
Abstract
Here we report on the first assessment of droplet digital PCR (ddPCR) for detection and absolute quantification of two quarantine plant pathogenic bacteria that infect many species of the Rosaceae and Solanaceae families: Erwinia amylovora and Ralstonia solanacearum. An open-source R script was written for the ddPCR data analysis. Analysis of a set of samples with known health status aided the assessment and selection of different threshold settings (QuantaSoft analysis, definetherain pipeline and manual threshold), which led to optimal diagnostic specificity. The interpretation of the E. amylovora ddPCR was straightforward, and the analysis approach had little influence on the final results and the concentrations determined. The sensitivity and linear range were similar to those for real-time PCR (qPCR), for the analysis of both bacterial suspensions and plant material, making ddPCR a viable choice when both detection and quantification are desired. With the R. solanacearum ddPCR, the use of a high global threshold was necessary to exclude false-positive reactions that are sometimes observed in healthy plant material. ddPCR significantly improved the analytical sensitivity over that of qPCR, and improved the detection of low concentrations of R. solanacearum in potato tuber samples. Accurate and rapid absolute quantification of both of these bacteria in pure culture was achieved by direct ddPCR. Our data confirm the suitability of these ddPCR assays for routine detection and quantification of plant pathogens and for preparation of defined in-house reference materials with known target concentrations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The basis of digital PCR (dPCR) is to quantify the absolute number of targets present in a sample using limiting dilutions, PCR and Poisson statistics, which is a concept that was first described by Sykes et al. in 1992 [1]. Several commercial dPCR platforms have been developed that partition a reaction into a large number of chambers or droplets [2]. After end-point PCR amplification, each partition is examined and defined as positive or negative and then used to calculate the concentration of the target copies [3]. An important advantage of dPCR over quantitative, or real-time, PCR (qPCR) is that it can be used to absolutely quantify a target’s concentrations without the need for calibration, which simplifies both experimentation and data comparability [4]. dPCR has been successfully applied to routine testing as well as to research analysis. Morisset et al. [5] showed that droplet dPCR (ddPCR) is useful for routine analysis of food and animal feed samples for the presence of DNA from genetically modified organisms. They concluded that for genetically modified organisms where both detection and quantification are desirable, ddPCR has the potential to provide improved throughput and cost-effectiveness compared to qPCR. ddPCR has also been used to detect and quantify viruses and bacterial pathogens. Roberts et al. [6] used ddPCR for the detection and quantification of ocular Chlamydia trachomatis in conjunctival swab samples, and Rothrock et al. [7] used ddPCR to quantify Salmonella spp., Campylobacter jejuni and Listeria monocytogenes in environmental water samples. ddPCR has also been shown to be less sensitive to inhibitors in several studies, including the analysis of inhibitors in clinical samples [8], food and animal feed [5] and environmental samples [9]. These promising results of ddPCR application in these fields prompted our investigation of its suitability for detection and quantification of bacterial plant pathogens in plant material.
We focused on two qPCR assays: (i) amsC assay for Erwinia amylovora detection in plant material of the Rosaceae family [10] and (ii) a broad-range assay for detection of the Ralstonia solanacearum species complex in potato (Solanum tuberosum) tubers [11]. Both of these bacteria have quarantine status in the European Union [12], as in some other regions. Thus systems for the sampling and testing for symptomatic and latent infections of E. amylovora and R. solanacearum have been defined in methods suggested and prescribed by the European and Mediterranean Plant Protection Organisation (EPPO; PM7/20 (2)) [13] and the European Commission (EC; Council Directive 2000/29/EC) [14]. The testing schemes are based on screening tests, where the results need to be confirmed by additional tests (based on a different biological principle or a different target DNA sequence), and on the isolation of the bacteria as pure cultures and their identification, followed by final confirmation of their pathogenicity on alternative host plants [13, 14]. For detection purposes, the concentration of the bacteria is of lesser importance, as here any positive confirmed result leads to phytosanitary management measures being taken. Thus, although qPCR is gaining acceptance as the first screening test and E. amylovora qPCR has been included in EPPO PM7/20 (2) [13], the results are usually interpreted only qualitatively i.e. the result is either positive or negative. Absolute quantification, however, provides additional information, which includes an indication of the likelihood of the sample being confirmed by the whole testing scheme, depending on the known analytical sensitivities of the individual tests used. Absolute quantification is also invaluable for research purposes, e.g. in studies of bacterial infection kinetics and determination of biologically relevant thresholds, and for characterisation of in-house reference materials when they are not otherwise available.
In the present study, we aimed to transfer two qPCR assays to ddPCR format and to assess the critical parameters of their performance in terms of detection and quantification of the target bacteria, in bacterial suspensions and in plant material. During this transfer, we also optimised the analysis steps through critical assessment and selection among different analysis approaches. As an aid to analysis, we developed an open-source R script for ddPCR data analysis that combines the calculation of the relevant parameters identified as essential in the digital Minimum Information for Publication of Quantitative Digital PCR Experiments (MIQE) [15] and the parameters that describe the quality of separation between the positive and negative droplets. The data obtained and the approaches developed should be applicable to detection and quantification of these and other organisms in plants and other materials.
Material and methods
Bacterial strains and plant material
The bacterial strain E. amylovora NIB Z 3 was isolated from Pyrus communis in the UK in 1959 (National Collection of Plant Pathogenic Bacteria [NCPPB] 683T) and the R. solanacearum biovar 2 strain NIB Z 30 was isolated from Solanum tuberosum cv. Bildstar in the Netherlands in 1995 (NCPPB 4156); they were both grown on King’s B medium [16] at 25 °C. Suspensions of bacteria were prepared in 10 mM phosphate buffer (pH 7.2) and standardised by turbidity measurements (DEN-1B McFarland densitometer, Biosan). The number of colony forming units (CFUs) was determined by plating on King’s B medium.
The plant material was collected by the Administration of the Republic of Slovenia for Food Safety, Veterinary and Plant Protection, Ministry of Agriculture and Environment and Phytosanitary Inspectorate, from 2003 to 2012. Extracts were prepared from the plant material of the Rosaceae family, and their health status was assessed according to the EPPO protocol for E. amylovora testing [13]. Extracts of plant material from potato tubers were prepared according to EC Directive 2000/29/EC for R. solanacearum latent testing [14], and their health status was assessed by immunofluorescence microscopy using the IACR-PS-278 anti-R. solanacearum (biovar 2) polyclonal rabbit primary antibody (Rothamstead Research), and using PCR [17] and qPCR [11]. Artificially contaminated samples were prepared by mixing selected negative plant extracts with known concentrations of target bacteria prior to the DNA extraction.
DNA extraction
DNA from the pure bacterial cultures and plant extracts was extracted and purified using magnetic-bead-based QuickPick™ SML Plant DNA kits (Bio-Nobile, Turku, Finland). This was automated on a KingFisher™ mL system (Thermo Labsystem), as described previously for E. amylovora [10], and with a minor modification (440 μL lysate used in the purification). The DNA was stored at or below −15 °C until analysis.
Real-time PCR
The qPCR assays used to detect E. amylovora and R. solanacearum were (i) an amsC assay that targets the amsC single-copy gene of E. amylovora [10] and (ii) an assay that uses RS-I-F and RS-II-R that flank the 16S rRNA region of R. solanacearum, and that has been suggested for broad-range detection of all of the R. solanacearum biovars [11]. The E. amylovora qPCR was previously validated according to the EPPO guidelines PM 7/98 [18]. These validation data have been deposited with the EPPO database on diagnostic expertise (http://dc.eppo.int/), and they include the performance criteria for the analytical specificity, analytical sensitivity, repeatability and reproducibility. Further data on the interlaboratory testing for E. amylovora is available [19, 20]. The analytical specificity and sensitivity for the R. solanacearum qPCR have also been reported previously [11, 21]. In this assay, several target sequences can be present per genome (see Electronic Supplementary Material, ESM 1).
All of the qPCR reactions were performed in triplicate on an ABI PRISM 7900 HT Sequence Detection system (Life Technologies) using the following universal cycling conditions: 2 min at 50 °C, 10 min at 95 °C, followed by 45 cycles of 15 s at 95 °C, and 1 min at 60 °C, using standard temperature ramping mode. The reaction volumes of 10 μL contained, as final concentrations, 900 nM primers, 250 nM FAM and BHQ-1 labelled probes (Integrated DNA Technologies), 1 × TaqMan Universal PCR Master Mix (Life Technologies) and 2 μL sample DNA. The qPCR data were analysed using the SDS software 2.4 (Life Technologies) with automatic baseline and a manual threshold of 0.1 for both assays. The qPCR data are given as Cq values (i.e. qPCR quantification cycle) and as the number of positives out of all of the tested replicates.
Droplet digital PCR
The DNA was detected and quantified using the QX100™ Droplet Digital™ PCR system (Bio-Rad, Pleasanton, CA, USA). ddPCR Supermix for probes (16 μL, Bio-Rad) and 4 μL sample DNA were used, with the primer and probe concentrations as for the qPCR. After droplet generation, 40 μL of the generated droplet emulsion was transferred to a new 96-well PCR plate (Eppendorf) and amplified in a T100™ Thermal Cycler (Bio-Rad). The amplification conditions were 10 min DNA polymerase activation at 95 °C, followed by 40–45 cycles of a two-step thermal profile of 30 s at 94 °C for denaturation, and 60 s at 60 °C for annealing and extension, followed by a final hold of 10 min at 98 °C for droplet stabilisation and cooling to 4 °C. The temperature ramp rate was set to 2.5 °C/s, and the lid was heated to 105 °C, according to the Bio-Rad recommendations. After the thermal cycling, the plates were transferred to a droplet reader (Bio-Rad). The software package provided with the ddPCR system (QuantaSoft 1.3.2.0, Bio-Rad) was used for data acquisition.
For optimisation of the primer concentrations in the ddPCR, primers with 5′ flaps [22] were tested, in addition to the original primers described by Weller et al. [11].
Analysis of the ddPCR data
The data from the droplet signals (amplitudes of individual droplets expressed in fluorescence units) were exported from the ddPCR (QuantaSoft 1.3.2.0, Bio-Rad) as .csv files. Different analysis approaches and threshold settings were then compared. Discrimination between droplets that did not contain the target (negatives) and those that did (positives) was based on the signals read by the QuantaSoft software (Bio-Rad), by applying (i) an automatic individual well analysis, using the proprietary software (QSi; QuantaSoft, Bio-Rad); (ii) a threshold determined by the k-nearest neighbour algorithm on a reference sample, using definetherain (DTRg) [23] and (iii) a manual global threshold (MTg) that was determined from the signals observed in no-template controls (NTCs). For the DTRg, the initial thresholds are determined on a sample with a target concentration within the linear range and with well-discriminated positive and negative droplets, as determined visually. The analyses were compared on a set of samples that included NTCs, negative plant material and serial tenfold dilutions of the target DNA and on a set of samples that contained a low, known amount of target DNA in the plant material that was close to the limit of detection.
The rejection criteria for the exclusion of a reaction from subsequent analysis included a low number of droplets measured (less than 10,000 per 20 μL PCR). A sample was interpreted as positive if the number of positive droplets was at least 2. The data from the ddPCR are given in target copies per microlitre of reaction or are calculated relative to the starting amount of sample before the DNA extraction and expressed as log (copies/mL). In the latter case, a correction factor was applied because only a part of the samples that were lysed were used in subsequent DNA purification steps (0.838 for BioNobile Quick Pick Plant DNA kit). However, 100 % efficiency of the DNA extraction was assumed.
Any bias in the determined concentrations was calculated by dividing the determined concentration (copies per microlitre of reaction) by a known (expected) concentration and expressing this as a ratio of overestimation/underestimation (e.g. a value of −0.33 means that the concentration was underestimated by 33 %).
To allow for higher-throughput analysis of the ddPCR data, an R script was written (see ESM 2) in the R language and environment [24]. The script analysed the data of the signals exported from the QuantaSoft software, with its automatic threshold defined individually for each reaction or with a selected, manually defined, threshold applied. This incorporates the calculation of (i) the basic parameters of the ddPCR (e.g. concentration, mean amplitudes of positive and negative droplets, with corresponding standard deviations); (ii) the mean copies per partition (lambda) and the total volume of the partitions measured (effective reaction size), as defined by the digital MIQE guidelines [15]; (iii) the parameters relating to the occurrence of “rain” droplets and (iv) the separability measures of the Bhattacharyya [25] and Jeffries–Matusita distance (R script adapted from [26]). In determining the effective reaction size, the volume of the droplets was assumed to be 0.91 nL, which was consistent with the instrument manufacturer’s software.
Results and discussion
Direct transfer of assays from qPCR to ddPCR
We examined the behaviour of the two qPCR assays that target these quarantined plant pathogenic bacteria using ddPCR: (i) the qPCR by Pirc et al. [10] for E. amylovora (Ams assay) and (ii) the broad-range qPCR for detection of the R. solanacearum species complex using the Rs-I-F and Rs-II-R primers, and the Rs-P probe, by Weller et al. [11]. Both of these qPCR assays were developed for and are routinely run on an ABI PRISM 7900 HT sequence detection system. The primer and probe concentrations in the ddPCR were as in the qPCR, although the ddPCR Supermix for probes (Bio-Rad 186–3026) and the suggested Bio-Rad protocol for the amplification were used.
The transfer of the optimised assay for E. amylovora was straightforward. Amplification of the NTCs and the negative plant material resulted in droplets with low and constant fluorescence (background) that could easily be distinguished from the positive amplification (Fig. 1).

With the assay targeting R. solanacearum, we occasionally experienced cross-reactions in the qPCR and weaker reactions with some isolates (higher Cq values at the same concentration) [21]. Also, during this study, the analytical sensitivity of the assay was low, as seen by the failure of the amplification in control samples with low concentrations of the target bacteria in the routine testing of potato tubers for latent infection (concentration of ca. 7 × 103 CFU/mL). The ddPCR results reflected these already known issues. In all of the plant samples, a discrete group of positive droplets was observed that had fluorescence slightly above the negative droplets (ca. 1,400 fluorescence units), which indicated that the assay reacted with the plant DNA or with a common microorganism present in all of the tested samples. In addition, more than eight droplets with a fluorescence level of up to 6,000 units were observed in four out of 10 negative potato tuber extracts of cvs. Adora, Désirée (one out of two tested samples) and Melody (two samples). Indeed, only two out of 10 of the assumed negative samples did not contain droplets above the negative signals. A similar pattern of amplification and an increased negative range was also observed in the presence of a cv. Carlingford extract (Fig. 1). The decision on the threshold setting was therefore more challenging in the case of the R. solanacearum ddPCR. This prompted further investigation of the influence of different threshold settings on the final data of the ddPCR for both of the examined ddPCR assays.
Assessment of analysis approaches and suitability of ddPCR assays for detection and quantification of the target bacteria
Assessment of analysis approaches
For the assessment of the analysis approaches and the suitability of the ddPCR assays for detection purposes, we combined the relevant aspects of the commonly reported approaches to ddPCR evaluation and the general guidelines on the validation of methods developed within the EPPO [18]. We chose a set of samples with known health status and assessed the approaches based on the resulting diagnostic specificity i.e. on their correct identification of positive and negative samples. In addition to the NTCs and the target DNA at various concentrations, the set included negative plant material and artificially prepared samples of plant material containing low concentrations of the relevant target organisms for routine diagnostics. These low concentrations were close to the theoretical and practical limits of detection of the examined qPCR assays.
The discrimination between droplets that did not contain target (negatives) and those that did (positives) was based on signals read by the QuantaSoft software (Bio-Rad), by applying (i) automatic individual well analysis using the proprietary software (QSi; QuantaSoft, Bio-Rad); (ii) global threshold determined by the k-nearest neighbour algorithm on a reference sample using definetherain (DTRg) [23] and (iii) application of a manual global threshold (MTg) determined from signals observed in the known negative samples. The analysis approaches differed in the group of droplets (negative or positive) used to define the threshold and in their treatment of the rain droplets i.e. droplets with intermediate fluorescence (Table 1).
In the QSi analysis, the majority of the rain droplets were considered as true positives. The thresholds determined depended on, and decreased with, the decreasing target concentration. In both the E. amylovora and R. solanacearum assays, the QSi failed to discriminate between the positive and negative droplets at the highest concentrations tested, which corresponded to an initial concentration of 108 CFU/mL. At these concentrations, there were many droplets with intermediate fluorescence and a low number of negative droplets, which appeared to cause the failure of the classifying algorithm and which resulted in an error message (“No Call”). At low concentrations, individual droplets with fluorescence close to the negative droplets were also sometimes classified as positive, as previously observed by Strain et al. [27]. In theory, this incorrect assignment of the individual droplets can lead to false-positive results. However, this did not occur with the E. amylovora assay, and all of the NTCs and negative plant samples were correctly identified as negative (Fig. 2). The QSi analysis, however, was not appropriate for detection and quantification of R. solanacearum using the developed assay, because of the increased fluorescence in the negative droplets that would be interpreted as cross-reactions in routine diagnostics, and which resulted in false-positive data (Fig. 2). In general, the QSi appears to represent a more conservative approach to the analysis, which requires a well-optimised assay, the presence of both negative and positive droplets, and good discrimination between these.

Comparison of the different thresholds on the serial decimal dilutions of the target DNA (a, c) and on the artificially prepared plant samples that contained low concentrations of the target bacteria (b, d) (as indicated). The positive and negative droplets as classified by the QuantaSoft automatic analysis of individual wells are shown in blue and grey, respectively. Thresholds determined by the definetherain analysis and the manual global thresholds are shown as horizontal lines
The DTRg analysis assumes that the majority of rain droplets (as identified in comparison with a representative reference sample) are not true positives, and consequently, this analysis excludes them from the calculation of the concentrations. The thresholds (the cut-off values for positive droplets) determined using the DTRg approach were the highest for both of the ddPCR assays (Table 1, Fig. 2), and these resulted in one false-negative result when testing for low target concentrations of E. amylovora in plant material (1/21 samples tested) (Tables 1 and 2).
In the MTg analysis approach, the threshold was set relative to the level and distribution of the signals in the NTCs and the negative plant material, i.e. minimally above the negative signal levels, which still led to correct results for the selected negative and positive samples. The threshold set thus reflects the behaviour of the individual assay and it allows for flexibility in the treatment of the rain droplets. A low threshold was determined for the well-optimised E. amylovora assay, and a high threshold was determined in the R. solanacearum assay, in which there were considerable background signals (false positives) (Fig. 2). The MTg threshold was defined as an averaged signal in the NTCs plus six times the standard deviation in the E. amylovora assay. This gave 2,322 fluorescence units (see ESM 3), which places this threshold somewhere in between the different thresholds set for the individual samples in the QSi (Fig. 2). Interestingly, the presence of plant material lowered the average fluorescence levels of the negative droplets, which appeared to be due to a combined effect of signal generation and inhibition. Nevertheless, the determined threshold did not differ much from that determined on the NTCs only (see ESM 3). In the R. solanacearum assay, the manual threshold could not be determined in the same way (see ESM 4) and it was manually set to a high level of 7,000 fluorescence units, which was above the signals in negative samples and close to the DTRg threshold.
Overall, the different analysis approaches of QSi, DTRg and MTg had little influence on the qualitative results and on the concentrations determined. The proprietary QuantaSoft analysis that defines a threshold for each sample individually led to the highest analytical sensitivity, which was on par or slightly below the analytical sensitivity of the qPCR in the testing for E. amylovora in necrotic plant material. For the R. solanacearum assay, the setting of a manual threshold was necessary.
The setting of a global threshold, as in DTRg and MTg approaches, simplifies the analysis. However, both of these approaches rely on the representative nature of one or more samples, respectively. Both of the DTRg and MTg approaches are thus expected to be sensitive to significant shifts in the signals (amplitudes) of negative or positive droplets that might be due to previously unobserved cross-reactions or the influence of inhibitors. When these approaches are used, the signals of positive and negative droplets should be followed through the runs.
Detection of E. amylovora with ddPCR
As expected, as a result of the low level of droplets with intermediate fluorescence, different thresholds or exclusion of such droplets had little effect on the final data and the determined concentrations in the E. amylovora ddPCR.
All of the negative samples and the samples of plant extracts containing low levels of target bacteria were correctly classified using the three different analysis approaches (100 % diagnostic specificity). The only exception was the DTRg analysis of one sample of M. domestica cv. Idared that contained low levels of E. amylovora that gave a false-negative result (Table 2). Using the QSi and MTg analyses, the number of positive droplets at low levels of target concentrations (corresponding to the 5 × 103 CFU/mL starting concentration in the plant material) resulted in 2–3 and 7–32 positive droplets, respectively (Table 2). This means that besides being suitable for E. amylovora detection, both the QSi and MTg analyses allowed for straightforward interpretation of the data even at these low target levels.
Lower sensitivity of ddPCR in comparison with qPCR has been reported previously for cytomegalovirus [28] and for Chlamydia trachomatis [29]. However, in our case, the lower concentrations tested were close to the theoretical limit of detection. It is known that in samples with very low copy numbers, random variations due to sampling errors (Poisson’s error) can become significant [30]. To confirm the observed differences between these two formats, higher numbers of samples should be analysed.
Quantification of E. amylovora with ddPCR
For quantification of the pure bacterial DNA, both DTRg and MTg analyses had a broader quantification range than for QSi. However, the quantification range was broader for DTRg and MTg if they also included the highest tested concentration that corresponded to the 108 CFU/mL starting concentration. From the detection point of view, this is less important than the sensitivity of the method, which was highest with the QSi analysis (Table 2).
The bias in the determined concentrations within the quantification range of the purified target DNA showed little variation. Within the linear range, the concentrations were slightly underestimated, independent of the analysis approach (Fig. 3a). This underestimation appears to be due to the inherent efficiency of the DNA extraction (i.e. below 100 %), although other factors might have also contributed to this, such as pipetting errors, binding of DNA to tubes and the efficiency of amplification in the ddPCR. Overall, the efficiency of the whole protocol leading to the quantification of pure E. amylovora DNA was approximately 19 %, as estimated from the data from one DNA extraction series.

Bias in the ddPCR quantification of E. amylovora and R. solanacearum as influenced by the analysis approach. The bias is expressed as the ratio of overestimation/underestimation of the concentrations in copies/reaction. Bias for E. amylovora (a, b) and R. solanacearum (c, d) are shown for pure cultures (a, c) and for artificial plant samples containing low levels of target DNA (b, d) (as indicated)
As expected, quantification of the same low concentration of E. amylovora in different plant extracts that mimicked natural infections showed more variable data and was more dependent on the analysis approach (Fig. 3b). Relatively high differences in the concentrations determined were seen in the repeated DNA extractions from the same plant material, which suggested that the influence of the plant material was lower than the combined effects of analysing lower concentrations (stochastic effects) and the variable efficiencies of the different series of DNA extractions. The QSi analysis approach was the most variable regarding the direction of bias. Overall, the bias ranged from −77 to −85 %. For quantification purposes for low levels of target DNA in the plant material, it was not possible to choose a single analysis approach that would guarantee the best result in the routine analysis of different samples. To determine if the analysis approaches indeed differ in their accuracy for low levels of target DNA in the plant material and to choose the optimal analysis approach a larger number of samples should be analysed, to compensate for the lower repeatability of the assay in the range examined.
E. amylovora ddPCR in comparison with qPCR
In comparison with qPCR, ddPCR showed similar levels of analytical sensitivity and a similar quantification range, in particular for the plant material (Table 2). This was further confirmed by analysis of three serial tenfold dilutions of E. amylovora in plant material of Malus domestica and Pyrus communis (Table 3). Both formats of the assay were suitable for detection and quantification of E. amylovora in plant material.
Detection of R. solanacearum with ddPCR
The R. solanacearum assay appears not to be optimal. This has been observed previously in qPCR as cross-reactions (false-positive signals) in plant material [21]. As confirmed in the present study, the analytical sensitivity of the assay is not always satisfactory, which is seen as a failure to detect the lower concentrations of bacteria in plant material (Table 4). In ddPCR this was seen in the broader distribution of the signals in the negative and positive samples, the higher frequency of droplets with intermediate fluorescence (rain) and the occurrence of droplets with high signals in negative plant samples (Fig. 1).
The QSi analysis algorithm often failed to correctly discriminate negative and positive droplets (Fig. 2, Table 4) and it was not suitable for analysis of ddPCR data in which negative and positive droplets are not well separated. As a result of amplifications in some negative plant extracts, it was also not possible to define a low global manual threshold that would allow the correct classification of negative and positive samples (Fig. 2; see also ESM 4). Setting a threshold manually at approximately 7,000 fluorescence units led to the correct classification of the negative samples (NTCs and negative plant material) and this was chosen for further analysis. It was expected that the high threshold might lead to lower assay sensitivity, which would hamper its usefulness for the detection of R. solanacearum in potato tuber extracts. However, when the threshold of 7,000 fluorescence units was applied to the serial dilutions of R. solanacearum DNA, this resulted in a broad linear range (log 1 to log 6) and high analytical sensitivity (log 3 CFU/mL of the starting concentration, resulting in six positive droplets). As expected, based on similar thresholds, the DTRg analysis with a cut-off for positive droplets set at 8,719 fluorecence units gave very similar result as the manual threshold (Table 4). Using the manual threshold of 7,000 fluorescence units, and the DTRg analyses for all NTCs, negative plant samples and plant samples with low amounts of target bacteria were correctly classified (100 % diagnostic specificity).
Quantification of R. solanacearum with ddPCR
For quantification of pure bacterial DNA, DTRg and the manual threshold analysis gave similar data within a broad quantification range and high analytical sensitivity (Table 4). However, within the quantification range, all of the analysis methods slightly overestimated the copy concentrations of the target when compared to the known starting concentration, expressed as CFU/mL (Fig. 3c). Bias was also observed in plant extracts with low concentrations of R. solanacearum added; this varied between overestimation and underestimation; however, this absolute bias was within 300 % (Fig. 3c). Some overestimation was expected, as the target copy is present in more than one copy per genome. No restriction of the DNA was used and it was therefore not expected that the target copies from one genome would be efficiently separated into discrete droplets. However, the target copies are distributed discretely in the genomes of R. solanacearum (ESM 1) and the DNA is unlikely to remain completely intact during the DNA extraction process. The shearing of DNA could lead to the target copies being distributed among different droplets, significantly contributing to the overestimation. The overestimation, however, was not consistently observed over all concentration levels (Fig. 3c). It is expected that enzymatic restriction of DNA would improve the repeatability of the target separation and the accuracy of quantification.
R. solanacearum ddPCR in comparison with qPCR
Compared to qPCR, the R. solanacearum ddPCR showed similar quantification range and analytical sensitivity in pure culture testing. However, ddPCR was far more efficient in detecting low target concentrations in plant material, with all of the tested samples (11/11) correctly classified as positive, compared with 5/11 samples that were correctly classified using qPCR. In samples positive in qPCR, the Cq values of 4/5 of these positive samples were in the range where it is difficult to differentiate them from non-specific amplification (Cq = 41.4–43.5), further complicating their interpretation. Compared to the overall signal in qPCR, ddPCR provided a more detailed insight into the individual components (reactions) that contributed to the final result.
On the basis of our data, we can conclude that despite its obvious shortcomings in design, the ddPCR format of the assay is more suited to the analysis of latently infected potato tuber samples than qPCR is. Further analyses are needed to see how the R. solanacearum ddPCR performs under conditions of mismatches in primers and probe i.e. in detection and quantification of different isolates. Also, the assay may warrant further optimization e.g. testing of different annealing temperatures.
An R script for exploratory analysis of ddPCR data, estimation of the rain and calculation of separability measures
To provide improved high-throughput and automated analysis of ddPCR at variable threshold settings, an R script was written (see ESM 2). This script analyses the data from the signals (amplitudes) and the cluster data as exported from the QuantaSoft software and applies a selected global threshold. It incorporates the calculation of (i) the basic parameters of the ddPCR (e.g. concentrations, mean amplitudes of positive and negative droplets, with corresponding standard deviations); (ii) the mean copies per partition (lambda), which is also used for calculation of the concentrations, and the total volume of the partitions measured (effective reaction size), as defined by the digital MIQE guidelines [15]; (iii) the parameters related to the occurrence of the rain droplets and (iv) the quality of the separation of negative and positive droplets via the separability measures of the Bhattacharya and Jeffries–Matusita distance. Examples of the data for sets of samples from Tables 2 and 4 using the manual global thresholds of 2,322 and 7,000 fluorescence units for E. amylovora and R. solanacearum, respectively, are available as a supplement (see ESM 6 and 7).
The data from the ddPCR assays and parameters calculated using the R script described above show that the average signals in the positive and negative droplets were relatively constant, i.e. independent of the target concentrations, except for the highest concentrations at which the rain droplets contributed to and increased the average signals in negative droplets. The effective reaction size calculated from the number of accepted droplets was not affected by the type of assay material (NTC, plant material, purified target DNA), cartridge or ddPCR run. On average, 63 % (±5 %) of the total reaction was analysed in both of the ddPCR assays. The number of accepted droplets was above 11,500 in all cases.
The R script also allowed calculation of the various parameters related to the occurrence of rain. For the purpose of this study, droplets of intermediate fluorescence (i.e. the rain) were defined as droplets with signals in the range between the mean amplitude of negative droplets plus three standard deviations and the mean amplitude of positive droplets minus three standard deviations. The R script provides the number of droplets outside these ranges and the number of droplets between the two groups (negative and positive droplets), as well as the percentages for the positive droplets and for all of the droplets. In general, the percentage of rain droplets increased with the target concentration. The rain occurred with both the E. amylovora and R. solanacearum assays; however, from the data of the negative and positive samples, it can be seen that the reasons are probably different. With the E. amylovora assay, the rain was related to the preparation of the DNA. There were higher numbers of rain droplets in the presence of inhibitors that arose from the DNA extraction or the bacterial components for direct ddPCR (Fig. 4). For the R. solanacearum assay, the droplets with high fluorescence above the negative droplets appear to be a consequence of non-specific reactions of primers and probes with the plant DNA or with the DNA of the microflora associated with potato (Fig. 1). On average, at low target concentrations, 0.2 and 1.2 % of all of the droplets had intermediate fluorescence with the E. amylovora and R. solanacearum ddPCR, respectively.

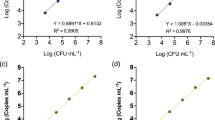
Accuracy of the CFU determinations in the direct ddPCR and the comparison of rain in direct ddPCR and amplification of extracted DNA. Accuracy of quantification with direct ddPCR is shown (a), as well as the distribution of droplets in amplification of extracted DNA and direct ddPCR and per cent of rain droplets for E. amylovora (b and c, respectively) and R. solanacearum (d and e, respectively)
The definetherain analysis is characterised by the exclusion of droplets with intermediate fluorescence (i.e. the rain) [23]. Based on our observations, a certain level of rain droplets is common in ddPCR, and these are likely to be caused by small differences in the amplification efficiencies or delayed amplification among droplets due to the Monte Carlo effect. Other possible reasons for the rain have been proposed, e.g. non-specific amplification [31], irregular droplet size [27], partial inhibition [8] or damaged positive droplets that emit a reduced signal, damaged negative droplets that emit an increased background fluorescence or a mixture of these two [23]. Both Strain et al. [27] and Jones et al. [23] propose exclusion of such droplets from the calculations of concentration. In our study the rain was minimal in the E. amylovora ddPCR, and the data collected did not provide justification for its exclusion from the analysis. The rain was more common in the R. solanacearum ddPCR and did not depend on the sample preparation (i.e. DNA extraction or direct ddPCR), which suggested that this rain was inherent to the assay. In the case of R. solanacearum, we distinguished between the rain and the droplets with signals just above negative signals, for which we hypothesise that they arise from the non-specific amplification of DNA from the plants or their microflora. Again, our data suggest that the rain is mostly caused by the Monte Carlo effect and that it is a useful indicator of the assay quality.
Of the separability measures calculated, the Bhattacharyya distance, which increases depending on the difference between the standard deviations of the two distributions (positive and negative droplets), had the highest resolution, which identifies small differences in the quality of the separation. Together with the percentage of rain droplets, the Bhattacharyya distance provides a useful way to describe the data quality and can be a useful parameter during the optimisation steps of ddPCR. In the assessment of the R. solanacearum ddPCR performance with variable primer concentrations, the Bhattacharyya distance was highest with the lowest primer concentrations. This indicates the best separation between negative and positive droplets despite the two clusters being closer to each other than with the higher primer concentrations used. On the contrary, at the lowest primer concentrations, the percentage of rain droplets out of the positive droplets was highest, which suggests that both of these parameters need to be taken into account in the selection of the optimal primer concentrations (see ESM 8).
The developed R script is open source and is thus available for further improvements through the incorporation of other possible parameters and/or analysis approaches.
Direct ddPCR for rapid and accurate determination of target concentrations
Direct ddPCR was performed on serial dilutions of the E. amylovora and R. solanacearum bacteria in 10 mM phosphate buffer by directly adding bacteria to the ddPCR reaction mixture. The data were assessed by comparing the concentrations determined with the concentrations expected i.e. with the starting concentrations in CFUs/mL. In direct ddPCR, we relied on the polymerase activation step (10 min at 94 °C) at the beginning of the PCR amplification to release the DNA from the bacterial cells. Inevitably, this leads to a higher level of interference in the droplets from the bacterial components in the PCR, the influence of which was seen as an increased number of droplets with intermediate fluorescence (Fig. 4). Interestingly, in the E. amylovora assay, the extracted DNA showed more rain than for the analysis of the comparable amount of DNA prepared by the dilution of a more concentrated DNA sample (Fig. 4). This suggests that the extraction itself introduces some inhibitors into the PCR amplification, either from the buffers used in the extraction and purification itself, or from more efficient co-extraction of bacterial components.
Despite the occurrence of rain, the estimations of the E. amylovora concentrations using direct ddPCR were closer to the true starting values, as expressed in CFU/mL, than the quantification following the DNA extraction (see ESM 5). This also means that the majority of the cells in the bacterial suspensions of E. amylovora were alive and formed colonies. The quantification range of the E. amylovora direct ddPCR extended over four log units (from 104 to 108 CFU/mL of the starting concentration; Fig. 4). The concentration of R. solanacearum in CFUs was accurately determined with the direct ddPCR (Fig. 4), although it was slightly overestimated through the analysis of the extracted DNA and its dilutions (see ESM 5). However, the positive bias was consistently the lowest in direct ddPCR. In direct ddPCR, the DNA is only released from the bacteria when they are already inside the droplets, and thus the multiple-copy targets from the same genome are not separated into discrete droplets. This suggests that the major part of the positive bias is probably due to the discrepancy between the initial number of CFUs and all of the cells present (including dead and viable but non-culturable cells), while the remaining positive bias might be due to multiple copies of the target sequence.
The strong correlation between the number of target DNA copies and the CFUs makes both of these direct ddPCR assays very useful for direct, rapid and accurate determination of bacterial/target concentrations in suspensions. These suspensions can then be used for preparation of in-house reference materials for various assays and to determine absolute DNA extraction efficiencies.
Conclusions
This is the first report on the suitability of ddPCR for routine detection of the quarantine plant pathogenic bacteria E. amylovora and R. solanacearum, which are causative agents of fire blight of rosaceous plants and potato brown rot, respectively. We report here on the assessment of the ddPCR performance, which combined the determination of the relevant ddPCR parameters with the selection of a suitable analysis approach for a defined set of samples with known health status, including (i) NTCs, (ii) negative plant material, (iii) serial dilutions of target DNA and (iv) artificially prepared samples with target concentrations relevant to routine testing (in this case, log 3 cells/mL plant extract).
The two assays examined differed in the target copy number per genome (one for E. amylovora and three for R. solanacearum) and their behaviour in qPCR. The E. amylovora assay shows good analytical sensitivity, and there were no cross-reactions during the extensive validation and its routine use. On the contrary, the R. solanacearum assay lacked the desired analytical sensitivity when used on samples of potato tubers and occasionally showed cross-reactions (see also [21]).
In the ddPCR format, the performance of the E. amylovora assay was comparable to its qPCR format, despite the reduced number of replicates (one in ddPCR compared with three in qPCR), which makes the ddPCR the first choice for characterisation of in-house reference materials and for any application where quantification is also required.
In contrast, for the R. solanacearum assay, where previous data and the data from this study indicate that its design might not be optimal, the ddPCR format significantly improved both its analytical and diagnostic sensitivity when analysed with a manual global threshold that was defined on a set of reference samples. Also, the data analysis and interpretation of the ddPCR was considerably simplified in comparison with qPCR. We therefore propose that the ddPCR format of this R. solanacearum assay is more suitable for routine analysis of samples than qPCR.
Overall, the optimal analysis approach depended on the assay characteristics. In general, the proprietary QuantaSoft analysis required data of high quality, and the algorithm often failed with the analysis of the R. solanacearum ddPCR data. In the quantification of E. amylovora, which has been well optimised already in the qPCR format, the data from the QuantaSoft analysis were similar to those obtained after the application of a global threshold set just above the negative droplets.
The R script enabled the automation of the ddPCR data analysis under different settings, thus significantly simplifying the optimisation of this step. The R script is also used to calculate additional parameters related to the occurrence of rain and to the quality of the separation of the negative and positive droplets via the Bhattacharyya distance. Using these parameters in optimisation of primer concentrations, we have generated apparently counter-intuitive data that show that the separation is better with lower primer concentrations (see also ESM 8). The R script is open source and can be used for exploratory analysis of ddPCR data in general. It also has the potential to be used for other analyses (e.g. clustering of droplets, as used in the definetherain analysis) and for the calculation of additional parameters. Specifically, the usefulness of the separability measures needs further investigation.
In addition to being suitable for detection and quantification of target copies in plant samples after the DNA extraction and purification step, both of these ddPCR assays were successfully performed on pure bacterial suspensions without any DNA extraction step. This direct ddPCR allowed the rapid determination of target concentrations that correlated well with the starting CFUs. This confirms the suitability of ddPCR for absolute quantification of E. amylovora in R. solanacearum preparations of in-house reference materials and for control samples for interlaboratory studies. This is of particular importance in the field of plant pathogen diagnostics, where no reference materials are commercially available.
Further improvements to the assays in the ddPCR format will be possible by analysing multiple reactions per sample and via multiplexing of assays that target different regions of the target pathogen DNA.
Abbreviations
- CFU:
-
colony forming units
- Cq:
-
quantification cycle in qPCR
- ddPCR:
-
droplet digital PCR
- dPCR:
-
digital PCR
- NCPPB:
-
UK National Collection of Plant Pathogenic Bacteria
- NTC:
-
no-template control
- PCR:
-
polymerase chain reaction
- qPCR:
-
quantitative/real-time PCR
- SD:
-
standard deviation
References
Sykes PJ, Neoh SH, Brisco MJ et al (1992) Quantitation of targets for PCR by use of limiting dilution. Biotechniques 13:444–449
Baker M (2012) Digital PCR hits its stride. Nat Methods 9:541–544. doi:10.1038/nmeth.2027
Pinheiro LB, Coleman VA, Hindson CM et al (2012) Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal Chem 84:1003–1011. doi:10.1021/ac202578x
Huggett JF, Whale A (2013) Digital PCR as a novel technology and its potential implications for molecular diagnostics. Clin Chem 59:1691–1693. doi:10.1373/clinchem.2013.214742
Morisset D, Štebih D, Milavec M et al (2013) Quantitative analysis of food and feed samples with droplet digital PCR. PLoS One 8:e62583. doi:10.1371/journal.pone.0062583
Roberts CH, Last A, Molina-Gonzalez S et al (2013) Development and evaluation of a next-generation digital PCR diagnostic assay for ocular Chlamydia trachomatis infections. J Clin Microbiol 51:2195–2203. doi:10.1128/JCM.00622-13
Rothrock MJ, Hiett KL, Kiepper BH et al (2013) Quantification of zoonotic bacterial pathogens within commercial poultry processing water samples using droplet digital PCR. Adv Microbiol 03:403–411. doi:10.4236/aim.2013.35055
Dingle TC, Sedlak RH, Cook L, Jerome KR (2013) Tolerance of droplet-digital PCR vs real-time quantitative PCR to inhibitory substances. Clin Chem. doi:10.1373/clinchem.2013.211045
Racki N, Morisset D, Gutierrez-Aguirre I, Ravnikar M (2014) One-step RT-droplet digital PCR: a breakthrough in the quantification of waterborne RNA viruses. Anal Bioanal Chem 406:661–667. doi:10.1007/s00216-013-7476-y
Pirc M, Ravnikar M, Tomlinson J, Dreo T (2009) Improved fireblight diagnostics using quantitative real-time PCR detection of Erwinia amylovora chromosomal DNA. Plant Pathol 58:872–881. doi:10.1111/j.1365-3059.2009.02083.x
Weller SA, Elphinstone JG, Smith NC et al (2000) Detection of Ralstonia solanacearum strains with a quantitative, multiplex, real-time, fluorogenic PCR (TaqMan) assay. Appl Environ Microbiol 66:2853–2858
EPPO (2014) PQR - EPPO database on quarantine pests. http://www.eppo.int. Accessed 26 May 2014
EPPO (2013) PM 7/20 (2) Erwinia amylovora. EPPO Bull 43:21–45. doi:10.1111/epp.12019
European Commission (1998) Council Directive 98/57/EC of 20 July 1998 on the Control of Ralstonia solanacearum (Smith) Yabuuchi et al. Off J 0001–0039
Huggett JF, Foy CA, Benes V et al (2013) The digital MIQE guidelines: minimum information for publication of quantitative digital PCR experiments. Clin Chem 59:892–902. doi:10.1373/clinchem.2013.206375
King EO, Ward MK, Raney DE (1954) Two simple media for the demonstration of pyocyanin and fluorescin. J Lab Clin Med 44:301–307
Pastrik K-H, Elphinstone JG, Pukall R (2002) Sequence analysis and detection of Ralstonia solanacearum by multiplex PCR amplification of 16S–23S ribosomal intergenic spacer region with internal positive control. Eur J Plant Pathol 108:831–842. doi:10.1023/A:1021218201771
EPPO (2010) PM 7/98 (1): specific requirements for laboratories preparing accreditation for a plant pest diagnostic activity. EPPO Bull 40:5–22. doi:10.1111/j.1365-2338.2009.02347.x
Dreo T, Pirc M, Ravnikar M (2012) Real-time PCR, a method fit for detection and quantification of Erwinia amylovora. Trees 26:165–178. doi:10.1007/s00468-011-0654-7
Reisenzein H, López MM, Duffy B et al (2010) Development and validation of innovative diagnostics tools for the detection of fire blight (Erwinia amylovora) (ERWINDECT): EUPHRESCO final report. 65
Lenarčič R, Morisset D, Pirc M et al (2014) Loop-mediated isothermal amplification of specific endoglucanase gene sequence for detection of the bacterial wilt pathogen Ralstonia solanacearum. PLoS One 9:e96027. doi:10.1371/journal.pone.0096027
Afonina I, Ankoudinova I, Mills A et al (2007) Primers with 5′ flaps improve real-time PCR. Biotechniques 43:770, 772, 774
Jones M, Williams J, Gärtner K et al (2014) Low copy target detection by Droplet4Digital PCR through application of a novel open access bioinformatic pipeline, ‘definetherain’. J Virol Methods 202:46–53
R Development Core Team (2008) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0. http://www.R-project.org
Bhattacharyya A (1943) On a measure of divergence between two statistical populations defined by their probability distributions. Bull Calcutta Math Soc 35:99–109
Alexandris N (2011) Burned area mapping via non-centered PCA using public domain data and free open source software. http://www.freidok.uni-freiburg.de/volltexte/8399/. Accessed 26 May 2014
Strain MC, Lada SM, Luong T et al (2013) Highly precise measurement of HIV DNA by droplet digital PCR. PLoS One 8:e55943. doi:10.1371/journal.pone.0055943
Hayden RT, Gu Z, Ingersoll J et al (2013) Comparison of droplet digital PCR to real-time PCR for quantitative detection of cytomegalovirus. J Clin Microbiol 51:540–546. doi:10.1128/JCM.02620-12
Schachter J (2013) Will droplet digital PCR become the test of choice for detecting and quantifying ocular Chlamydia trachomatis infection? Maybe not. Expert Rev Mol Diagn 13:789–792. doi:10.1586/14737159.2013.847792
Picard C, Silvy M, Gabert J (2006) Overview of real-time RT-PCR strategies for quantification of gene rearrangements in the myeloid malignancies. Methods Mol Med 125:27–68
Witwer KW, McAlexander MA, Queen SE, Adams RJ (2013) Real-time quantitative PCR and droplet digital PCR for plant miRNAs in mammalian blood provide little evidence for general uptake of dietary miRNAs: limited evidence for general uptake of dietary plant xenomiRs. RNA Biol 10:1080–1086. doi:10.4161/rna.25246
Acknowledgments
This study was financed by the Slovenian Research Agency (contract P4-0165) and by the Euphresco Phytosanitary ERA-NET. The ddPCR equipment used in this study was financed by the Metrology Institute of the Republic of Slovenia (MIRS) with financial support from the European Regional Development Fund. The plant material used in this study was collected by the Administration of the Republic of Slovenia for Food Safety, Veterinary and Plant Protection, of the Ministry of Agriculture and Environment and Phytosanitary Inspectorate. The authors would like to acknowledge Nejc Rački for critical discussions, Lidija Matičič and Špela Prijatelj Novak for technical assistance, and Chris Berrie for language editing and proofreading.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the topical collection Nucleic Acid Quantification with guest editors Hendrik Emons and Philippe Corbisier.
Rights and permissions
About this article

Cite this article
Dreo, T., Pirc, M., Ramšak, Ž. et al. Optimising droplet digital PCR analysis approaches for detection and quantification of bacteria: a case study of fire blight and potato brown rot. Anal Bioanal Chem 406, 6513–6528 (2014). https://doi.org/10.1007/s00216-014-8084-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-014-8084-1