSAR, ANN, and molecular docking has potential to accelerate the discovery of novel bioactive peptides from milk proteins. They can allow rapid screening of the large amount of information generated during LC-MS/MS peptide identification. While to date, QSAR does not seem to generally provide a precise prediction of peptide potency, it has allowed the ranking of peptides based on their predicted potency. In this way, QSAR modeling has facilitated the identification of potent, novel, and bioaccessible bioactive peptides from milk proteins. The extent of the information currently available on bioavailable peptides from milk proteins is relatively low. However, new studies have been conducted in this area using information from a broad range of food proteins. e.g., fish and soy [for review, see: 29]. Because some of these sequences identified are relatively short (di- and tripeptides), they may also be found within the milk proteome. The development of targeted strategies to release bioavailable peptides during milk protein hydrolysis may therefore be the key for the scientific validation of health-promoting ingredients for humans.
Most studies conducted on the identification of bioactive peptides are qualitative in the sense that they report the presence of specific peptide sequences. Generally, the amount of different peptides within milk protein hydrolysates is not determined. Quantification of peptides within milk protein hydrolysates represents a major challenge due to the number of sequences that are generally present within hydrolysates (hundreds to thousands of sequences may be identified). However, when selected target peptide sequences are known, their quantification may be carried out in a more systematic manner [135]. Optimization of the yield of a target ACE inhibitory peptide from bovine β-CN has been successfully carried out during enzymatic hydrolysis [114]. The preparation of milk protein hydrolysates maximally enriched in bioactive peptides is key to minimizing the dose required while still maintaining a physiological effect in humans. This will ensure compliance with the current legislative framework, which requires, for example according to EFSA Regulations, that bioactive compounds/foods should be ingested in amounts consistent with those consumed as part of a balanced diet.
The use of enzyme combinations may in certain instances allow increased yield for specific bioactive peptides. This was the case during the generation of CPPs released from a commercial peptic CN hydrolysate (Hyvital CPPs, FrieslandCampina Domo, Amersfoort, The Netherlands), which was further hydrolyzed with trypsin and thermolysin [136]. Enzyme combinations have also been described to achieve debittering of milk protein hydrolysates [137, 138]. The utilization of sequential hydrolysis with different enzyme preparations may allow the release of unique bioactive peptides and generate low-bitterness hydrolysates from milk proteins. However, to date, the availability of in silico methods allowing the selection of specific enzyme combinations during the targeted generation of specific milk protein hydrolysis does not seem to exist.
Other areas of research can make the generation of bioactive peptide more relevant to humans. These would include the development of targeted release of bioactive peptides in an integrated fashion, leading to the generation of sequences having the ability to be (1) bioavailable, (2) multifunctional (i.e., target multiple markers involved in the aetiology of metabolic diseases), and (3) palatable (i.e., non-bitter).
References
Korhonen H, Pihlanto A (2006) Bioactive peptides: production and functionality. Int Dairy J 16:945–160
Anusha R, Bindhu OS. Bioactive peptides from milk. In: Gigli I, Ed. Milk Proteins - From Structure to Biological Properties and Health Aspects. Rijeka: InTech. DOI: 10.5772/62993. Available from: https://www.intechopen.com/books/milk-proteins-from-structure-to-biological-properties-and-health-aspects/bioactive-peptides-from-milk; 2016.
Nongonierma AB, O’Keeffe MB, RJ FG. Milk Protein Hydrolysates and Bioactive Peptides. Advanced Dairy Chemistry. New York: Springer; 2016. p. 417–82.
Nongonierma AB, FitzGerald RJ. Biofunctional properties of caseinophosphopeptides in the oral cavity. Caries Res. 2012;46:234–67.
Nongonierma AB, FitzGerald RJ. The scientific evidence for the role of milk protein-derived bioactive peptides in humans: A review. J Funct Foods. 2015;640:640–56.
Nongonierma AB, FitzGerald RJ. Bioactive properties of milk proteins in humans: A review. Peptides. 2015;73:20–34.
Boutrou R, Henry G, Sanchez-Rivera L. On the trail of milk bioactive peptides in human and animal intestinal tracts during digestion: A review. Dairy Sci Technol. 2015:1–15.
Egger L, Ménard O. Update on bioactive peptides after milk and cheese digestion. Curr Opin Food Sci. 2017;14:116–21.
Kaiser S, Martin M, Lunow D, Rudolph S, Mertten S, Möckel U, et al. Tryptophan-containing dipeptides are bioavailable and inhibit plasma human angiotensin-converting enzyme in vivo. Int Dairy J. 2016;52:107–14.
Foltz M, Meynen EE, Bianco V, van Platerink C, Koning TMMG, Kloek J. Angiotensin converting enzyme inhibitory peptides from a lactotripeptide-enriched milk beverage are absorbed intact into the circulation. J Nutr. 2007;137:953–8.
Caira S, Pinto G, Vitaglione P, Dal Piaz F, Ferranti P, Addeo F. Identification of casein peptides in plasma of subjects after a cheese-enriched diet. Food Res Int. 2016;84:108–12.
Boutrou R, Gaudichon C, Dupont D, Jardin J, Airinei G, Marsset-Baglieri A, et al. Sequential release of milk protein-derived bioactive peptides in the jejunum in healthy humans. Am J Clin Nutr. 2013;97:1314–23.
Pripp AH. Initial proteolysis of milk proteins and its effect on formation of ACE-inhibitory peptides during gastrointestinal proteolysis: a bioinformatic, in silico, approach. Eur Food Res Technol. 2005;221:712–6.
Udenigwe CC. Bioinformatics approaches, prospects and challenges of food bioactive peptide research. Trends Food Sci Technol. 2014;36:137–43.
Nongonierma AB, FitzGerald RJ. Strategies for the discovery, identification and validation of milk protein-derived bioactive peptides. Trends Food Sci Technol. 2016;50:26–43.
Li-Chan EC. Bioactive peptides and protein hydrolysates: Research trends and challenges for application as nutraceuticals and functional food ingredients. Curr Opin Food Sci. 2015;1:28–37.
Contreras MM. Hernández-Ledesma B, Amigo L, Martín-Álvarez PJ, Recio I. Production of antioxidant hydrolyzates from a whey protein concentrate with thermolysin: Optimization by response surface methodology. LWT Food Sci Technol. 2011;44:9–15.
van der Ven C, Gruppen H, de Bont DBA, Voragen AGJ. Optimization of the angiotensin converting enzyme inhibition by whey protein hydrolysates using response surface methodology. Int Dairy J. 2002;12:813–20.
Iwaniak A, Minkiewicz P, Darewicz M, Protasiewicz M, Mogut D. Chemometrics and cheminformatics in the analysis of biologically active peptides from food sources. J Funct Foods. 2015;16:334–51.
Udenigwe CC, Gong M, Wu S. In silico analysis of the large and small subunits of cereal RuBisCO as precursors of cryptic bioactive peptides. Process Biochem. 2013;48:1794–9.
Udenigwe CC. Towards rice bran protein utilization: In silico insight on the role of oryzacystatins in biologically-active peptide production. Food Chem. 2016;191:135–8.
Wang T-Y, Hsieh C-H, Hung C-C, Jao C-L, Lin P-Y, Hsieh Y-L, et al. A study to evaluate the potential of an in silico approach for predicting dipeptidyl peptidase-IV inhibitory activity in vitro of protein hydrolysates. Food Chem. 2017;234:431–8.
Lacroix IME, Li-Chan ECY. Evaluation of the potential of dietary proteins as precursors of dipeptidyl peptidase (DPP)-IV inhibitors by an in silico approach. J Funct Foods. 2012;4:403–22.
Minkiewicz P, Dziuba J, Michalska J. Bovine meat proteins as potential precursors of biologically active peptides - a computational study based on the BIOPEP database. Food Sci Technol Int. 2011;17:39–45.
Vercruysse L, Van Camp J, Smagghe G. ACE inhibitory peptides derived from enzymatic hydrolysates of animal muscle protein: a review. J Agric Food Chem. 2005;53:8106–15.
Gu Y, Majumder K, Wu J. QSAR-aided in silico approach in evaluation of food proteins as precursors of ACE inhibitory peptides. Food Res Int. 2011;44:2465–74.
Fu Y, Wu W, Zhu M, Xiao Z. In silico assessment of the potential of patatin as a precursor of bioactive peptides. J Food Biochem. 2016;40:366–70.
Rajendran SRCK, Mason B, Udenigwe CC. Peptidomics of peptic digest of selected potato tuber proteins: Post-translational modifications and limited cleavage specificity. J Agric Food Chem. 2016;64:2432–7.
Nongonierma AB, FitzGerald RJ. Strategies for the discovery and identification of food protein-derived biologically active peptides. Trends Food Sci Technol. 2017;69:289-305.
Capriotti AL, Cavaliere C, Piovesana S, Samperi R, Laganà A. Recent trends in the analysis of bioactive peptides in milk and dairy products. Anal Bioanal Chem. 2016;408:2677–85.
El-Salam MA, El-Shibiny S. Bioactive peptides of buffalo, camel, goat, sheep, mare, and yak milks and milk products. Food Rev Int. 2013;29:1–23.
Mati A, Senoussi-Ghezali C, Zennia SSA, Almi-Sebbane D, El-Hatmi H, Girardet J-M. Dromedary camel milk proteins, a source of peptides having biological activities–A review. Int Dairy J. 2017;73:25–37.
Vincenzetti S, Pucciarelli S, Polzonetti V, Polidori P. Role of proteins and of some bioactive peptides on the nutritional quality of donkey milk and their impact on human health. Beverages. 2017;3:34.
Agarwal S, Beausire RLW, Patel S, Patel H. Innovative uses of milk protein concentrates in product development. J Food Sci. 2015;80:A23–A9.
de la Fuente MA, Hemar Y, Tamehana M, Munro PA, Singh H. Process-induced changes in whey proteins during the manufacture of whey protein concentrates. Int Dairy J. 2002;12:361–9.
Le Maux S, Nongonierma AB, FitzGerald RJ. Peptide composition and dipeptidyl peptidase IV inhibitory properties of β-lactoglobulin hydrolysates having similar extents of hydrolysis while generated using different enzyme-to-substrate ratios. Food Res Int. 2017;99:84–90.
Nongonierma AB, FitzGerald RJ. Tryptophan-containing milk protein-derived dipeptides inhibit xanthine oxidase. Peptides. 2012;37:263–72.
Kalyankar P, Zhu Y, O’Keeffe M, O’Cuinn G, RJ FG. Substrate specificity of glutamyl endopeptidase (GE): Hydrolysis studies with a bovine α-casein preparation. Food Chem. 2013;136:501–12.
Lacroix IM, Li-Chan ECY. Inhibition of dipeptidyl peptidase (DPP)-IV and α-glucosidase activities by pepsin-treated whey proteins. J Agric Food Chem. 2013;61:7500–6.
Fan F, Tu M, Liu M, Shi P, Wang Y, Wu D, et al. Isolation and characterization of lactoferrin peptides with stimulatory effect on osteoblast proliferation. J Agric Food Chem. 2017;65:7179–85.
Cheison SC, Schmitt M, Leeb E, Letzel T, Kulozik U. Influence of temperature and degree of hydrolysis on the peptide composition of trypsin hydrolysates of β-lactoglobulin: Analysis by LC–ESI-TOF/MS. Food Chem. 2010;121:457–67.
Nongonierma AB, FitzGerald RJ. Enzymes exogenous to milk in dairy technology | Proteinases. In: Fuquay JW, editor. Encyclopedia of Dairy Sciences. 2nd ed. San Diego: Academic Press; 2011. p. 289–96.
Tavano OL. Protein hydrolysis using proteases: an important tool for food biotechnology. J Mol Catal B Enzym. 2013;90:1–11.
Stepaniak L. Dairy enzymology. Int J Dairy Technol. 2004;57:153–71.
Sanchón J, Fernández-Tomé S, Miralles B, Hernández-Ledesma B, Tomé D, Gaudichon C, et al. Protein degradation and peptide release from milk proteins in human jejunum. Comparison with in vitro gastrointestinal simulation. Food Chem. 2018;239:486–94.
Lemes AC, Sala L, Ores JDC, ARC B, Egea MB, Fernandes KF. A review of the latest advances in encrypted bioactive peptides from protein-rich waste. Int J Mol Sci. 2016;17:950.
Toldrá F, Reig M, Aristoy MC, Mora L. Generation of bioactive peptides during food processing. Food Chem. 2017.
Mullally MM, O'Callaghan DM, FitzGerald RJ, Donnelly W, Dalton JP. Proteolytic and peptidolytic activities in commercial pancreatic protease preparations and their relationship to some whey protein hydrolyzate characteristics. J Agric Food Chem. 1994;42:2973–81.
Spellman D, Kenny P, O'Cuinn G, FitzGerald RJ. Aggregation properties of whey protein hydrolysates generated with Bacillus licheniformis proteinase activities. J Agric Food Chem. 2005;53:1258–65.
Kilcawley KN, Wilkinson MG, Fox PF. Determination of key enzyme activities in commercial peptidase and lipase preparations from microbial or animal sources. Enzym Microb Technol. 2002;31:310–20.
Merz M, Eisele T, Berends P, Appel D, Rabe S, Blank I, et al. Flavourzyme, an enzyme preparation with industrial relevance: Automated nine-step purification and partial characterization of eight enzymes. J Agric Food Chem. 2015;63:5682–93.
Merz M, Appel D, Berends P, Rabe S, Blank I, Stressler T, et al. Batch-to-batch variation and storage stability of the commercial peptidase preparation Flavourzyme in respect of key enzyme activities and its influence on process reproducibility. Eur Food Res Technol. 2016;242:1005–12.
Butré CI, Sforza S, Gruppen H, Wierenga PA. Determination of the influence of substrate concentration on enzyme selectivity using whey protein isolate and Bacillus licheniformis protease. J Agric Food Chem. 2014;62:10230–9.
Butré CI, Sforza S, Wierenga PA, Gruppen H. Determination of the influence of the pH of hydrolysis on enzyme selectivity of Bacillus licheniformis protease towards whey protein isolate. Int Dairy J. 2015;44:44–53.
NCBI. https://www.ncbi.nlm.nih.gov/protein/. Accessed 19 Sept 2017.
UniProt. www.uniprot.org/. Accessed 19 Sept 2017.
Dziuba J, Minkiewicz P, Nałecz D, Iwaniak A. Database of biologically active peptide sequences. Food/Nahrung. 1999;43:190–5.
BIOPEP. http://www.uwm.edu.pl/biochemia/index.php/en/biopep. Accessed 18 Sept 2017.
MBPDB. http://mbpdb.nws.oregonstate.edu. Accessed 19 Sept 2017.
Nielsen SD, Beverly RL, Qu Y, Dallas DC. Milk bioactive peptide database: A comprehensive database of milk protein-derived bioactive peptides and novel visualization. Food Chem. 2017;232:673–82.
Vermeirssen V, van der Bent A, Van Camp J, van Amerongen A, Verstraete W. A quantitative in silico analysis calculates the angiotensin I converting enzyme (ACE) inhibitory activity in pea and whey protein digests. Biochimie. 2004;86:231–9.
Dziuba M, Dziuba B, Iwaniak A. Milk proteins as precursors of bioactive peptides. Acta Sci Pol Technol Aliment. 2009;8:71–90.
Nongonierma AB, FitzGerald RJ. An in silico model to predict the potential of dietary proteins as sources of dipeptidyl peptidase IV (DPP-IV) inhibitory peptides. Food Chem. 2014;165:489–98.
Dziuba J, Iwaniak A, Minkiewicz P. Computer-aided characteristics of proteins as potential precursors of bioactive peptides. Polimery. 2003;48:50–3.
Tulipano G, Sibilia V, Caroli AM, Cocchi D. Whey proteins as source of dipeptidyl dipeptidase IV (dipeptidyl peptidase-4) inhibitors. Peptides. 2011;32:835–8.
Dziuba B, Dziuba M. New milk protein-derived peptides with potential antimicrobial activity: An approach based on bioinformatic studies. Int J Mol Sci. 2014;15:14531–45.
Nongonierma AB, FitzGerald RJ. Dipeptidyl peptidase IV inhibitory and antioxidative properties of milk-derived dipeptides and hydrolysates. Peptides. 2013;39:157–63.
Nongonierma AB, FitzGerald RJ. Inhibition of dipeptidyl peptidase IV (DPP-IV) by proline containing peptides. J Funct Foods. 2013;5:1909–17.
ExPASy. http://web.expasy.org/peptide_cutter/peptidecutter_enzymes.html#Peps. Accessed 18 Sept 2017.
MEROPS. https://www.ebi.ac.uk/merops/. Accessed 18 Sept 2017.
O’Loughlin IB, Murray BA, Kelly PM, FitzGerald RJ, Brodkorb A. Enzymatic hydrolysis of heat-induced aggregates of whey protein isolate. J Agric Food Chem. 2012;60:4895–904.
Vorob’ev MM, Butré CI, Sforza S, Wierenga PA, Gruppen H. Demasking kinetics of peptide bond cleavage for whey protein isolate hydrolyzed by Bacillus licheniformis protease. J Mol Catal B: Enzym. 2017.
Panchaud A, Affolter M, Kussmann M. Mass spectrometry for nutritional peptidomics: How to analyze food bioactives and their health effects. J Proteome. 2012;75:3546–59.
Sánchez-Rivera L, Martínez-Maqueda D, Cruz-Huerta E, Miralles B, Recio I. Peptidomics for discovery, bioavailability and monitoring of dairy bioactive peptides. Food Res Int. 2014;63(Part B):170–81.
Nongonierma AB, Le Maux S, Hamayon J, FitzGerald RJ. Strategies for the release of dipeptidyl peptidase IV (DPP-IV) inhibitory peptides in an enzymatic hydrolyzate of α-lactalbumin. Food Funct. 2016;7:3437–43.
Kalyankar P, Zhu Y, O’Cuinn G, FitzGerald RJ. Investigation of the substrate specificity of glutamyl endopeptidase using purified bovine β-casein and synthetic peptides. J Agric Food Chem. 2013;61:3193–204.
Norris R, Poyarkov A, O’Keeffe MB, FitzGerald RJ. Characterization of the hydrolytic specificity of Aspergillus niger derived prolyl endoproteinase on bovine β-casein and determination of ACE inhibitory activity. Food Chem. 2014;156:29–36.
Norris R, O’Keeffe MB, Poyarkov A, FitzGerald RJ. Peptide identification and angiotensin converting enzyme (ACE) inhibitory activity in prolyl endoproteinase digests of bovine αs-casein. Food Chem. 2015;188:210–7.
Tulipano G, Faggi L, Nardone A, Cocchi D, Caroli AM. Characterization of the potential of β-lactoglobulin and α-lactalbumin as sources of bioactive peptides affecting incretin function: in silico and in vitro comparative studies. Int Dairy J. 2015;48:66–72.
Guinane CM, Kent RM, Norberg S, O'Connor PM, Cotter PD, Hill C, et al. Generation of the antimicrobial peptide caseicin A from casein by hydrolysis with thermolysin enzymes. Int Dairy J. 2015;49:1–7.
Zhang Y, Chen R, Zuo F, Ma H, Zhang Y, Chen S. Comparison of dipeptidyl peptidase IV-inhibitory activity of peptides from bovine and caprine milk casein by in silico and in vitro analyses. Int Dairy J. 2016;53:37–44.
Nongonierma AB, Paolella S, Mudgil P, Maqsood S, FitzGerald RJ. Dipeptidyl peptidase IV (DPP-IV) inhibitory properties of camel milk protein hydrolysates generated with trypsin. J Funct Foods. 2017;34:49–58.
Nongonierma AB, FitzGerald RJ. Susceptibility of milk protein-derived peptides to dipeptidyl peptidase IV (DPP-IV) hydrolysis. Food Chem. 2014;145:845–52.
Butré CI, Buhler S, Sforza S, Gruppen H, Wierenga PA. Spontaneous, non-enzymatic breakdown of peptides during enzymatic protein hydrolysis. Biochim Biophysica Acta (BBA) - Proteins Proteomics. 2015;1854:987–94.
Pina AS, Roque ACA. Studies on the molecular recognition between bioactive peptides and angiotensin-converting enzyme. J Mol Recognit. 2009;22:162–8.
Norris R, Casey F, FitzGerald RJ, Shields DC, Mooney C. Predictive modeling of angiotensin converting enzyme inhibitory dipeptides. Food Chem. 2012;133:1349–54.
Pripp AH. Docking and virtual screening of ACE inhibitory dipeptides. Eur Food Res Technol. 2007;225:589–92.
Nongonierma AB, Mooney C, Shields DC, FitzGerald RJ. Inhibition of dipeptidyl peptidase IV and xanthine oxidase by amino acids and dipeptides. Food Chem. 2013;141:644–53.
Vukic VR, Vukic DV, Milanovic SD, Ilicic MD, Kanuric KG, Johnson MS. In silico identification of milk antihypertensive di- and tripeptides involved in angiotensin I–converting enzyme inhibitory activity. Nutr Res. 2017;46:22–30.
Nongonierma AB, Mooney C, Shields DC, FitzGerald RJ. In silico approaches to predict the potential of milk protein-derived peptides as dipeptidyl peptidase IV (DPP-IV) inhibitors. Peptides. 2014;57:43–51.
Fernández-Tomé S, Martínez-Maqueda D, Girón R, Goicoechea C, Miralles B, Recio I. Novel peptides derived from αs1-casein with opioid activity and mucin stimulatory effect on HT29-MTX cells. J Funct Foods. 2016;25:466–76.
Nongonierma AB, FitzGerald RJ. Learnings from quantitative structure-activity relationship (QSAR) studies with respect to food protein-derived bioactive peptides: A review. RSC Adv. 2016;6:75400–13.
Udenigwe CC, Li H, Aluko RE. Quantitative structure–activity relationship modeling of renin-inhibiting dipeptides. Amino Acids. 2011;42:1379–86.
Wu J, Aluko RE. Quantitative structure-activity relationship study of bitter di- and tri-peptides including relationship with angiotensin I-converting enzyme inhibitory activity. J Pept Sci. 2007;13:63–9.
Wu J, Aluko RE, Nakai S. Structural requirements of angiotensin I-converting enzyme inhibitory peptides: Quantitative structure-activity relationship study of di-and tripeptides. J Agric Food Chem. 2006;54:732–8.
Wang J-H, Liu Y-L, Ning J-H, Yu J, Li X-H, Wang F-X. Is the structural diversity of tripeptides sufficient for developing functional food additives with satisfactory multiple bioactivities? J Mol Struct. 2013;1040:164–70.
Pripp AH, Isaksson T, Stepaniak L, Sørhaug T. Quantitative structure-activity relationship modeling of ACE-inhibitory peptides derived from milk proteins. Eur Food Res Technol. 2004;219:579–83.
Nongonierma AB, FitzGerald RJ. Structure activity relationship modeling of milk protein-derived peptides with dipeptidyl peptidase IV (DPP-IV) inhibitory activity. Peptides. 2016;79:1–7.
Maruyama S, Miyoshi S, Kaneko T, Tanaka H. Angiotensin I-converting enzyme inhibitory activities of synthetic peptides related to the tandem repeated sequence of a maize endosperm protein. Agric Biol Chem. 1989;53:1077–81.
Mooney C, Haslam NJ, Pollastri G, Shields DC. Towards the improved discovery and design of functional peptides: common features of diverse classes permit generalized prediction of bioactivity. PLoS One. 2012;7:e45012.
Zenezini Chiozzi R, Capriotti AL, Cavaliere C, La Barbera G, Piovesana S, Samperi R, et al. Purification and identification of endogenous antioxidant and ACE-inhibitory peptides from donkey milk by multidimensional liquid chromatography and nanoHPLC-high resolution mass spectrometry. Anal Bioanal Chem. 2016;408:5657–66.
Rani S, Pooja K, Pal GK. Exploration of potential angiotensin converting enzyme inhibitory peptides generated from enzymatic hydrolysis of goat milk proteins. Biocatalysis Agric Biotechnol. 2017;11:83–8.
Nongonierma AB, Le Maux S, Esteveny C, FitzGerald RJ. Response surface methodology (RSM) applied to the generation of casein hydrolysates with antioxidant and dipeptidyl peptidase IV (DPP-IV) inhibitory properties. J Sci Food Agric. 2017;97:1093–101.
Naik L, Mann B, Bajaj R, Sangwan R, Sharma R. Process optimization for the production of bio-functional whey protein hydrolysates: adopting response surface methodology. Int J Peptide Res Therapeut. 2013;19:231–7.
Zhao XH, Wu D, Li TJ. Preparation and radical scavenging activity of papain-catalyzed casein plasteins. Dairy Sci Technol. 2010;90:521–35.
Zheng H, Shen X, Bu G, Luo Y. Effects of pH, temperature and enzyme-to-substrate ratio on the antigenicity of whey protein hydrolysates prepared by Alcalase. Int Dairy J. 2008;18:1028–33.
Liu X, Luo Y, Li Z. Effects of pH, temperature, enzyme-to-substrate ratio and reaction time on the antigenicity of casein hydrolysates prepared by papain. Food Agric Immunol. 2012;23:69–82.
Guo Y, Pan D, Tanokura M. Optimization of hydrolysis conditions for the production of the angiotensin-I converting enzyme (ACE) inhibitory peptides from whey protein using response surface methodology. Food Chem. 2009;114:328–33.
Tavares TG, Contreras MM, Amorim M, Martín-Álvarez PJ, Pintado ME, Recio I, et al. Optimization, by response surface methodology, of degree of hydrolysis and antioxidant and ACE-inhibitory activities of whey protein hydrolysates obtained with cardoon extract. Int Dairy J. 2011;21:926–33.
Vermeirssen V, Van Camp J, Devos L, Verstraete W. Release of angiotensin I converting enzyme (ACE) inhibitory activity during in vitro gastrointestinal digestion: from batch experiment to semicontinuous model. J Agric Food Chem. 2003;51:5680–7.
Pan D, Guo Y. Optimization of sour milk fermentation for the production of ACE-inhibitory peptides and purification of a novel peptide from whey protein hydrolysate. Int Dairy J. 2010;20:472–9.
Zhao X-H, Li Y-Y. An approach to improve ACE-inhibitory activity of casein hydrolysates with plastein reaction catalyzed by Alcalase. Eur Food Res Technol. 2009;229:795–805.
Uluko H, Li H, Cui W, Zhang S, Liu L, Chen J, et al. Response surface optimization of angiotensin converting enzyme inhibition of milk protein concentrate hydrolysates in vitro after ultrasound pretreatment. Innovative Food Sci Emerg Technol. 2013;20:133–9.
Quirós A, Hernández-Ledesma B, Ramos M, Martín-Álvarez PJ, Recio I. Short communication: Production of antihypertensive peptide HLPLP by enzymatic hydrolysis: Optimization by response surface methodology. J Dairy Sci. 2012;95:4280–5.
Nongonierma AB, Lalmahomed M, Paolella S, FitzGerald RJ. Milk protein isolate (MPI) as a source of dipeptidyl peptidase IV (DPP-IV) inhibitory peptides. Food Chem. 2017;231:202–11.
Nongonierma AB, Mazzocchi C, Paolella S, FitzGerald RJ. Release of dipeptidyl peptidase IV (DPP-IV) inhibitory peptides from milk protein isolate (MPI) during enzymatic hydrolysis. Food Res Int. 2017;94:79–89.
Asledottir T, Le TT, Petrat-Melin B, Devold TG, Larsen LB, Vegarud GE. Identification of bioactive peptides and quantification of β-casomorphin-7 from bovine β-casein A1, A2 and I after ex vivo gastrointestinal digestion. Int Dairy J. 2017;71:98–106.
Cunsolo V, Muccilli V, Saletti R, Foti S. Applications of mass spectrometry techniques in the investigation of milk proteome. Eur J Mass Spectrom. 2011;17:305–20.
Le TT, Deeth HC, Larsen LB. Proteomics of major bovine milk proteins: Novel insights. Int Dairy J. 2017;67:2–15.
Giacometti J, Buretić-Tomljanović A. Peptidomics as a tool for characterizing bioactive milk peptides. Food Chem. 2017;230:91–8.
Nongonierma AB, Gaudel C, Murray BA, Flynn S, Kelly PM, Newsholme P, et al. Insulinotropic properties of whey protein hydrolysates and impact of peptide fractionation on insulinotropic response. Int Dairy J. 2013;32:163–8.
Lahrichi SL, Affolter M, Zolezzi IS, Panchaud A. Food Peptidomics: Large scale analysis of small bioactive peptides - A pilot study. J Proteome. 2013;88:83–91.
Morifuji M, Koga J, Kawanaka K, Higuchi M. Branched-chain amino acid-containing dipeptides, identified from whey protein hydrolysates, stimulate glucose uptake rate in L6 myotubes and isolated skeletal muscles. J Nutr Sci Vitaminol. 2009;55:81–6.
Dallas DC, Guerrero A, Parker EA, Robinson RC, Gan J, German JB, et al. Current peptidomics: Applications, purification, identification, quantification, and functional analysis. Proteomics. 2015;15:1026–38.
Le Maux S, Nongonierma AB, FitzGerald RJ. Improved short peptide identification using HILIC-MS/MS: Retention time prediction model based on the impact of amino acid position in the peptide sequence. Food Chem. 2015;175:847–54.
Harscoat-Schiavo C, Nioi C, Ronat-Heit E, Paris C, Vanderesse R, Fournier F, et al. Hydrophilic properties as a new contribution for computer-aided identification of short peptides in complex mixtures. Anal Bioanal Chem. 2012;403:1939–49.
Le Maux S, Nongonierma AB, Murray B, Kelly PM, FitzGerald RJ. Identification of short peptide sequences in the nanofiltration permeate of a bioactive whey protein hydrolysate. Food Res Int. 2015;77:534–9.
Sagardia I, Iloro I, Elortza F, Bald C. Quantitative structure–activity relationship based screening of bioactive peptides identified in ripened cheese. Int Dairy J. 2013;33:184–90.
Nongonierma AB, Paolella S, Mudgil P, Maqsood S, Fitzgerald RJ. Identification of novel dipeptidyl peptidase IV (DPP-IV) inhibitory peptides in camel milk protein hydrolysates. Food Chem. 2017;Submitted.
Murray BA, Walsh DJ, FitzGerald RJ. Modification of the furanacryloyl-L-phenylalanylglycylglycine assay for determination of angiotensin-I-converting enzyme inhibitory activity. J Biochem Biophys Methods. 2004;59:127–37.
Shan L, Marti T, Sollid LM, Khosla C. Comparative biochemical analysis of three bacterial prolyl endopeptidases: Implications for coeliac sprue. Biochem J. 2004;383:311–8.
Lacroix IM, Li-Chan EC. Comparison of the susceptibility of porcine and human dipeptidyl-peptidase IV to inhibition by protein-derived peptides. Peptides. 2015;69:19–25.
Devabhaktuni A, Elias JE. Application of de novo sequencing to large-scale complex proteomics data sets. J Proteome Res. 2016;15:732–42.
Tagliazucchi D, Shamsia S, Helal A, Conte A. Angiotensin-converting enzyme inhibitory peptides from goats' milk released by in vitro gastro-intestinal digestion. Int Dairy J. 2017;71:6–16.
Zhu Y-s. Kalyankar P, FitzGerald RJ. Relative quantitation analysis of the substrate specificity of glutamyl endopeptidase with bovine α-caseins. Food Chem. 2015;167:463–7.
Rocha-Martin J, Fernández-Lorente G, Guisan JM. Sequential hydrolysis of commercial casein hydrolysate by immobilized trypsin and thermolysin to produce bioactive phosphopeptides. Biocatal Biotransform. 2017:1–16.
Cheung LK, Aluko RE, Cliff MA, Li-Chan EC. Effects of exopeptidase treatment on antihypertensive activity and taste attributes of enzymatic whey protein hydrolysates. J Funct Foods. 2015;13:262–75.
Barry C, O'Cuinn G, Harrington D, O'Callaghan D, Fitzgerald R. Debittering of a tryptic digest of bovine b-casein using porcine kidney general aminopeptidase and X-prolydipeptidyl aminopeptidase from Lactococcus lactis subsp. cremoris AM2. J Food Sci. 2000;65:1145–50.
Acknowledgements
The work described herein was supported by Enterprise Ireland under grant number TC2013-0001. The authors thank Dr. Sara Paolella for her insightful discussions on peptide bond selectivity during enzymatic hydrolysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interests
The authors declare that they have no conflict of interest.
Additional information
Published in the topical collection Discovery of Bioactive Compounds with guest editors Aldo Laganà, Anna Laura Capriotti, and Chiara Cavaliere.
Rights and permissions
About this article

Cite this article
Nongonierma, A.B., FitzGerald, R.J. Enhancing bioactive peptide release and identification using targeted enzymatic hydrolysis of milk proteins. Anal Bioanal Chem 410, 3407–3423 (2018). https://doi.org/10.1007/s00216-017-0793-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-017-0793-9