
Abstract
A method for the simultaneous determination of seven commonly used artificial sweeteners in water is presented. The analytes were extracted by solid phase extraction using Bakerbond SDB 1 cartridges at pH 3 and analyzed by liquid chromatography electrospray ionization tandem mass spectrometry in negative ionization mode. Ionization was enhanced by post-column addition of the alkaline modifier Tris(hydroxymethyl)amino methane. Except for aspartame and neohesperidin dihydrochalcone, recoveries were higher than 75% in potable water with comparable results for surface water. Matrix effects due to reduced extraction yields in undiluted waste water were negligible for aspartame and neotame but considerable for the other compounds. The widespread distribution of acesulfame, saccharin, cyclamate, and sucralose in the aquatic environment could be proven. Concentrations in two influents of German sewage treatment plants (STPs) were up to 190 μg/L for cyclamate, about 40 μg/L for acesulfame and saccharin, and less than 1 μg/L for sucralose. Removal in the STPs was limited for acesulfame and sucralose and >94% for saccharin and cyclamate. The persistence of some artificial sweeteners during soil aquifer treatment was demonstrated and confirmed their environmental relevance. The use of sucralose and acesulfame as tracers for anthropogenic contamination is conceivable. In German surface waters, acesulfame was the predominant artificial sweetener with concentrations exceeding 2 μg/L. Other sweeteners were detected up to several hundred nanograms per liter in the order saccharin ≈ cyclamate > sucralose.

Some artificial sweeteners are excreted unchanged and in particular acesulfame is a perfect tracer for municipal waste water
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Artificial sweeteners (Table 1) are used as sugar substitutes in remarkable amounts in food and drinks but also in drugs and sanitary products [1]. They provide no or insignificant energy as they are not decomposed as carbohydrates. Insulin level is not affected by their consumption, which makes them suitable for diabetics. They can help to reduce calorie intake as their sweetness is much higher than that of table sugar. Consequently, these “high-intensity sweeteners” are used in comparably small amounts, making the calorie intake insignificant, even for those sweeteners that are metabolized [2]. Artificial sweeteners can prevent potential dental caries as most of them are not metabolized like sugars or only fermented slightly by the mouth microflora [3, 4]. They can develop an artificial, metallic, or licorice-like aftertaste. Therefore, they often can be found blended in food to overcome this disadvantage.
Since the beginning of use, there is an ongoing discussion about potential health risks of artificial sweeteners in gray literature as well as on a scientific base. Numerous internet forums, newspaper reports, and scientific publications deal with possible risks and other safety issues [1, 2, 5–7].
Five artificial sweeteners are approved by the US Food and Drug Administration (FDA) and are “generally recognized as safe” (GRAS) in the USA: acesulfame-K, aspartame, neotame, saccharin, and sucralose [8]. In the European Union, the use of neotame in foodstuffs is not allowed, but contrary to the USA, neohesperidin dihydrochalcone (NHDC) and cyclamate are additionally approved [9, 10].
Cyclamate is banned in the USA since 1970. Oser et al. [11] accused cyclamate of causing bladder cancer in rats, which prompted the US Department of Health, Education and Welfare to remove cyclamate from the GRAS list. Further studies, however, did not show any relation between cyclamate and cancer. Cyclamate is still banned in the USA but waiting for its reapproval by the FDA [12].
Acesulfame is commercially used as potassium salt and also known as acesulfame-K. It is 200 times sweeter than table sugar and provides the common benefits of artificial sweeteners mentioned above but has also a bitter aftertaste. Acesulfame-K is used in about 90 countries and, according to Kroger and co-authors [2], no health problems associated with its consumption have been reported in scientific literature. However, in 2008, a study was published where DNA damage due to acesulfame exposure was reported. The authors suggested restricting the use of some artificial sweeteners [6]. Acesulfame-K, cyclamate, and saccharin are excreted mainly unchanged through the renal system [13].
In the 1980s, studies showed an increasing risk for bladder cancer in rats when applying high doses of saccharin in the animals' diet [14, 15]. Therefore, saccharin was prohibited in Canada. In the USA, products containing saccharin had to be labeled with a warning that saccharin “has to be determined to cause cancer in laboratory animals.” In 2001, saccharin was removed from the list of potential carcinogens in the USA as the mechanism causing bladder cancer in rats is not relevant for humans [16]. In Canada, authorities have received a submission to reinstate saccharin as a food additive, and in the EU, an acceptable daily intake of 0–5 mg/kg body weight is approved [17].
Aspartame provides, like sugar, 4 cal/g. Since it is about 180 times sweeter than sugar, only small amounts of aspartame are needed to sweeten food and drinks. Contrary to acesulfame, aspartame is not heat-stable and degrades in liquids when stored over a longer period of time. Aspartame is made up of phenylalanine, aspartic acid, and methanol. For people with a seldom genetic disorder, the generated phenylalanine does carry some risk as their body cannot metabolize the degradation product. As a consequence, all products containing aspartame have to be labeled to point out the presence of a phenylalanine source. In scientific literature, aspartame is the most controversially discussed artificial sweetener regarding health aspects. Numerous publications with contrary results about possible adverse effects of aspartame like neurological disturbances [18–20] or even cancer in rats [21, 22] are available. Nevertheless, FDA and the European Union consider the compound as safe based on toxicological and clinical studies.
One of the latest outcomes of the research for new artificial sweeteners is neotame. Its structure is closely related to aspartame on which a branched hydrocarbon chain is attached. It is 7,000 to 13,000 times sweeter than sucrose, and like aspartame, it is metabolized, but phenylalanine release is insignificant. Products containing neotame are not required to be labeled as possible phenylalanine sources [23].
Neohesperidin dihydrochalcone is about 1,500 times sweeter than sugar but is also used as a flavor enhancer. It is produced by hydrogenation of a flavonoid found in citrus fruits. To overcome its licorice and menthol-like aftertaste, it is often found blended with other artificial sweeteners. NHDC is metabolized by intestinal microflora and excreted via urine [24]. Antioxidant properties have been proven [25].
Sucralose has a disaccharide structure where three hydroxyl groups are replaced by chlorine atoms. It is thermally stable and excreted unchanged with the feces [26]. More than hundred safety studies have been conducted on sucralose and proved its safety for human consumption [5], but it is also discussed as a migraine trigger [27, 28]. Due to its half-life in water of several years and a missing environmental review, its relevance in the aquatic environment is discussed. The compound is reported to pass sewage treatment plants (STP) and was found up to several micrograms per liter in STP influents and effluents and up to several hundred nanograms per liter in surface waters in Sweden [29]. In an EU-wide monitoring program, concentrations up to 1 μg/L sucralose were found in European surface waters. The compound was mainly detected in samples from Western Europe and Scandinavia. In samples from Germany and Eastern Europe, minor concentrations were reported [30].
Findings of sucralose suggest its main distribution in Western Europe, likely based on the use of other artificial sweeteners in countries where sucralose is not predominant. If excreted unchanged and if artificial sweeteners should prove to be persistent during waste water treatment, their ubiquitous distribution in the aquatic environment is likely. Robust analytical methods for clarifying their environmental fate are crucial. To the best of our knowledge, there is no scientific report on occurrence of artificial sweeteners in the aquatic environment other than for sucralose. This paper intends to provide first information on that topic.
Materials and methods
Chemicals
All reference compounds (Table 1) were of high purity (>98%). Acesulfame potassium, saccharin, aspartame, and sucralose were purchased from Dr. Ehrenstorfer GmbH (Augsburg, Germany), sodium cyclamate from Supelco (Bellefonte, PA, USA), neotame from USP Reference Standards (Rockville, MD, USA), neohesperidin dihydrochalcone from European Pharmacopoeia Reference Standard (Strasbourg, France), and sucralose-d6 from Campro Scientific GmbH (Berlin, Germany). Individual stock solutions were prepared by dissolving the compounds in methanol. Concentrations of the stock solutions were between 0.2 to 0.8 g/L. All stock solutions were stored at −18 °C. Standard mixtures containing all analytes were prepared by diluting the stock solutions with methanol to concentrations of 0.1 and 0.01 mg/L. A standard solution of sucralose-d6 was prepared by diluting the stock solution with methanol to a concentration of 0.1 mg/L.
High-performance liquid chromatography (HPLC)-grade methanol and acetone as well as formic acid and hydrochloric acid (32%) were supplied by Merck (Darmstadt, Germany). Purities of all organic solvents were higher than 99.8%. Ammonium acetate (purity >98%) was purchased from Sigma-Aldrich (Steinheim, Germany) and Tris(hydroxymethyl)amino methane (TRIS) from Carl Roth GmbH (Karlsruhe, Germany). Ultrapure water was provided by an Arium 611 laboratory water purification system (Sartorius AG, Göttingen, Germany).
The nitrogen used for drying the solid-phase cartridges and for evaporation of solvents was of 99.999% purity and was purchased from Air Liquide (Düsseldorf, Germany).
Sampling sites and protocol
The STP in Eggenstein-Leopoldshafen (STP 1) is applying conventional, i.e., mechanical and biological treatment. It has a capacity of 20,000 population equivalents (PE) with about 15,000 inhabitants living in the catchment area and treats ca. 2,500 to 3,500 m3 per day of domestic waste water. Hydraulic retention time is about 5 hours with an average sludge retention time of 20 days. The STP of the city of Karlsruhe (STP 2) treats 40 million cubic meters (capacity 875,000 PE) per year for about 350,000 inhabitants. It is applying mechanical treatment with additional phosphate precipitation, followed by biological treatment with a denitrification/nitrification unit, equipped with a trickling filter. Hydraulic retention time is about 1 day for dry weather conditions. Samples for both STPs were corresponding 24-h composite samples.
The soil aquifer treatment (SAT) site is located in a Mediterranean country and treats secondary effluent from a STP that processes over 100 million cubic meters waste water per year. Treatment includes primary mechanical treatment (bar screen and grit removal) followed by conventional activated sludge treatment including nitrification/denitrification and a limited biological phosphorous removal. The secondary STP effluent is spread in percolation basins, where infiltration through an unsaturated zone, up to 40 m in depth, takes place. The effluent flows lateral in the saturated zone to observation and recovery wells located in a circle of up to 1,000 m in the periphery of percolation basins. Influence of waste water was assessed using chloride as hydrogeological tracer. The residence time in the aquifer exceeds 1.5 years. The recharge operation is carried out by intermittent flooding up to 1 day and 2 days drying. Dilution with local groundwater is very limited. Samples from the SAT site were the STP effluent used for aquifer recharge, a sampling point located vertically below the percolation basin (well 1), two sampling points in the periphery (well 2 and well 3), and one from a private drinking water well supposed not to be influenced by waste water.
Grab samples from major German rivers were collected in 1 L brown glass bottles. If no immediate analysis was possible, samples were stored at 4 °C in the dark for a maximum of 3 days after sampling. No preservation agents were added. Filtration proved to be unnecessary in preliminary tests.
Solid-phase extraction
For solid-phase extraction (SPE), styrol-divinylbenzene cartridges were used (Bakerbond SDB 1, 200 mg/6 mL from J.T. Baker, Deventer, The Netherlands). Other cartridges tested were Isolute ENV+ and C18 material (IST, Mid Glamorgan, UK), Varian Bond Elut PPL (Varian, Lake Forest, CA, USA), Strata X and Strata X-AW (Phenomenex, Aschaffenburg, Germany) and Oasis HLB, WAX, MAX, and MCX (Waters, Milford, MA, USA). For detailed results for all cartridges tested, see Table S1, Electronic Supplementary Material.
A vacuum manifold set from IST equipped with 60 mL reservoirs from Supelco (Bellefonte, PA, USA) was used for SPE. Prior to SPE, the sample pH was adjusted with hydrochloric acid. Several sample pH values were tested to optimize the SPE procedure. For method optimization and validation, water samples (50 mL) were spiked with known amounts of each analyte. Cartridges were conditioned with 3 × 3 mL of methanol followed by 3 × 3 mL of ultra-pure water set to the pH of the corresponding sample. Subsequently, the water samples were passed through the cartridges, and the loaded sorbent materials were completely dried by a gentle nitrogen stream. If the sorbent had no anion or cation exchange capacity, the analytes were eluted with 3 × 3 mL of methanol. The strong cation exchanger material (MCX) and the weak anion exchanger materials (X-AW and WAX) were eluted with methanol/NH4OH (98:2, v/v). The weak cation exchange material Strata X-CW was eluted with 2% formic acid in methanol (v/v). SPE extracts were evaporated to dryness in a stream of nitrogen and reconstituted with 400 μL of solvent A and 100 μL of solvent B used for liquid chromatography.
Liquid chromatography electrospray tandem mass spectrometry analysis
Liquid chromatography (LC) analysis was carried out using a model 1200 SL HPLC system from Agilent Technologies (Waldbronn, Germany) equipped with a solvent cabinet, a micro vacuum degasser, a binary pump, a high-performance autosampler with two 54 vial plates, and a temperature-controlled column compartment.
Several reversed phase (RP) and hydrophilic interaction chromatographic columns were tested. Chromatographic retention and separation was achieved using a Zorbax Eclipse XDB-C8 column (150 × 4.6 mm; 5 μm) from Agilent Technologies connected to a C18 guard column (4 × 2 mm) from Phenomenex. The extra-dense bonding (XDB) of organo-silane ligands and the double endcapping deactivates the column's silica and makes it especially useful for retention of highly polar compounds in RP liquid chromatography. For separation, a gradient consisting of (A) 20 mM ammonium acetate in water and (B) 20 mM ammonium acetate in methanol was used. Acetonitrile was also suitable for chromatographic separation, but with respect to the current worldwide shortage of acetonitrile, methanol was used for method development. The gradient program started with 98% of eluent A, decreased to 25% A in 13 min, kept isocratic for 4 min, and then returned to initial conditions within 1 min. Before each injection, the column was re-equilibrated for 7 min. Separation took place at 40 °C with a flow rate of 0.8 mL/min. This flow was chosen for optimum chromatographic performance during method development and kept constant thereafter. For the purpose of independent confirmation, a Hypercarb column (150 × 2.1 mm; 5 μm) from Thermo Fisher Scientific (Waltham, MA, USA) was used as a second chromatographic column.
Injection volume was 15 μL, and injector needle and injection port were automatically washed with methanol after each injection to avoid potential carryover. Instrumental and sample preparation contaminations were controlled by measuring injector and SPE blanks at regular intervals of every ten injections.
The HPLC system was connected to an API 4000 Q-Trap triple-quadrupole mass spectrometer (Applied Biosystems/MDS Sciex Instruments, Concord, ON, Canada) with an electrospray interface operated in negative ionization mode. Retention time (RT) windows were defined for every single compound in order to use dwell times which enabled optimized peak-to-noise ratios. All RT windows were set to the following mass spectrometer (MS) parameters: ion spray voltage, −4.5 kV; heater temperature, 550 °C; collision gas, medium; ion source gas 1/2, 60/75 psi, and curtain gas, 40 psi. Outside the RT windows, solvent flow was directed to waste to prevent the interface from any unnecessary contamination.
The two most intensive transitions between precursor ion and product ions were used for identification and quantification in multiple reaction monitoring (MRM) mode. For cyclamate, only one transition could be obtained. For sucralose, only the isotopic pattern obtained two transitions adequate as qualifier and quantifier. For the results presented, the average of the concentrations calculated for the two transitions (where possible) is reported. Declustering potential, collision energy, and cell exit potential were optimized for each ion transition. Results of this optimization procedure are summarized in Table 2. Analyst 1.4 software was used to record and evaluate the obtained chromatographic data.
Quantification
For the quantification of sweetener levels, both environmental samples and fortified tap water samples for calibration were subjected to the entire analytical procedure. Surface water samples were analyzed as sampled. STP influent samples were diluted at least by a factor of 10 and STP effluent samples at least by a factor of 5 with Karlsruhe tap water to obtain a matrix, which approximately matches the calibration matrix. All sweeteners, except for sucralose, were quantified by external standard calibration of the entire analytical procedure. The results of the external standard evaluation were not corrected for recoveries deviating from 100%. A similar approach was applied by Hernando et al. [31] for the analysis of beta blockers and lipid lowering agents from waste water. These authors demonstrated that a dilution of 1:5 (v/v) and 1:10 (v/v) resulted in a complete elimination of ion suppression. If weak matrix effects could not have been avoided by sample dilution, the reported environmental levels in this study represent minimal values. Sucralose was quantified by internal standard calibration. For this purpose, the original or diluted water samples were spiked at a level of 200 ng/L of the internal standard (IS) sucralose-d6 prior to SPE. By this means, matrix effects affecting the quantification of sucralose were corrected. The other analytes under investigation possess significantly different chemical structures compared to sucralose and elute at different retention times (eluent compositions) and, thus, under different ionization conditions. Therefore, sucralose-d6 was not used as an IS for other sweeteners than sucralose.
Method validation
An external calibration with directly injected standards ranging from 0.1 ng/mL to 1 μg/mL was set up to determine linearity of detection. An 11-point calibration curve from 1 to 1,500 ng/L was established with spiked tap water samples (50 mL, Karlsruhe tap water, free of any contamination with artificial sweeteners), which were subjected to the entire analytical protocol including SPE. For quantification in the lower range of the calibration, the highest calibration points were excluded. The limits of detection (LOD) and limits of quantification (LOQ) were calculated as three or six times the signal-to-noise ratio, respectively. If waste water samples or other highly contaminated samples were diluted, the reduced sample volume was taken into account when calculating the LOQ.
Recoveries were determined for drinking water (Karlsruhe tap water), surface water (Rhine river at Karlsruhe), and waste water (municipal STP effluent of STP 1) at two different levels, one in the lower (200 ng/L) and one in the upper part (1 μg/L) of the linear calibration range. The recoveries for the entire sample preparation were calculated by comparing peak areas obtained from samples spiked prior to SPE to peak areas derived from a direct injected standard solution. External standards were prepared by evaporation and reconstitution of the same amount of analytes used for SPE. If native contaminations of artificial sweeteners were present in the original sample matrix, calculated recoveries were corrected for these background contaminations.
For method validation, the quantification of matrix effects for different sample matrices is crucial. Many publications do not differentiate to what extent a higher matrix burden effects the SPE yield or ionization in the interface of the MS or both. To determine if matrix impact on recoveries were due to signal suppression/enhancement or inappropriate SPE conditions, samples were (1) spiked prior to SPE and reconstituted as described above or (2) spiked after SPE by reconstituting the dry residue in a buffer solution containing the same absolute amount of analytes. Both final solutions for HPLC-electrospray tandem mass spectrometry (ESI-MS/MS) measurement contained the same matrix burden, but samples derived from (2) did not undergo changes in analyte concentration during sample enrichment. As a consequence, reduced recoveries for (2) are attributed to signal suppression in the interface.
Results and discussion
Optimization and validation of LC-MS/MS conditions
The Zorbax Eclipse XDB-C8 (150 × 4.6 mm; 5 μm) column provided excellent retention and separation of all analytes under investigation. For faster sample analyses, the optimized method was transferred to a rapid resolution column with similar column characterictics. The Zorbax Eclipse XDB-C18 RRHT (50 × 4.6 mm; 1.8 μm) is packed with a microparticulate C18 material for high-speed RP-HPLC. The column enabled to reduce the time of analysis more than by half to 9 min with a slightly different gradient (see Tables S2 and S3, Electronic Supplementary Material, for retention times and gradient programs and Fig. S1 for the corresponding chromatogram).
Even though in MRM a definite precursor/product ion relationship exists and complete separation is not absolutely necessary in LC-MS/MS, retention time is still an important confirmation tool. An adequate separation is still desired as analytes like acesulfame and cyclamate produced rather unspecific product ions during fragmentation. For difficult environmental matrices like waste water, we used the Hypercarb column, which provides a completely different retention mechanism (see Fig. S2, Electronic Supplementary Material) to confirm positive results obtained with the Zorbax Eclipse XDB-C18 RRHT.
For all analytes, negative electrospray ionization was used (Table 2), but even after optimization of the MS parameters, fragmentation and sensitivity remained poor for sucralose, aspartame, and NHDC. As the chromatographic conditions were already in an optimum, we added TRIS post-column in order to increase the ionization yield. TRIS, as a strong base, facilitates deprotonation of the weakly acidic analytes. Higher intensity by the addition of TRIS buffer is explained by the high gas-phase proton affinity and the high proton consumption by TRIS in the buffer system [32]. TRIS buffer was introduced in the flux with a conventional syringe pump connected with a T-piece directly to the interface of the mass spectrometer. With a concentration of 20 mM TRIS and a flow rate of 5 μL/min, signal enhancement ranging from 30% for NHDC to 290% for saccharin was achieved. Higher buffer concentrations increased the signal even three to four times for some compounds (see Table S4, Electronic Supplementary Material, for signal enhancement by addition of TRIS buffer). Gomides Freitas et al. observed an improvement in sensitivity for herbicides and their metabolites even by a factor of 13–22 when applying TRIS in ESI negative mode [33]. In our case, 20 mM TRIS resulted in sufficient signal enhancement for the poorly ionizable compounds. For further experiments, we did not exceed this concentration to prevent the interface of the mass spectrometer from unnecessary contamination.
Method performance: linearity, recoveries, LOD, and LOQ
The calibration with directly injected standards was linear up to 2.25 ng on column for all analytes. All correlation coefficients of both calibration with directly injected standards for the evaluation of detector linearity and calibration including the entire analytical protocol were higher than 0.995.
For five of seven analytes under investigation, method recoveries >75% were obtained for tap water (50 mL) with Bakerbond SDB 1 cartridges, which were eluted with methanol (see Table S5, Electronic Supplementary Material, for recoveries of artificial sweeteners in different matrices). For aspartame and NHDC, recoveries were 41% and 59%, respectively, which was sufficient for screening purposes. As these two compounds were not detected in any of the analyzed environmental samples, no further optimization of their method recoveries was necessary. In surface water, recoveries decreased slightly for acesulfame and saccharin. For all other compounds, the method proved to be robust for surface water. In waste water, recoveries for acesulfame could not be determined, as the native concentration in undiluted STP effluent was still too high to obtain meaningful values for a spike amount of only 1 μg/L. For cyclamate, aspartame, and sucralose, recoveries were higher than 50%, whereas recoveries for saccharin decreased to 30%. Neotame was very robust against any matrix effect.
Except for aspartame, recoveries determined by spiking the sample prior to SPE (method recoveries) or during the reconstitution step (ionization recovery, Table S5, Electronic Supplementary Material) were similar for all analytes. Thus, reduced recoveries for six of the seven tested artificial sweeteners are rather due to ion suppression in the ESI process than due to losses during sample extraction.
In contrast to the other analytes, the method recoveries of aspartame between 41% and 55% in all three matrices tested are mainly a consequence of losses during the extraction step.
Contrary to Loos et al. [30], who reported signal suppression for sucralose of 80% for waste water and 65% for river water extracted at pH 3, no severe ion suppression for this compound was observed with our method. Higher method recoveries for sucralose in our study (88% compared to 62% [30]) at a spiking level of 1 μg/L in tap water can be attributed to more suitable SPE material and/or to a reduced sample volume. We observed similar method recoveries for sucralose as the above-mentioned study of about 60% for Oasis HLB cartridges at pH 7 but no severe decrease at reduced pH. Furthermore, we studied the effect of a higher sample volume on method recovery. When increasing the sample volume by a factor of 4 to 200 mL, acesulfame and cyclamate partly did break through the sorbent material. Method recoveries for saccharin and sucralose decreased with an increase of interfering substances, if larger sample volumes were extracted.
Based on a sample volume of 50 mL and applying 20 mM TRIS post column with a flow rate of 5 μL/min, limits of quantification were 1 ng/L for neotame, 2 ng/L for acesulfame and saccharin, 5 ng/L for cyclamate and aspartame, and 10 ng/L for sucralose and NHDC.
Occurrence and behavior in environmental samples
Behavior of sweeteners in waste water treatment
In the two German STPs investigated in this study, four of seven artificial sweeteners were detected (Fig. 1). Influent concentrations were comparable in both STPs for each of the compounds. Concentrations ranged from 34 to 50 μg/L for acesulfame and saccharin, up to 190 μg/L for cyclamate and below 1 μg/L for sucralose. Elimination of acesulfame and sucralose is low. Acesulfame was removed up to 41% in STP 1 and was discharged at concentrations higher than 20 μg/L into the receiving waters. Sucralose was eliminated only by about 20% in both STPs. Our findings support the results obtained by Brorström-Lundén et al. [29], who reported removal efficiency <10% for sucralose in corresponding waste water samples. Saccharin and cyclamate were eliminated >90% in both STPs, but due to high influent concentrations, STP effluent concentrations were still up to 2.8 μg/L. The trickling filter in STP 2 had no additional benefit for the poorly eliminated acesulfame and sucralose but contributed to the overall removal of saccharin and cyclamate. The results found in two STPs clearly show that, due to incomplete elimination during waste water treatment, acesulfame, saccharin, cyclamate, and sucralose are introduced via STPs into rivers and streams used as receiving waters. Other artificial sweeteners were not found in concentrations above the LOQ as they are metabolized in the body.

Influent and effluent concentrations of artificial sweeteners in two German municipal sewage treatment plants; samplings were in February 2009 for STP 1 and in March 2009 for STP 2
Based on the average flow of the two STPs, daily influent and effluent loads for artificial sweeteners were calculated (Table 3). Taking into account the influent concentrations and the number of inhabitants living in the catchment area of the two STPs, overall annual inputs into German STPs comprising 82 million inhabitants were extrapolated. Assuming no degradation in the sewer system, the figures give a rough estimation of the consumption of acesulfame, saccharin, cyclamate, and sucralose in Germany.
Behavior of sweeteners during soil aquifer treatment
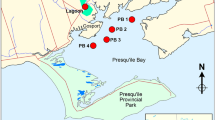
In order to compare conventional waste water treatment as applied in the two German STPs and advanced waste water treatment by soil aquifer treatment, the behavior of artificial sweeteners was also studied at a SAT site in a Mediterranean country. Like in the German STP effluents, also in the effluent used for SAT, the highest concentrations of artificial sweeteners were found for acesulfame (Fig. 2 and Table S6, Electronic Supplementary Material). However, the acesulfame/sucralose ratio in STP effluents in Germany was about 40, whereas it was only 3 for the waste water used for SAT. Assuming similar removal efficiency in conventional treatment in both countries, this finding suggests a significantly different usage pattern of both sweeteners. Acesulfame appeared to be more persistent during SAT than in conventional waste water treatment. In all three sampling campaigns, it was still found downgradient of the percolation basin in well 3 after a residence time of about 1.5 years after discharge at a level of more than 30 μg/L (Fig. 2). In the STP effluent used for SAT, sucralose concentrations were remarkably higher than in the two German STP effluents. A significant decrease of the sucralose concentration in the aquifer occurred, but it was still present at a level of 1.4 μg/L at well 3 after more than 1.5 years in the subsurface. The results for acesulfame and sucralose are remarkable compared to the results of previous studies dealing with the overall removal efficiency of the SAT process for other organic trace pollutants [34–36]. Besides acesulfame and sucralose, which were shown here to resist SAT to a certain extent, only few other compounds like carbamazepine and primidone are known to persist during long-term SAT [34]. At the sampled SAT site, total organic carbon and most organic trace pollutants were eliminated to about ≥90% already within the variably saturated vadose zone and are found only in traces in well 1, right below the percolation basin.

Occurrence of acesulfame and sucralose in samples from three sampling campaigns (June 2008, December 2008, February 2009) from a soil aquifer treatment site. Detention times to wells 1, 2, and 3 are approximately 1.5 months, 1 year, and >1.5 years (n = 3 for recharge effluent, well 1, and well 3, n = 2 for well 2 and well 4)
The slow concentration decrease of sucralose is consistent with the slow and incomplete mineralization of sucralose in lake water and in sewage under aerobic conditions. Under anaerobic conditions, little or no mineralization was observed [37, 38]. The recalcitrant character of acesulfame and sucralose suggests their use as tracers for anthropogenic contamination of natural waters. Of the remaining studied sweeteners, only cyclamate and saccharin were found at levels up to 400 ng/L in the STP effluent used for SAT but were detected only in traces in the observation wells. As expected, in well 4, which is known to be separated by a hydraulic barrier from any waste water influence, none of the artificial sweeteners was detected.
Occurrence of sweeteners in German surface waters
In all German surface waters analyzed, acesulfame, saccharin, cyclamate, and sucralose were detected, which proved the observed incomplete removal in STPs (Fig. 3 and Table S7, Electronic Supplementary Material). Sweetener levels in the investigated German rivers correspond to STP effluent concentrations when taking into account a dilution approximately between a factor of 10 and a factor of 100. Acesulfame was found in several samples in concentrations higher than 2 μg/L and, in most cases, occurred in about tenfold higher concentrations than other sweeteners. Saccharin and cyclamate were detected at levels between 50 and 150 ng/L in the majority of the river water samples. Findings of sucralose in German rivers were in excellent correlation to the values obtained for Germany in the EU-wide monitoring program [30]. Most samples showed sucralose concentrations between 60 and 80 ng/L with only one value exceeding 100 ng/L. Aspartame, neotame, and NHDC were again not detected in any analyzed sample.

Concentrations of four artificial sweeteners in German surface waters (Rhine, Neckar, Danube, Main; n = 23)
Conclusions
The method developed allows the simultaneous extraction and analysis of seven artificial sweeteners from difficult environmental matrices, such as waste water and surface water. Accurate quantification could be achieved by the use of a deuterated standard and sample dilution. Application of this method to waste water samples, samples obtained from a soil aquifer site, and surface water samples demonstrated incomplete removal of some of these compounds during waste water purification. Due to their use as food additives, the occurrence of artificial sweetener traces in the aquatic environment might become a primary issue of consumer acceptance, especially as the aspect of drinking water quality, which might be negatively influenced by potential metabolites of these trace pollutants, is completely unknown yet.
References
Weihrauch MR, Diehl V (2004) Ann Oncol 15:1460–1465
Kroger M, Meister K, Kava R (2006) Compr Rev Food Sci Food Safety 5:35–47
Imfeld T (1993) Caries Res 27:50–55
Strubig W (1988) Oralprophylaxe 10:68–73
Grice HC, Goldsmith LA (2000) Food Chem Toxicol 38(Supplement 2):1–6
Bandyopadhyay A, Ghoshal S, Mukherjee A (2008) Drug Chem Toxicol 31:447–457
Magnuson BA, Burdock GA, Doull J, Kroes RM, Marsh GM, Pariza MW, Spencer PS, Waddell WJ, Walker R, Williams GM (2007) Crit Rev Toxicol 37:629–727
FDA (2006) FDA Consum Mag 40:27–28
EU (1994) European Parliament and Council Directive 94/35/EC of 30 June 1994 on sweeteners for use in foodstuffs
EU (2003) European Parliament and Council Directive 2003/115/EC of 22 December 2003 amending Directive 94/35/EC on sweeteners for use in foodstuffs
Oser BL, Carson S, Cox GE, Vogin EE, Sternberg SS (1975) Toxicology 4:315–330
CFSAN/Office of Food Additive Safety (2009) U.S. Food and Drug Administration, Silver Spring http://www.foodsafety.gov/∼dms/opa-abey.html. Accessed 12 Jan 2009
Renwick AG (1986) Xenobiotica 16:1057–1071
Taylor JM, Weinberger MA, Friedman L (1980) Toxicol Appl Pharmacol 54:57–75
Squire RA (1985) Food Chem Toxicol 23:491–497
Cohen SM, Arnold LL, Emerson JL (2008) Agro Food Ind Hi Tec 19:24
Information document on the proposal to reinstate saccharin for use as a sweetener in foods in Canada (2007) Health Canada, Ottawa http://www.hc-sc.gc.ca/fn-an/alt_formats/hpfb-dgpsa/pdf/securit/saccharin_prop-eng.pdf. Accessed 14 Feb 2009
Shaywitz BA, Anderson GM, Novotny EJ, Ebersole JS, Sullivan CM, Gillespie SM (1994) Ann Neurol 35:98–103
Tsakiris S, Giannoulia-Karantana A, Simintzi I, Schulpis KH (2006) Pharmacol Res 53:1–5
Simintzi I, Schulpis KH, Angelogianni P, Liapi C, Tsakiris S (2007) Pharmacol Res 56:155–159
Soffritti M, Belpoggi F, Esposti DD, Lambertini L, Tibaldi E, Rigano A (2006) Environ Health Perspec 114:379–385
Soffritti M, Belpoggi F, Tibaldi E, Esposti DD, Lauriola M (2007) Environ Health Perspec 115:1293–1297
Duffy VB, Sigman-Grant M (2004) J Am Diet Assoc 104:255–275
Varnam AH, Sutherland JM (1994) Beverages: technology. Chemistry and microbiology. Aspen, Gaithersburg
Choi JM, Yoon BS, Lee SK, Hwang JK, Ryang R (2007) Biol Pharm Bull 30:324–330
Roberts A, Renwick AG, Sims J, Snodin DJ (2000) Food Chem Toxicol 38(Supplement 2):31–41
Bigal ME, Krymchantowski AV (2006) Headache 46:515–517
Patel RM, Sarma R, Grimsley E (2006) Headache 46:1303–1304
Brorström-Lundén E, Svenson A, Victor T, Woldegiorgis A, Remberger M, Kaj L, Dye C, Bjerke A, Schlabach M (2008) Measurements of sucralose in the Swedish screening program 2007
Loos R, Gawlik BM, Boettcher K, Locoro G, Contini S, Bidoglio G (2009) J Chromatogr A 1216:1126–1131
Hernando MD, Petrovic M, Fernandez-Alba AR, Barcelo D (2004) J Chromatogr A 1046:133–140
Shen JC, Lu SG, Zhuang ZX, Wang XR, Lee FSC (2005) Int J Mass Spectrom 243:163–169
Freitas LG, Gotz CW, Ruff M, Singer HP, Muller SR (2004) J Chromatogr A 1028:277–286
Drewes JE, Heberer T, Reddersen K (2002) Water Sci Technol 46:73–80
Quanrud DM, Hafer J, Karpiscak MM, Zhang HM, Lansey K, Arnold RG (2003) Water Res 37:3401–3411
Yoo HH, Miller JH, Lansey K, Reinhard M (2006) J Environ Eng-Asce 132:674–682
Labare MP, Alexander M (1993) Environ Toxicol Chem 12:797–804
Labare MP, Alexander M (1994) Appl Microbiol Biotechnol 42:173–178
Acknowledgments
This study was financially supported by the German Ministry of Education and Research as part of the project 02WA0901. The assistance by our project partners in organizing and execution of the sampling campaigns in the SAT field is kindly acknowledged. We thank the municipalities of Eggenstein-Leopoldshafen and Karlsruhe for providing waste water samples for this study. Furthermore, we thank Doreen Richter for the careful corrections and fruitful discussions on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM
(DOC 339 kb)
Rights and permissions
About this article
Cite this article
Scheurer, M., Brauch, HJ. & Lange, F.T. Analysis and occurrence of seven artificial sweeteners in German waste water and surface water and in soil aquifer treatment (SAT). Anal Bioanal Chem 394, 1585–1594 (2009). https://doi.org/10.1007/s00216-009-2881-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-009-2881-y