
Abstract
A two-dimensional LC (2D-LC) method, based on the work of Erni and Frei in 1978, was developed and coupled to an ion mobility-high-resolution mass spectrometer (IM-MS), which enabled the separation of complex samples in four dimensions (2D-LC, ion mobility spectrometry (IMS), and mass spectrometry (MS)). This approach works as a continuous multiheart-cutting LC system, using a long modulation time of 4 min, which allows the complete transfer of most of the first - dimension peaks to the second - dimension column without fractionation, in comparison to comprehensive two-dimensional liquid chromatography. Hence, each compound delivers only one peak in the second dimension, which simplifies the data handling even when ion mobility spectrometry as a third and mass spectrometry as a fourth dimension are introduced. The analysis of a plant extract from Ginkgo biloba shows the separation power of this four-dimensional separation method with a calculated total peak capacity of more than 8700. Furthermore, the advantage of ion mobility for characterizing unknown compounds by their collision cross section (CCS) and accurate mass in a non-target approach is shown for different matrices like plant extracts and coffee.

Principle of the four-dimensional separation
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The ambition to determine as many compounds as possible in very complex samples qualitatively and quantitatively leads to more complex and powerful analytical methods. For this purpose, comprehensive two-dimensional chromatographic techniques (GC×GC or LC×LC) are coupled to modern high-resolution mass spectrometers [1, 2]. The introduction of ion mobility spectrometry (IMS), separating compounds according to their shape-to-charge ratio [3, 4], obtains the possibility to add a further separation dimension to such an instrumental setup.
Erni and Frei implemented a two-dimensional liquid chromatography method in 1978 [5]. They coupled a gel permeation chromatography column and a reversed phase column online to separate a senna glycoside extract collecting the whole sample in seven fractions of 1.5 mL within 10 h. This approach can be described as a multiple heart-cutting LC with very large fractions. Based on this, in 1990 Bushey and Jorgensen [6] introduced comprehensive two-dimensional liquid chromatography (LC×LC). The main difference is that in LC×LC, the separation of the first dimension column is preserved by using a very short sampling period (modulation time), so that each peak coming from the first dimension is modulated at least three or four times [7], which is a major characteristic of LC×LC according to the nomenclature published by Marriott et al. [8]. The resulting separation power of LC×LC is outstanding, and therefore, this technique is often used today for the analysis of complex samples [1, 9, 10]. The method developed by Erni and Frei was not used again. LC×LC leads to a powerful separation, but also to more complex data, because each compound is cut in several peaks. For quantitative analysis in LC×LC, each peak has to be integrated separately and integration errors sum up [11, 12]. In a multiple heart-cutting approach, complete peaks are transferred to the second dimension and each compound reaches the detector only once.
The coupling of ion mobility spectrometry (IMS) to mass spectrometry (MS) started in the 1960s and experienced a strong development since the 1990s [4, 13]. The first commercially available ion mobility mass spectrometer (IM-MS) was the Synapt HDMS system, a quadrupole/travelling-wave IMS/TOF (TWIMS) system introduced in 2006 by Waters [14]. Since 2014, the Agilent 6560 ion mobility quadrupole time-of-flight (IM-qTOF), a system using drift time IMS (DTIMS), is available [15]. IMS separates ions drifting through a tube filled with buffer gas in a weak electric field according to their shape-to-charge ratio and therefore has the ability to measure collision cross sections (CCS or Ω) of the ions [16]. The CCS is a specific value for an ion and can, as additional parameter next to exact mass, increase certainty in the identification of compounds. Furthermore, IM-MS obtains the possibility to separate isobaric species with the same sum formula and exact mass when they differ in their size and shape. Even the best high-resolution MS would deliver only one peak for two isobaric ions because they have the same mass-to-charge (m/z) ratio. In comparison to TWIMS, where a calibration has to be performed to determine CCS because a non-uniform electric field is used and therefore, the Mason-Schamp equation cannot be directly applied [17], DTIMS allows to calculate CCS directly from the observed drift times [3, 18]. Values for CCS are available in the literature for different peptides [18–21]. Furthermore, data can be found for other substances like N-glycans [17], drug-like compounds [16], metabolites [22], lipids [23], or different biomolecules [24].
Different applications show that the coupling of IM-MS with HPLC offers a powerful tool for the analysis of complex samples like plasma [25] or saliva [26]. The coupling of LC×LC with IM-MS would facilitate a powerful separation technique in four dimensions (2D-LC, IMS, and MS) where even coeluting and isobaric compounds can be separated by their CCS. But although this is theoretically possible, there is a big challenge for data evaluation because currently no available software can simplify the data into a readable plot. After the first chromatographic dimension, each substance would be fractionated into three to four parts and each of these fractions is then injected onto the second dimension column before being transferred to the IM-MS instrument, which leads to very complex data in four dimensions. To evaluate complex samples after an LC×LC-IM-MS analysis, a feature analysis considering all four separation dimensions is needed and it is not clear whether and if so, when such software will be available. To overcome this and to simplify data evaluation with available software when IMS as a further separation dimension is introduced, we developed a method, here called LC + LC, similar to the one of Erni and Frei [5] with a long modulation time, realizing that peaks are mostly not—or for a few compounds only one time—modulated so that most of the compounds are only observed in a single second dimension chromatogram. This allows to view the data of a 2D-LC separation on a single time axis like in a simple 1D chromatogram. The LC + LC separation described above was coupled to IM-qTOF-MS to demonstrate the ability of this system using four separation dimensions (2D-LC, IMS, and MS) for the analysis of complex samples. CCS were measured in a real sample separated by this system, and a developed database was used to characterize compounds detected in those samples.
Material and methods
Chemicals
All solvents and mobile phases were used as LC-MS grade. Methanol was purchased from VWR (Leuven, Belgium) and formic acid was from Merck (Darmstadt, Germany). Ultrapure water was generated with a water purification system from Sartorius Stedim (Goettingen, Germany). Standard substances of colchicine, d-fructose, and l-phenylalanine were purchased from Sigma-Aldrich (St. Louis, MO, USA). Rutin-trihydrate was from Alfa Aesar (Karlsruhe, Germany). All other standards for the establishment of a CCS database were collected from different working groups. For mass calibration of the MS as well as for direct CCS calibration, a low concentration tuning mix (G1969-85000, Agilent Technologies, Santa Clara, CA, US) was used.
Instrumental setup
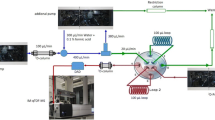
An Agilent 1290 Infinity two-dimensional liquid chromatography system was used, consisting of a 1290 Infinity Binary Pump (G4220A) using a Jet Weaver V35 mixer for each the first and the second dimension, a 1290 Infinity Flexible cube solvent management module (G4227A), a 1290 Infinity HiP sampler (G4226A), a 1290 Infinity Thermostatted Column compartment (G1316C) with a 2D-LC-Quick Change Valve (2 Pos/4 Port duo valve, G4236A), and a 1290 Infinity Diode Array detector (G4212A). The interface between first and second dimension was set up with a dilution and flow splitting system using an additional 1290 Infinity Binary Pump (Fig. 1). Pump Head B is used for diluting the effluent (100 μL/min) of the first dimension column with 0.1 % formic acid in water (300 μL/min). The flow goes into a Jet Weaver V35 mixer to get a proper mixing of the eluate and the water. After this, a T-piece and Pump Head A of the additional binary pump are used for an active flow splitting, resulting in a total flow of 20 μL/min transferred to the loops. With a modulation time of 4 min and a loop volume of 100 μL, the loops are filled up to 80 % with the diluted fractions coming from the first dimension column before injection onto the second dimension.

Interface for the LC + LC system with an additional pump for dilution and split of the first dimension column effluent. DAD diode array detector, IM-qTOF ion mobility quadrupole time-of-flight mass spectrometer
In the first dimension, a 150 × 2.0 mm Luna CN column packed with 3.0-μm particles was used with water containing 0.1 % formic acid and acetonitrile as mobile phase (gradient from 5 to 80 % ACN, see red line in Fig. 2). The second dimension column was a 50 × 3.0 mm Kinetex C18 with 2.6-μm core-shell particles. The mobile phase consisted of water and methanol, both containing 0.1 % formic acid. For the second dimension with a flow of 500 μL, a shifted gradient was used, starting with a gradient from 10 to 60 % methanol in the first modulation and reaching a gradient from 50 to 90 % methanol in the last modulation (see blue line in Fig. 2).

Gradients used for LC + LC: gradient in the first dimension (red line) (a) with eluent A: water with 0.1 % formic acid, and eluent B: acetonitrile, flow rate 100 μL/min, and shifted gradient in the second dimension (blue line). Gradient program of the second dimension in the first modulation (b) with eluent A: water with 0.1 % formic acid, and eluent B: methanol with 0.1 % formic acid, flow rate 500 μL/min
IM-qTOF measurements were performed with an Agilent 6560 IM-qTOF system, equipped with a Dual Agilent Jet Stream electrospray ionization (AJS ESI) source. Nitrogen was used as a buffer gas in the drift tube at a pressure of about 3.95 Torr. The instrument was used in two different modes: in qTOF-only mode, the trapping gate is off and ions are just transferred through the drift tube to the rear funnel so that no ion mobility separation occurs and the system is used like a conventional qTOF instrument.
In IM mode, ions are collected in the trapping funnel when a voltage is applied to the trapping gate for a certain time (trapping time) and released in packages to be separated in the drift tube before they are transferred and pulsed to the TOF mass analyzer. Instrument parameters are given in Table 1.
Measurement of CCS
To collect collision cross-section values of different compounds for establishing a database, at the moment 300 standards were measured and CCS were calculated with the following method. Standard solutions of 10 mg/L in methanol/water (50/50) were injected eight times to the IM-qTOF instrument using the HiP sampler and the binary pump of the HPLC for an in-flow injection. For each of the eight injections, different drift tube entrance voltages between 1000 and 1700 V (in 100 V steps) were applied. With a constant drift tube exit voltage of 250 V and a drift tube length of 80 cm, the resulting electric fields were between 9.375 and 18.125 V/cm. Corrected drift times td were calculated according to [24] by subtracting the flight time through the interfacing IM-MS ion optics and MS from the total observed drift times.
The calculation of the CCS for all observed adducts in positive and negative mode, such as [M + H]+, [M + Na]+, and [M + NH4]+, according to Mason-Schamp [3, 18] was done with the Agilent IM-MS Browser B.07.01 software. For a direct CCS calculation from the observed drift times in real samples, a direct CCS calibration with a known tuning mix (G1969-85000, Agilent Technologies, Santa Clara, CA, US) was performed as described in detail in [27] on the same day.
Sample preparation
Leaves of Ginkgo biloba were collected in Essen (Germany) and dried at 60 °C. For extraction, the dried leaves were powdered and 300 mg of the homogenized powder were extracted in 3 mL methanol for 40 min in an ultrasonic bath. The extract was filtered via a 0.2-μm syringe filter and stored at −20 °C until usage. Before LC + LC analysis, the methanol extract was diluted 1:10 with water and colchicine was added as an internal standard to a final concentration of 100 μg/L.
Results and discussion
Setup of the LC + LC method for coupling to IM-qTOF
For the coupling of two-dimensional liquid chromatography to IM-qTOF, a method called LC + LC was developed as an intermediate of comprehensive and heart-cut two-dimensional LC (continuous multiheart-cutting LC). A longer modulation time of 4 min compared to comprehensive LC×LC (e.g., 1 min [28, 29]) has the effect that most of the peaks coming from the first dimension are not or only one time (when they elute between two modulation steps) modulated. On the contrary, in LC×LC, all peaks from the first dimension are modulated at least three or four times. This longer modulation time leads to a loss of resolution in the first dimension because all substances separated in 4 min in the first dimension are pooled again, but according to the longer analysis time available in the second dimension, an increase in peak capacity in the second dimension is achieved. However, with LC + LC, we have a better peak capacity as a one-dimensional LC and more or less no fractionation. Therefore, each substance appears only once in the two LC dimensions, which allows a simple data handling and feature analysis with a commercially available software from the MS manufacturer. Only for a few compounds, which will be modulated one time—because they elute from the first dimension between two modulation steps—we get two signals, which does not allow an automated data analysis. For all other compounds, an automated data analysis (qualitative and quantitative) is possible. In this work, we used a modulation time of 4 min and fractions coming from the first dimension were collected in loops of 100 μL volume. A large injection volume on the second dimension column and a high proportion of organic solvent in the fractions can lead to broad peaks because of high elution power as described by Stoll et al. [30]. To avoid such problems, we set up a system with a dilution and flow splitting system between the first dimension column and the loops (Fig. 1) according to the concepts of Filgueira et al. and Stoll et al., which allows to optimize the first and the second dimension independently and results in narrower peaks due to the focusing of the injected fraction on the second dimension column [30, 31].
Different column combinations were tested to optimize the LC + LC system using HILIC, anion exchange, CN, and C18 columns (results not shown). The best result for our sample was obtained using a CN column in the first dimension and a C18 column in the second dimension. This column combination gives the best separation power and peak capacity when a shifted gradient is used (Fig. 2), which was shown in our previous work [29].
The contour plot in Fig. 3 shows an extract from G. biloba analyzed by LC + LC-qTOF (instrument used in qTOF only mode) as described above (TIC, blank was subtracted) with a large number of separated compounds over a relatively wide retention space. The same sample was analyzed using the instrument in IM mode. Here, the IM provides an additional separation dimension to the two chromatographic dimensions of the LC + LC method. This results in an increased separation power and the data evaluation benefits from the fact that in our LC + LC method, peaks from the first dimension are not modulated several times as is common in LC×LC. This allows to view the two-dimensional LC + LC separation as a 1D chromatogram vs. the drift time in ion mobility spectrometry because each compound should appear only once or not more than twice. This heat map of retention time vs. drift time is shown in Fig. 4 for the G. biloba extract (a). Substances coeluting even after the two separation dimensions of the LC + LC system are now further separated in the IM dimension. The zoom in Fig. 4b shows the separation on the second dimension column between 8 and 12 min, where the second fraction, which is collected in the loop between 4 and 8 min, is transferred and separated in the second dimension. After the two chromatographic dimensions, substances are separated, according to their CCS, in the IM drift tube and subsequently by their m/z in TOF-MS. A 2D plot of m/z vs. drift time extracted from 9.63 to 9.76 min (Fig. 4c) shows that in this short retention range, still several substances are contained, resulting in a variety of peaks in the drift spectrum and in the mass spectrum. A closer zoom of this plot (d) reveals a compound showing one peak in the mass spectrum with m/z 379.1001 that gives two separate peaks in the IM drift spectrum (23.12 and 24.85 ms). This is an example for the ability of the IM dimension to differentiate between isobaric compounds that are not chromatographic separated and have the same m/z in mass spectrometry.

Separation of an extract from Ginkgo biloba by LC + LC-qTOF with a modulation time of 4 min (gradients see Fig. 2). TIC, blank subtracted. Dashed lines show the area considered as A eff for peak capacity calculation

Heat map of an LC + LC-IM-qTOF measurement of a Ginkgo biloba extract (a). The separation on the second dimension column of one fraction collected between 4 and 8 min (eluting from the second dimension between 8 and 12 min) from the sample is zoomed in (b). 2D plot of m/z vs. drift time extracted from 9.63 to 9.76 min (c). The zoomed in image shows the separation of two peaks with the same m/z in the IM dimension (d)
Repeatability
The same sample of G. biloba extract was injected several times under the same conditions to determine the repeatability of the described method. Retention times of the peaks of colchicine (internal standard) and rutin, a flavonoid found as a major active compound in G. biloba [32], in the LC + LC chromatogram were evaluated on three different days with three analyses per day. According to the LC + LC setup, final retention times given in the chromatograms in this work are the sum of the first dimension retention time (4-min modulation time multiplicated with the fraction number) and the retention time in the second dimension. The retention times are highly reproducible for several injections and from day to day. The average retention time for rutin was 22.50 min with a relative standard deviation (RSD) of 0.03 %. In a methanolic extract of Castanea sativa, rutin was found at a retention time of 22.52 min (Table S3 in the Electronic Supplementary Material, ESM), which shows that retention times are also reproducible for other sample matrices. For colchicine, the RSD was 0.01 % at an average retention time of 69.97 min. In addition to those two compounds, eight randomly chosen peaks eluting between 9.46 and 111.03 min with m/z ratios between 205.0972 and 763.2044 have been inspected. Relative standard deviations between 0.01 and 0.07 % could be determined.
For four consecutive measurements, the ratio of the peak areas observed for the internal standard colchicine (m/z 400.1755) and a single peak that elutes close to it (m/z 763.1849) were calculated and the peak ratio shows a relative standard deviation of 5.1 %, which is not as good as in 1D methods, but still shows that the presented method can be used for quantitative analysis.
Peak capacity
To determine the total peak capacity of this method, P total, the peak capacity of the two chromatographic dimensions and the ion mobility dimension have to be combined. The product of the peak capacity in the LC + LC separation LC+LC n and in the IM dimension IM n should give the maximum possible number of separated peaks [33]:
The peak capacity for 2D-LC separations can be calculated for optimal conditions, where the two dimensions are completely independent of each other, as the product of the first and second dimension peak capacity 1 n and 2 n:
For the first LC dimension, where a loss of resolution occurs because of the long modulation time of 4 min, instead of a real peak capacity, the number of fractions transferred to the second dimension within the total analysis time t total (120 min) will be used:
Integrating Eq. 3 in Eq. 2 gives the formula for the LC + LC peak capacity if the two dimensions are completely orthogonal:
Since complete orthogonality in two-dimensional chromatography is rarely possible [34, 35], the peak capacity will be corrected by a factor for the orthogonality O A , which is here calculated as the percentage of the effective distribution area A eff, where peaks are distributed, in the total area A of the 2D counter plot.
Here, the effective area A eff is determined according to the approach in our previous work [28], as the area of the two rectangles visibly containing peaks as shown in Fig. 3a (dashed lines). With A eff = 174 and A = 480, we receive a factor of O A = 0.36 for the orthogonality. The peak capacity n in one dimension can be approximated as the quotient of the separation time t and the average peak width at base w b [36]:
For the second dimension and the IM dimension, the peak width at base was calculated as the average of three peaks taken from the beginning, the middle, and the end of one separation cycle of the G. biloba analysis (w b(2D) = 0.13 min and w b (IM) = 0.83 ms). With a separation time t in the second dimension of 4 min, the peak capacity here can be calculated to 2 n = 31. In the IM dimension, only the time range between 14 and 36 ms, where peaks are detected (compare Fig. 4a), was considered (t = 22 ms), which results in a peak capacity of IM n = 27. After all, we achieve a peak capacity for the LC + LC separation of LC+LC n = 324 according to Eq. 5 The total peak capacity of the LC + LC-IM-qTOF-MS coupling can be roughly calculated with Eq. 1 to P total = 8748.
The number of 29 fractions transferred to the second dimension is very low because of the long modulation time as described above, which results in a peak capacity LC+LC n of the LC + LC method that is lower compared to LC×LC methods, but still better than in 1D-LC methods (for comparison with the peak capacity of 1D-LC, it should be mentioned that in our calculation, only the part of the contour plot with peaks was used). The addition of ion mobility as a third separation dimension is a powerful method to increase the maximum number of possible peaks, so that a total peak capacity of 8748 could be achieved.
This calculation is a rough estimate because some peaks in a complex sample will also be one time modulated, which leads to two peaks for one compound. The fact that some compounds form different adducts or fragments in the ion source and therefore can have more than one drift peak in the IM dimension should also be kept in mind. This rough calculation should only show the potential of this instrumental setup.
Measurement of CCS
Collision cross sections for different standards were measured following the procedure described above to generate a database (data not shown). Table 2 shows measured CCS in nitrogen as a drift gas for only some substances including the calculated exact m/z for each adduct. Where available, the literature values for CCS are given to show that the obtained data are comparable to values measured on another instrument in another lab. Deviations from the literature are always below 1 %.
The G. biloba extract was analyzed with LC + LC-IM-qTOF and, using the IM-MS Browser B.07.01 software (Agilent, Santa Clara, USA), a feature analysis was performed. The resulting list contains information about retention time, drift time, CCS (calculated directly from drift times), m/z, and abundance for each feature. These CCS values (±1 %) in combination with the m/z ratio (±5 ppm) were automatically compared with the CCS values and m/z ratios contained in a home-made CCS database with more than 300 standards. This database search leads to seven hits (one is the added standard colchicine, Table 3). Two retention times for gallocatechine were determined (features 577 and 1968) with the same mass and CCS value. Possibly here two isobaric compounds with very similar CCS are separated chromatographically in the second dimension, but at the moment, only one of them is available in our database and, thus, only one result is found. This emphasizes the need to continue the work on the database. For confirmation, standards were measured with the same method and gallocatechine was detected with a retention time of 9.2 min. Hence, only the first of the two peaks in the sample can be assigned to gallocatechine. This shows the importance of combining the powerful IM-MS technique with a good chromatographic separation. For rutin, four features were found (features 77, 228, 1526, and 2553), two of them belonging to the H+ (features 228 and 2552) and two to the Na+ (features 77 and 1526) adducts. This can easily be explained by calculating the difference in the retention times (26.45 minus 22.51 min) of 3.94 min, which is the time of one modulation (a little less than 4 min because a shift gradient was used in the second dimension, which leads to slightly lower retention times for the second fraction). In this case, due to the modulation of the peak after the first dimension, the area of both peaks must be summed up for quantitative analysis.
With exactly the same chromatographic conditions and IM and MS parameters, we have also analyzed waste water (data not shown), coffee, and C. sativa extract samples and compared the results with our database (see ESM).
These examples demonstrate that with this LC + LC-IM-qTOF-MS system, we have now a powerful instrumental setup for non-target analysis of different samples with a minimum of method development (sample preparation), and the only limitation in the identification of the analytes in a complex sample is the size of the CCS database.
Conclusion
A two-dimensional HPLC method with a longer modulation time called LC + LC was implemented. There is a loss of resolution in the first dimension separation compared to comprehensive LC×LC, but the long modulation time leads to a higher peak capacity in the second dimension. This method allows to transfer peaks completely from the first to the second dimension column without fractionation or modulating them only once. In total, the peak capacity of LC + LC is between 1D-LC and LC×LC. The coupling of LC + LC to IM-qTOF-MS was then realized to add ion mobility as a further separation dimension, so that with LC + LC and TOF-MS now four dimensions are available to separate complex samples. The analysis of a G. biloba extract shows that the addition of IM reveals a higher complexity of the sample and that now, even coeluting isobaric compounds can be separated. The developed method shows a good reproducibility of retention times and of peak areas when related to an internal standard and it results in a total peak capacity of more than 8700. It is possible to determine directly CCS of compounds separated with this method. The CCS can then help to identify those compounds with a CCS database, which has a high potential in non-target analysis.
References
Elsner V, Laun S, Melchior D, Köhler M, Schmitz OJ. Analysis of fatty alcohol derivatives with comprehensive two-dimensional liquid chromatography coupled with mass spectrometry. J Chromatogr A. 2012;1268:22–8. doi:10.1016/j.chroma.2012.09.072.
Marriott PJ, Chin ST, Maikhunthod B, Schmarr HG, Bieri S. Multidimensional gas chromatography. Trends Anal Chem. 2012;34:1–20. doi:10.1016/j.trac.2011.10.013.
Kanu AB, Dwivedi P, Tam M, Matz L, Hill HHJ. Ion mobility–mass spectrometry. J Mass Spectrom. 2008;43:1–22. doi:10.1002/jms.
Mukhopadhyay R. IMS/MS: its time has come. Anal Chem. 2008;80:7918–20. doi:10.1021/ac8018608.
Erni F, Frei RW. Two-dimensional column liquid chromatographic technique for resolution of complex mixtures. J Chromatogr. 1978;149:561–9.
Bushey MM, Jorgenson JW. Automated instrumentation for comprehensive two-dimensional high-performance liquid chromatography of proteins. Anal Chem. 1990;62:161–7.
Murphy RE, Schure MR, Foley JP. Effect of sampling rate on resolution in comprehensive two-dimensional liquid chromatography. Anal Chem. 1998;70:1585–94. doi:10.1021/ac971184b.
Marriott PJ, Wu Z, Schoenmakers P. Nomenclature and conventions in comprehensive multidimensional chromatography—an update. LCGC Eur. (2012);25
Hu L, Chen X, Kong L, Su X, Ye M, Zou H. Improved performance of comprehensive two-dimensional HPLC separation of traditional Chinese medicines by using a silica monolithic column and normalization of peak heights. J Chromatogr A. 2005;1092:191–8. doi:10.1016/j.chroma.2005.06.066.
Stoll DR, Carr PW. Fast, comprehensive two-dimensional HPLC separation of tryptic peptides based on high-temperature HPLC. J Am Chem Soc. 2005;127:5034–5. doi:10.1021/ja050145b.
De La Mata P, Harynuk JJ. Limits of detection and quantification in comprehensive multidimensional separations. 1. A theoretical look. Anal Chem. 2012;84:6646–53. doi:10.1021/ac3010204.
Thekkudan DF, Rutan SC, Carr PW. A study of the precision and accuracy of peak quantification in comprehensive two-dimensional liquid chromatography in time. J Chromatogr A. 2010;1217:4313–27. doi:10.1016/j.chroma.2010.04.039.
Lapthorn C, Pullen F, Chowdhry BZ. Ion mobility spectrometry-mass spectrometry (IMS-MS) of small molecules: separating and assigning structures to ions. Mass Spectrom Rev. 2013;32:43–71. doi:10.1002/mas.
Pringle SD, Giles K, Wildgoose JL, Williams JP, Slade SE, Thalassinos K, et al. An investigation of the mobility separation of some peptide and protein ions using a new hybrid quadrupole/travelling wave IMS/oa-ToF instrument. Int J Mass Spectrom. 2007;261:1–12. doi:10.1016/j.ijms.2006.07.021.
May JC, McLean JA. Ion mobility-mass spectrometry: time-dispersive instrumentation. Anal Chem. 2015;87:1422–36. doi:10.1021/ac504720m.
Campuzano I, Bush MF, Robinson CV, Beaumont C, Richardson K, Kim H, et al. Structural characterization of drug-like compounds by ion mobility mass spectrometry: comparison of theoretical and experimentally derived nitrogen collision cross sections. Anal Chem. 2012;84:1026–33. doi:10.1021/ac202625t.
Hofmann J, Struwe WB, Scar CA, Scrivens JH, Harvey DJ, Pagel K. Estimating collision cross sections of negatively charged N-glycans using traveling wave ion mobility-mass spectrometry. Anal Chem. 2014;86:10789–95. doi:10.1021/ac5028353.
Tao L, McLean JR, McLean JA, Russell DH. A collision cross-section database of singly-charged peptide ions. J Am Soc Mass Spectrom. 2007;18:1232–8. doi:10.1016/j.jasms.2007.04.003.
Counterman AE, Valentine SJ, Srebalus CA, Henderson SC, Hoaglund CS, Clemmer DE. High-order structure and dissociation of gaseous peptide aggregates that are hidden in mass spectra. J Am Soc Mass Spectrom. 1998;9:743–59. doi:10.1016/S1044-0305(98)00052-X.
Valentine SJ, Counterman AE. A database of 660 peptide ion cross sections: use of intrinsic size parameters for bona fide predictions of cross sections. J Am Soc Mass Spectrom. 1999;10:1188–211. doi:10.1016/S1044-0305(99)00079-3.
Bush MF, Hall Z, Giles K, Hoyes J, Robinson CV, Ruotolo BT. Collision cross sections of proteins and their complexes: a calibration framework and database for gas-phase structural biology. Anal Chem. 2010;82:9557–65. doi:10.1021/ac1022953.
Paglia G, Williams JP, Menikarachchi LC, Thompson JW, Tyldesley-Worster R, Halldórsson S, et al. Ion mobility-derived collision cross-sections to support metabolomics applications. Anal Chem. 2014;86:3985–93. doi:10.1021/ac500405x.
Paglia G, Angel P, Williams JP, Richardson K, Olivos HJ, Thompson JW, et al. Ion mobility-derived collision cross section as an additional measure for lipid fingerprinting and identification. Anal Chem. 2015;87:1137–44. doi:10.1021/ac503715v.
May JC, Goodwin CR, Lareau NM, Leaptrot KL, Morris CB, Kurulugama RT, et al. Conformational ordering of biomolecules in the gas phase: nitrogen collision cross sections measured on a prototype high resolution drift tube ion mobility-mass spectrometer. Anal Chem. 2014;86:2107–16. doi:10.1021/ac4038448.
Liu X, Valentine SJ, Plasencia MD, Trimpin S, Naylor S, Clemmer DE. Mapping the human plasma proteome by SCX-LC-IMS-MS. J Am Soc Mass Spectrom. 2007;18:1249–64. doi:10.1016/j.jasms.2007.04.012.
Malkar A, Devenport NA, Martin HJ, Patel P, Turner MA, Watson P, et al. Metabolic profiling of human saliva before and after induced physiological stress by ultra-high performance liquid chromatography-ion mobility-mass spectrometry. Metabolomics. 2013;9:1192–201. doi:10.1007/s11306-013-0541-x.
Kurulugama RT, Darland E, Kuhlmann F, Stafford G, Fjeldsted J. Evaluation of drift gas selection in complex sample analyses using a high performance drift tube ion mobility-QTOF mass spectrometer. Analyst (Cambridge, U K). 2015;140:6834–44. doi:10.1039/c5an00991j.
Dück R, Sonderfeld H, Schmitz OJ. A simple method for the determination of peak distribution in comprehensive two-dimensional liquid chromatography. J Chromatogr A. 2012;1246:69–75. doi:10.1016/j.chroma.2012.02.038.
Li D, Schmitz OJ. Use of shift gradient in the second dimension to improve the separation space in comprehensive two-dimensional liquid chromatography. Anal Bioanal Chem. 2013;405:6511–7. doi:10.1007/s00216-013-7089-5.
Stoll DR, Talus ES, Harmes DC, Zhang K Evaluation of detection sensitivity in comprehensive two-dimensional liquid chromatography separations of an active pharmaceutical ingredient and its degradants. Anal Bioanal Chem; 2014. 265–277. doi: 10.1007/s00216-014-8036-9.
Filgueira M, Huang Y, Witt K, Castells C, Carr PW. Improving peak capacity in fast on-line comprehensive two-dimensional liquid chromatography with post first dimension flow-splitting. Anal Chem. 2011;83:9531–9. doi:10.1021/ac202317m.
Ding S, Dudley E, Plummer S, Tang J, Newton RP, Brenton G. Quantitative determination of major active components in Ginkgo biloba dietary supplements by liquid chromatography/mass spectrometry. Rapid Commun Mass Spectrom. 2006;20:2753–60. doi:10.1002/rcm.
Giddings JC. Two-dimensional separations: concept and promise. Anal Chem. 1984;56:1258A–70A. doi:10.1021/ac00276a003.
Gilar M, Olivova P, Daly AE, Gebler JC. Orthogonality of separation in two-dimensional liquid chromatography. Anal Chem. 2005;77:6426–34. doi:10.1021/ac050923i.
Liu Z, Patterson Jr D. Geometric approach to factor analysis for the estimation of orthogonality and practical peak capacity in comprehensive two-dimensional separations. Anal Chem. 1995;67:3840–5. doi:10.1021/ac00117a004.
Dolan JW, Snyder LR, Djordjevic NM, Hill DW, Waeghe TJ. Reversed-phase liquid chromatographic separation of complex samples by optimizing temperature and gradient time I. Peak capacity limitations. J Chromatogr A. 1999;857:1–20. doi:10.1016/S0021-9673(99)00765-7.
Taylor L, Tichy S. Ion mobility technology for LC/MS reveal greater detail. http://www.agilent.com/cs/library/eseminars/public/Ion Separation - Innovations in Mobility Techniques.pdf. 2013 Accessed 22 Jan 2016.
Acknowledgments
We are thankful to Agilent for the third Infinity pump system and Phenomenex for the HPLC columns.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 530 kb)
Rights and permissions
About this article

Cite this article
Stephan, S., Jakob, C., Hippler, J. et al. A novel four-dimensional analytical approach for analysis of complex samples. Anal Bioanal Chem 408, 3751–3759 (2016). https://doi.org/10.1007/s00216-016-9460-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-016-9460-9