
Abstract
Artificial sweeteners are food additives employed as sugar substitutes which are now considered to be emerging organic contaminants. In the present study, a method is developed for the determination of a group of artificial sweeteners in environmental waters. Considering the polar and hydrophilic character of these compounds, hydrophilic interaction liquid chromatography is proposed for their separation as an alternative to traditional reversed-phase liquid chromatography. Two stationary phases with different chemistry were compared for this purpose. For the detection of the analytes, high-resolution mass spectrometry (Orbitrap) was employed to take advantage of its benefits in terms of reliable quantification and confirmation for the measurement of accurate masses. Solid-phase extraction was chosen as the sample treatment, in which the extract in a mixture of NH4OH:MeOH:ACN (1:4:15) was directly injected into the chromatographic system, simplifying the analytical procedure. The optimized method was validated on river and waste water samples. For example, in the case of effluent water samples, limits of detection ranged from 0.002 to 0.7 μg/L and limits of quantification ranged from 0.004 to 1.5 μg/L. Apparent (whole method) recoveries ranged from 57 to 74 % with intra-day precision (%RSD, n = 5) ranging from 6 to 25 %. The method was successfully applied to water samples from different rivers in Catalonia and different waste water treatment plants in Tarragona. Acesulfame, cyclamate, saccharine and sucralose were found in several samples.

Illustrative figure of the SPE/HILIC-HRMS method proposed
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Artificial sweeteners are a class of food additives used as sugar substitutes in food, beverages, sanitary products and pharmaceuticals [1]. Among the list of artificial sweeteners (which comprise various classes of polar compounds), those approved by the European Union (EU) are acesulfame, aspartame, cyclamate, saccharin, sucralose and neohesperidin dihydrochalcone [2]. Moreover, stevioside and glycyrrhizic acid are sweeteners of natural origin of which consumption has recently increased [3, 4]. The use of steviol glycosides as sweeteners has only been permitted in the EU since 2011 [3].
The use of these additives rather than sugar is preferred because they do not provide calories, their sweetness is quite intense, they do not cause blood glucose levels to rise and are also tooth-friendly. The consumption of artificial sweeteners can help to control obesity and diabetes but their use is often controversial due to suspicions of adverse health effects [1, 2, 5]. Therefore, to prevent potential risk to human health, regulations have been developed for these compounds, setting an upper limit on their concentration in different products, while some of them are even banned [2]. For example, cyclamate and neohesperidine dihydrochalcone are not included on the list of artificial sweeteners allowed in the USA [1, 6]. Furthermore, recent studies have documented their widespread occurrence in the aquatic environment, becoming a new class of emerging organic contaminants (EOCs) in water samples. For these reasons, there is a need for accurate and reliable analytical methods to determine sweeteners not only in food samples, but also in environmental samples [7].
A large number of the existing analytical methods for the determination of artificial sweeteners have been developed for foodstuffs and beverages. However, the sensitivity of these methods is not suitable for environmental samples [2]. Considering that the list of artificial sweeteners that are employed in the industry is long, and some of the compounds are mixed to provide enhanced sweetness or improved taste, the simultaneous determination of several sweeteners in environment water samples is necessary [5, 8]. The first liquid chromatography-tandem mass spectrometry (LC-MS/MS) method to determine a group of seven sweeteners in environmental water samples was presented by Scheurer et al. [1, 5]. Since then, the number of methods developed has increased but the need for validated multi-analyte methods for artificial sweeteners is still unaddressed [8].
The most common approach employed for determining these compounds is LC-MS/MS either directly injecting the sample or after a solid-phase extraction (SPE) procedure [2, 5]. To the best of our knowledge, there are only two methods in the literature that employ hydrophilic interaction liquid chromatography (HILIC) coupled to MS/MS using QqQ as an analyser, and only one of these has actually been validated and applied to environmental water samples [5]. HILIC has been employed for polar and hydrophilic compounds due to the advantages that it offers. This chromatographic mode uses polar stationary phases (providing alternative selectivity to traditional reversed-phase LC) and mobile phases are aqueous/organic mixtures, in which the organic solvent is in a higher proportion. The high organic content of the mobile phase favours its coupling with MS detection because it facilitates spraying and desolvation in the interface, offers low backpressures in the LC system and enables the direct injection of organic extracts. The number of studies that have employed high-resolution mass spectrometry (HRMS) is lower compared to MS/MS, and most of these studies focus only on determining sucralose [9–13]. Using HRMS instruments, accurate masses from both molecular ions and fragments are observed, which is an advantage in complex matrices as a more accurate identification of the target analytes is possible [14].
In this work, the development and validation of a SPE/HILIC-HRMS method is described to determine a large group of commonly used sweeteners in environmental waters. To the best of our knowledge, this is the first HILIC-HRMS method developed for this group of analytes. HILIC conditions are optimized by testing two different stationary phases and different parameters affecting chromatographic separation of the analytes. In addition, SPE conditions are optimized to enable the direct injection of the organic extract into the HILIC system. Finally, the validated method is applied to environmental water samples.
Experimental
Reagents and standards
Potassium acesulfame (ACE), sodium cyclamate (CYC), saccharin (SAC), aspartame (ASP), sucralose (SUC), neohesperidine dihydrochalcone (NHDC), stevioside (STE), and glycyrrhizic acid (GLY) ammonium salt were purchased from Sigma-Aldrich (St. Louis, USA). Stock solutions of 1 mg/mL were prepared by dissolving each solid standard in methanol (MeOH) and were stored at −20 °C. Only in the case of stevioside, a proportion of water (1:10 H2O/MeOH) is needed to completely dissolve the solid. Working solutions of a mixture of all compounds were prepared in acetonitrile (ACN) and were stored at 4 °C in the dark. The chemical structures, CAS number and LogP and pK a values of the analytes are shown in Table 1.
Ultrapure water was provided by a water purification system (Veolia, Sant Cugat del Vallès, Spain) and HPLC-grade MeOH and ACN were purchased from J. T. Baker (Deventer, the Netherlands). Analytical grade ammonium acetate (CH3COONH4) and ammonium formate (HCOONH4) were supplied by Sigma-Aldrich. Acetic acid (CH3COOH) from SDS (Peypin, France), formic acid (HCOOH) from Sigma-Aldrich, hydrochloric acid (HCl) from Scharlab (Barcelona, Spain) and ammonium hydroxide (NH4OH) from Panreac (Barcelona, Spain) were used to adjust the pH of the mobile phase and the samples. Mobile phase was filtered through a 0.22-μm nylon filter (Scharlab) before use.
Sampling
Waste waters were collected from three urban treatment plants located in Tarragona, Reus and Vila-seca, all of which are in the Tarragona region in NE Spain. Influent and effluent samples were collected from Tarragona and Reus sewage treatment plants (STPs), which operate with a primary and a secondary treatment. Vila-seca STP operates with an additional tertiary treatment (a reverse osmosis process) and so influent and effluent samples from this tertiary treatment were also collected. River samples were collected from the Ebre, Segre, Ter, Llobregat and Francolí rivers. Water samples were collected in pre-cleaned bottles and were then stored at −20 °C. Prior to analysis, the samples were filtered through a 0.45-μm glass-fibre filter (Fisherbrand, Loughborough, UK). The pH of the samples was adjusted to three with HCl before the SPE procedure.
Solid-phase extraction
The extraction procedure was adapted from a previous study [15]. Oasis HLB cartridges (500 mg) supplied by Waters (Milford, MA, USA) were employed. STP water samples (50 mL for influent waters and 100 mL for both secondary and tertiary effluent and river samples) adjusted to pH 3 were loaded. A washing step with 10 mL of H2O (pH = 3) was included in order to remove salts and highly polar compounds, followed by drying under vacuum. The analytes were eluted with 5 mL of a mixture of NH4OH:MeOH:ACN (1:4:15). The extract was filtered with a 0.22 μm PTFE syringe filter and directly injected into the HILIC-HRMS instrument.
Hydrophilic interaction LC-high-resolution mass spectrometry
All experiments were performed using an Accela 1250 UHPLC system from Thermo Scientific (Bremen, Germany), equipped with a quaternary pump (1250 bar), an Accela Autosampler automatic injector and a column oven. The LC system was connected to an Exactive OrbitrapTM mass spectrometer from Thermo Scientific. The interface employed was a heated electrospray ionization (HESI) source, operating in negative ionization mode. The instrument was equipped with a HCD collision cell in order to fragment the analytes for confirmation purposes.
The bare silica stationary phase Atlantis HILIC Silica (100 mm × 2.1 mm i.d., 5 μm particle size) supplied by Waters and the zwitterionic sulfoalkylbetaine phase Syncronis (100 mm × 2.1 mm i.d., 5 μm particle size) supplied by Thermo Scientific were tested to evaluate the retention of the sweeteners. The optimal mobile phase was a mixture of solvent A (100 mM HCOONH4/HCOOH buffer adjusted to pH 3.75) and solvent B (ACN), for both silica and zwitterionic stationary phases. The gradient profile started with 98 % ACN and was held for 2 min, before being reduced to 90 % in 3 min, and subsequently reduced to 70 % in 3 min and held for 4 min. A period of 5 min was included in the profile in order to return to the initial conditions, which were then held for 8 min to equilibrate the column for the subsequent analysis. The optimal flow rate was 0.5 mL min−1 and temperature of the column oven was set at 25 °C.
To optimize the HRMS conditions, a solution of each compound was introduced into the source by direct infusion (via syringe pump) together with a flow of mobile phase with 70 % of ACN through a T connection. The signal of the negative molecular ions [M]− or [M-H]− of the analytes was monitored to optimize interface conditions in order to obtain the highest response for all of the compounds. Conditions were optimized in full scan at high resolution (50,000 FWHM) over a mass range of 100 to 1000 m/z. The optimal parameters were: spray voltage, 3.5 kV; sheath gas, 40 AU (adimensional units); auxiliary gas, 5 AU; skimmer voltage, −26 V; capillary voltage, −25 V; tube lens voltage, −90 V; heater temperature, 350 °C; capillary temperature, 300 °C; and probe position adjustment: side to side, 0, vertical C and micrometer, 0.75. Two windows were used in negative mode with different collision voltages for fragmentation. Ions from characteristic fragments (Table 1) of the analytes were selected for confirmation and the HCD cell voltage was optimized to obtain the highest response for the fragment. The first window (0 to 7 min) used a full scan at 50,000 FWHM with 250 ms of injection time over a mass range of 100 to 1000 m/z and a fragmentation scan at 10,000 FWHM with 50 ms of injection time over a mass range of 60 to 900 m/z at 20 eV in the HCD cell. The second window (7–25 min) used the previous scan events plus a third scan of fragmentation at 40 eV in the HCD cell using the previous fragmentation parameters. The molecular ions were measured for quantification (with a mass extraction window of 5 ppm) and fragments and the corresponding ion ratios were used for confirmation purposes. All of this information is shown in Table 1.
Results and discussion
Optimization of HILIC-HRMS conditions
The HILIC columns tested were a bare silica stationary phase and a zwitterionic material which has a quaternary amine and a sulfonic acid group in the same ligand, separated by a short alkyl chain. These two columns were tested because they have different chemistry and are commonly used in HILIC applications. For both stationary phases, a HCOONH4/HCOOH buffer adjusted to pH 3.75 and a CH3COONH4/CH3COOH buffer adjusted to pH 4.75 were tested at different salt concentrations (10, 50 and 100 mM). Additionally, a 100 mM HCOONH4/HCOOH solution adjusted to pH 4.75 and a 100 mM CH3COONH4/CH3COOH buffer adjusted to pH 3.75 were also assayed. In the case of the bare silica phase, a strong distortion and broadening of the peak corresponding to NHDC was observed for all mobile phase conditions, except for the 100 mM HCOONH4/HCOOH buffer. Under these conditions, higher retention of all the analytes was observed in comparison with lower concentrations of the same buffer, except for ASP. Furthermore, higher retention of all the analytes and best peak shape for NHDC was observed for the 100 mM HCOONH4/HCOOH buffer adjusted to pH 3.75 compared to the same buffer adjusted to pH 4.75. However, for all of the conditions tested, ACE and SAC were poorly retained and separation between STE and ASP was not satisfactory enough.
Meanwhile, when the zwitterionic sulfoalkylbetaine stationary phase was tested with buffers at high concentrations, peak shape and retention of the analytes were better. The optimal pH value for 100 mM buffers was that which had the highest buffering capacity for each buffer. When comparing buffer type, higher retention and better peak shape were observed for HCOONH4/HCOOH solutions. For the reasons stated above, a 100 mM HCOONH4/HCOOH buffer adjusted to pH 3.75 was selected as optimal. Applying these mobile phase conditions, flow rate and temperature were optimized by comparing 0.3, 0.5, 0.8 and 1.0 mL min−1 and 25, 35, 45 and 55 °C. A flow of 0.5 mL min−1 was selected because, at higher flow rates, ACE and CYC overlapped and ASP showed peak distortion, while at 0.3 mL min−1, analysis time was longer. The optimal temperature was 25 °C because higher temperatures caused overlapping and peak distortion. A chromatogram of the final separation is shown in Fig. 1.

Chromatogram of the separation of a mixture of the analytes under optimal conditions
In general, retention was higher in the zwitterionic stationary phase compared to the silica material. Better separation between ASP and STE was achieved. Surprisingly, the elution order was practically the same, which might indicate that water layer conditions are more important than direct interaction between the analytes and the phase with respect to retention.
Better results were obtained compared to a previous study in which the HILIC column employed (the solid core bare silica Ascentis Express HILIC) did not give greater separation than that observed for the RP stationary phases evaluated, because four of the compounds eluted at void volume for the mobile phase conditions employed [15]. When comparing the present separation with the RP separation obtained in that study, a shorter analysis time was observed for the HILIC approach.
Optimal HRMS parameters (described in “Hydrophilic interaction LC-high-resolution mass spectrometry”) were chosen as a compromise between the highest responses obtained for each analyte. Ranges tested for each parameter were: spray voltage, 2 to 5 kV; sheath gas, 20 to 80 AU; auxiliary gas, 0 to 40 AU; skimmer voltage, −5 to −50 V; capillary voltage, 10 to 100 V; tube lens voltage, −50 to −200 V; heater and capillary temperature, 200 to 400 °C and probe position adjustment including side to side position, −1 to +1, vertical, C or D and micrometer, 1–2. The range of HCD cell collision voltage tested was 5 to 70 eV. With respect to optics, tube lens voltage was the parameter that affected the response of the analytes most differently and for which the selection of a compromise was hardest. For the gases flow rate optimization, mobile phase flow rate requirements were considered together with the effect on the response, which was similar for all of the compounds.
The entire acquisition was carried out in negative mode because negative molecular ions [M]− or [M-H]− were selected for quantification. STE displayed a formate adduct [M + HCOO]−, as HCOONH4 was added to the mobile phase. For this compound, the adduct was selected for quantification as its response was more abundant than [M-H]−.
With respect to fragmentation, some fragments were clearly observed at low collision voltages (10 to 20 e.V.), while others were present at higher energies (>35 e.V.), as is the case for STE and NHDC. For this reason, two scans were selected at two different collision voltages (20 and 40 e.V.), which were selected as a compromise between the highest responses of all the fragments present at each voltage. One fragment was selected for each analyte, which results in at least four identification points, fulfilling European guidelines for residues [16]. Details of the selected fragments are shown in Table 1. For ACE, CYC and SAC, sulphur fragments (SO3 for example) were selected. Even when these fragments are less characteristic, the response obtained was high, enabling distinction from noise. For NHDC and STE, more characteristic fragments were observed because of their more fragmentable structure. The fragment selected for STE might be the result of losing one of the six atom rings substituted with four hydroxyl groups. In the case of NHDC, the fragment consists of the two aromatic rings of the structure, as the molecule breaks from the two hydroxyl substituted six atom rings through the carbon oxygen bond. These fragments have been observed and reported in the literature when MS/MS is employed [7, 8, 17–19].
Under these conditions, low instrumental LODs (0.02–10 μg/L) and LOQs (0.05–15 μg/L) were achieved. The limits obtained are in the same order of magnitude as those in other studies where MS/MS with QqQ was employed [2, 5]. The highest limits were obtained for SAC but only because the selected fragment ion did not present a high response compared to the molecular ion.
Optimization of the SPE
For the SPE procedure, conditioning and loading at pH 3 with HCl was adapted from a previous study in which satisfactory results were reported when using Oasis HLB cartridges [15]. However, elution was optimized for the organic extract to be compatible with the HILIC mobile phase. Thus, the idea was to eliminate the evaporation step and take advantage of the compatibility of HILIC with the organic extract. For this reason, the suitability of the injecting solvent was tested, as peak distortion or changes in retention time may be observed. It was observed that a percentage of MeOH higher than 25 % in the sample solvent caused peak distortion in the HILIC separation. Thus, the use of MeOH solutions required subsequent evaporation to redissolve the sample in ACN. Therefore, 25 mL of a solution with the analytes were loaded into the cartridge and then eluted with 5 mL of different solutions: (1) ACN, (2) 10 % NH4OH/ACN, (3) 1:9 MeOH:ACN and (4) 1:5:16 NH4OH:MeOH:ACN. Other solutions for which evaporation would be needed were also tested for comparison purposes: (5) MeOH, (6) 5 % NH4OH/MeOH, (7) 10 % NH4OH/MeOH.
The use of pure MeOH or ACN or a mixture of the two was not sufficient to elute all of the analytes. The use of NH4OH was needed to achieve higher recoveries for NEO and STE. This can be explained by the formation of hydrogen bonds between the sorbent and the hydroxyl groups of these two compounds, which is only possible when the –OH is in its protonated form, as sorbent polar functionalities do not have acidic hydrogens, which are not later present at basic pH. Thus, NH4OH/MeOH and NH4OH/ACN mixtures were the best option. Of the mixtures mentioned above, ACN solution does not need evaporation and, for this reason, it was selected for eluting the analytes from the cartridge. The only care that must be taken when using this elution solvent is that ASP is not stable in the solution more than 1 h, so the extract must be injected immediately. Alternatively, dry cartridges loaded with sample can be stored at −4 °C when preservation in the middle of the process is needed. In addition, the solubility of GLY in organic solvents is limited so it is preferable to include a portion of water when injected into the LC-HRMS system. However, solutions with this proportion of water distorted the peak shape of the rest of the compounds. As an alternative, a portion of the extract can be evaporated and redissolved in a mixture of H2O:MeOH (1:4) with the same volume. However, considering that using this alternative involves injecting the redissolved solution for the sole purpose of quantifying this analyte (since the peaks of some of the other compounds would be distorted), it was decided to eliminate this compound from the target list of the present study. In any case, the chromatographic separation proposed herein is suitable for separating GLY. If GLY determination is desired, the present procedure can be used by changing the injection solvent, as explained above.
Several volumes of eluting solvent (2 to 10 mL) were tested in order to optimize this parameter. It was found that 5 mL of NH4OH:MeOH:ACN (1:5:16) was optimal for completely eluting the analytes, matching the volume randomly used for the optimization of other SPE parameters. For optimization of the washing step, the solutions tested were (1) 10 % MeOH in H2O, (2) H2O adjusted to pH 3 and (3) H2O without adjusting the pH. The best option was the ultrapure water adjusted to pH 3 because losses of analytes were not observed and the matrix effect was lower, probably due to the elimination of salts and highly polar compounds from the matrix [20].
The sample volume was optimized by passing 50, 100 and 250 mL through the cartridges. Up to 250 mL of H2O could be passed through the cartridges, obtaining recoveries greater than 77 % for all of the compounds, except for ASP for which 61 % is recovered. However, recoveries of ACE, CYC and ASP for this volume (77 % for both ACE and CYC, and 61 % for ASP) were slightly lower compared to 100 mL (100 and 97 % for ACE and CYC, respectively, and 76 % for ASP), due to losses through percolation, because these analytes are quite polar. For this reason, higher volumes were not tested further. When extracting 100 mL of the samples, recoveries for river samples and secondary effluent waste waters were satisfactory, unlike primary influent waters, for which 50 mL were selected to reduce the high matrix effect found. Extraction recoveries (%R SPE) were higher than 76 % for secondary effluent water and higher than 74 % for all of the compounds in primary influent water (Table 2). %R apparent for primary influent waters ranged from 51 % (SAC) to 101 % (ASP), while values ranged from 57 % (ACE) to 74 % (SAC) and from 69 to 92 % for secondary effluent and river water samples, respectively. Apparent recoveries (%R apparent) shown in Table 2 were calculated from the interpolation of the signal of a sample spiked at the beginning of the analytical procedure in a calibration curve prepared in NH4OH:MeOH:ACN (1:4:15).
The matrix effect (%ME) shown in Table 2 was calculated from the interpolation of the signal of an extracted sample spiked just before injection in the calibration curve mentioned. The concentration obtained (C exp) from this calibration curve was replaced in the following formula %ME = (C exp/C theo × 100 %) − 100 %. When calculated this way, ME is expressed as the percentage of response that increases or decreases. It must be pointed out that unspiked samples were always considered to subtract blank signals.
In the case of primary influent waters, the ME ranged from −42 to 2 % (Table 2), while for secondary effluent and river waters, it ranged from −40 to −21 % and from −43 to 5 %, respectively. Values indicated the presence of ion suppression due to the complexity of the matrices studied. Enhancement of the signal was scarcely observed in contrast to the results of the HILIC method reported by Kokotou et al. [5], in which all analytes displayed response enhancement, except for neotame (not included in the present study), SAC and CYC [5]. Even when ion suppression was observed, the ME was lower than in other studies. For instance, in a reversed-phase liquid chromatography (RPLC) approach, Scheurer et al. [1] reported ME values between −77 and 56 % for effluent waters when 50 mL was percolated and later reconstituted with 0.5 mL of a 20 mM CH3COONH4 in 4:1 H2O:ACN solution. ME values reported by Ordoñez et al. [2] ranged from −93 to −31 % when 50 mL of influent water was preconcentrated to 2 mL and then injected into a HILIC system. The RPLC approach of the same study gave ME values similar on average to the percentages obtained in the present method (Table 2).
Method validation
Validation of the method proposed was performed by evaluating the linear range, LOQ, LOD and precision (intra-day and inter-day) of the results obtained.
Precision of the concentration values were expressed as relative standard deviation (%RSD, n = 5). Extractions were assessed for two levels of concentration within the same day (intra-day) and on consecutive days (inter-day) for the river samples and at one concentration for the primary influent and secondary effluent samples, due to the ubiquity of some of the compounds in these matrices at relatively high concentrations. Repeatability (Table 2) expressed as %RSD (n = 5) evaluated on the same day and on consecutive days was <25 % in all cases.
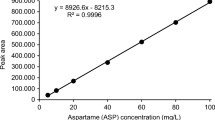
In order to correct the ME, matrix-matched calibration was used for all of the compounds except for ACE, which was present at relatively high concentrations in all of the samples analysed. Due to ACE ubiquity in the samples, quantification at low levels with the matrix-matched calibration was not possible, so an external calibration was considered. For the matrix-matched calibration curves, the optimal volume for each sample (influent, effluent and river water) was spiked with different concentrations, extracted and subsequently injected. Five points were considered with spiked concentrations ranging between LOQs and 100 μg/L. Matrix-matched calibration curves showed satisfactory determination coefficients (0.9903 < R 2 < 0.9998) when the linearity of the response was evaluated.
Limits were established as the concentration at which the signal-to-noise ratio was 3 and 10 for the LODs and the LOQs, respectively. When noise was not observable, a signal of magnitude ∼5 × 103 was selected as the LOD, and LOQs were selected as the lowest point of the calibration curve. Detection of the selected confirmatory fragment was always taken into account. Due to the presence of some of the analytes in the blanks, determination of LODs and LOQs of these compounds was difficult. In these cases (ACE, CYC, SAC and SUC), LODs and LOQs were estimated from the instrumental limits taking into account the %R apparent, and results are shown in Table 3. For primary influent and secondary effluent waters, LODs ranged between 0.002 and 0.7 μg/L, with LOQs between 0.004 and 1.5 μg/L. Lower values were obtained for river water samples, with LOQs not exceeding 0.5 μg/L. These values are of the same order of magnitude as those reported in other studies [14, 19, 21]. Another study could not be compared with the values presented above because validation was performed for tap water only [1].
Application to environmental samples
Five samples of both primary influent and secondary effluent STP waters, three samples of both influent and effluent waters of the tertiary process, and five samples of river waters were analysed in duplicate. Detailed information of the concentrations obtained can be found in Table 4. ACE and SUC were quantified in all of the influent waters in both the primary and tertiary samples. CYC and SAC were found in half of the primary influent samples from the Reus and Tarragona treatment plants. Table 4 also shows the mass error for confirmation purposes and as it can be seen it was lower than 5 ppm in all cases. Levels reported in this paper are similar to those reported in the literature. Kokotou and Thomaidis [5] reported concentration ranges from 12 to 25 μg/L for ACE, 15 to 25 μg/L for SUC, 6 to 58 μg/L for CYC and 15 to 45 μg/L for SAC in influent waters from STPs in Athens. Ordoñez et al. [2] reported levels ranging from 25.0 to 26.7 μg/L for ACE, 3.0 to 5.3 μg/L for SUC, 25.9 to 36.4 μg/L for CYC and 18.4 to 22.3 μg/L for SAC in waste waters from NW Spain. Scheurer et al. [1] reported concentrations ranges from 34 to 50 μg/L for ACE and SAC, up to 190 μg/L for CYC and below 1 μg/L for SUC in influent waste waters from Germany. Other similar results can be found in several studies [19, 21, 22]. The results of the present study were slightly different from those obtained in a previous work, in which the levels reported for ACE, SUC and SAC were higher and CYC concentrations were lower [15]. This can be attributed to differences in the sample collection period or differences in the consumption of the sweeteners. A chromatogram of an influent water sample is shown in Fig. 2 where the concentration and mass error for each compound is also detailed.

Chromatogram obtained when a blank influent water sample (Influent Reus) was analysed by SPE/HILIC-HRMS. Left molecular ions. Right fragments
In secondary effluents, concentrations ranged from 5.4 to 16.6 μg/L for ACE and 2.7 to 6.7 μg/L for SUC. CYC and SAC were not found in secondary effluent waters, which might be attributed to the removal of these two compounds during the treatment process. This fact has been reported in several studies [1, 5, 22]. In the case of ACE and SUC, they were poorly eliminated in the treatment process. The results obtained in this study regarding elimination of ACE and SUC are consistent with literature [1, 13, 23, 24].
With regard to river waters, in all of the samples analysed, ACE, CYC, SAC and SUC were detected below LOQs. ASP, NHDC and STE were not detected in any sample. Only one river sample from the Llobregat River displayed quantifiable levels of ACE, CYC and SUC between 0.03 and 0.57 μg/L. The presence of ACE and CYC in the river may be explained by an accumulation of the low quantities coming from STPs or direct industrial disposal. These levels are similar to those found in some studies. For instance, Gan et al. [17] found concentrations of ACE between 2.7 and 4.7 μg/L and between 0.02 and 0.7 μg/L for the rest of the compounds. Other studies have reported values ranging from 0.05 to 5.8 μg/L [1, 2]. In a European screening of SUC including 120 river samples in 23 countries, the concentration of this sweetener was confirmed with concentrations up to around 1 μg/L [11].
Conclusions
A method was successfully developed and validated for the determination of a group of artificial sweeteners employing HILIC coupled to HRMS in river and waste water samples. Stevioside, which has never been determined in multi-analyte methods, was included in the method. The zwitterionic stationary phase gave better retention for ACE and SAC compared to the bare silica material under optimized conditions.
Apparent recoveries (overall method) achieved were higher than 51 % using Oasis HLB cartridges with a satisfactory matrix effect. Fast sample treatment was possible, as cartridges were eluted with a mixture of NH4OH:MeOH:ACN (1:4:15) and directly injected into the HILIC-HRMS system achieving great detection and quantification limits.
Four analytes (ACE, CYC, SAC and SUC) were found in different samples from the Tarragona area, mostly present in influent samples.
References
Scheurer M, Brauch H-J, Lange FT (2009) Anal Bioanal Chem 394(6):1585–1594
Ordóñez EY, Benito Quintana J, Rodil R, Cela R (2013) J Chromatogr A 1320:10–16
Morlock GE, Meyer S, Zimmermann BF, Roussel J-M (2014) J Chromatogr A 1350:102–111
Chen X-H, Zhao Y-G, Shen H-Y, Jin M-C (2012) J Chromatogr A 1263:34–42
Kokotou MG, Thomaidis NS (2013) Anal Methods 5(16):3825–3833
Zygler A, Wasik A, Namiesnik J (2009) TrAC Trend Anal Chem 28(9):1082–1102
Lange FT, Scheurer M, Brauch H-J (2012) Anal Bioanal Chem 403(9):2503–2518
Perkola N, Sainio P (2014) Environ Pollut 184:391–396
Neset T-SS, Singer H, Longree P, Bader H-P, Scheidegger R, Wittmer A, Andersson JCM (2010) Sci Total Environ 408(16):3261–3269
Ferrer I, Zweigenbaum JA, Thurman EM (2013) Anal Chem 85(20):9581–9587
Loos R, Gawlik BM, Boettcher K, Locoro G, Contini S, Bidoglio G (2009) J Chromatogr A 1216(7):1126–1131
Mawhinney DB, Young RB, Vanderford BJ, Borch T, Snyder SA (2011) Environ Sci Technol 45(20):8716–8722
Batchu SR, Quinete N, Panditi VR, Gardinali PR (2013) Chem Cent J 7
Ferrer I, Thurman EM (2010) J Chromatogr A 1217(25):4127–4134
Arbeláez P, Borrull F, Pocurrul E, Marcé RM (to be submitted)
Commission Decision (EU) of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results, 2002 L221:8
Gan Z, Sun H, Wang R, Feng B (2013) J Chromatogr A 1274:87–96
Berset J-D, Ochsenbein N (2012) Chemosphere 88(5):563–569
Ngoc Han T, Hu J, Ong SL (2013) Talanta 113:82–92
Fontanals N, Marcé RM, Borrull F (2011) J Chromatogr A 1218(35):5975–5980
Wolf L, Zwiener C, Zemann M (2012) Sci Total Environ 430:8–19
Buerge IJ, Buser H-R, Kahle M, Mueller MD, Poiger T (2009) Environ Sci Technol 43(12):4381–4385
Scheurer M, Storck FR, Brauch H-J, Lange FT (2010) Water Res 44(12):3573–3584
Sang Z, Jiang Y, Tsoi Y-K, Leung KS-Y (2014) Water Res 52:260–274
Acknowledgments
The authors would like to thank the Ministry of Economy and Competitiveness (Project CTQ2011-24179) for the financial support given. D. Salas would also like to thank the Ministry of Economy and Competitiveness for a grant (BES-2012-057792).
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the topical collection Advances in LC-MS/MS Analysis with guest editors Damià Barceló and Mira Petrovic.
Rights and permissions
About this article

Cite this article
Salas, D., Borrull, F., Fontanals, N. et al. Hydrophilic interaction liquid chromatography coupled to high-resolution mass spectrometry to determine artificial sweeteners in environmental waters. Anal Bioanal Chem 407, 4277–4285 (2015). https://doi.org/10.1007/s00216-014-8330-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-014-8330-6