
Abstract
Fire blight, a devastating disease of pome fruit trees continues to pose threat to agricultural production. Detection of its causative agent, bacterium Erwinia amylovora, is usually straightforward in symptomatic samples. Methods with increased sensitivity however, are sometimes needed for detection of E. amylovora and real-time PCR assays have been shown to have required sensitivity and reliability. Here we summarize our previous results on real-time PCR detection of fire blight and present new, fast and sensitive real-time PCR assay based on amsC gene performed on SmartCycler® instrument. The setting is optimal for analysis of small number of samples in the laboratory or for on-site detection. Many advantages of real-time PCR assays warrant their use in detection and diagnosis of E. amylovora, particularly in detection of low concentrations of target bacteria e.g. in testing for latent infections. It is to be expected that the use of real-time PCR will increase in both diagnostics and in research, as a tool for target detection and quantification as well as for gene expression analysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction: fire blight and real-time PCR
Fire blight is a devastating disease of pome fruit trees and many other plants of Rosaceae family (van der Zwet and Beer 1995). Crataegus, Cotoneaster, Photinia davidiana, Sorbus spp. have been found especially important for the spread of E. amylovora under European conditions and their new planting is restricted or prohibited in some areas (Gianetti et al. 2004; Duffy et al. 2005). The disease is thought to originate from indigenous American plants, such as hawthorn (Crataegus), native crab apples (Malus), mountain ash (Sorbus) and perhaps others (Thomson 2000) and became a major concern when European apples and pears were introduced to Northern America. The disease, caused by a bacterium Erwinia amylovora (Burrill 1882) Winslow et al. (1920) can decimate apple and pear orchards in a single season and can disrupt orchard production for several years (Bonn and van der Zwet 2000). Bacterium induces necrosis of plant tissues and can rapidly migrate through cortical parenchyma. It can affect all parts of trees, causing blossom blight, spur and shoot blight, cankers on twigs and trunks, rootstock blight and fire blight of fruit. Most conspicuous symptoms include bacterial ooze on wilting green shoots and shepherd’s crook (van der Zwet and Beer 1995). In the absence of efficient chemical control, removal of infected material is a necessary part of fire blight management, ranging from cutting of infected trees to uprooting whole orchards.
While being a poor epiphyte, E. amylovora can be present in plants causing no visible symptoms for longer periods. Such latently infected or symptomless plant material has been recognized as an important means of its spread to pathogen-free areas through infected bud wood or trees (Bonn and van der Zwet 2000). Despite efforts to limit it, the disease continues to spread to new geographical areas (van der Zwet 2006).
Faced with high economic consequences of fire blight there is a need for highly reliable laboratory methods of disease detection and identification of the causative bacteria. In case of symptomatic plants identification is usually straightforward as bacteria are present in large numbers and grow well on artificial media. Reliable diagnosis, however, can be difficult when bacteria are hindered in their growth on artificial media, or the population size is low due to latent infections. E. amylovora populations are also low in dormant cankers in winter and spring, when plant inspectors and farmers control their plants and do cutting and pruning.
The European and Mediterranean Plant Protection Organization (EPPO) have published guidelines compiling methods that allow detection, identification and confirmation of E. amylovora in plant material as a diagnostic protocol (EPPO 2004). Since its publication, new methods became available or were further developed. In addition to many PCR methods, several real-time PCR assays are now available (Salm and Geider 2004; De Bellis et al. 2007; Lehman et al. 2008; Mohammadi et al. 2009; Pirc et al. 2009; Svircev et al. 2009), previously reviewed by Dreo (2009). In this review, we complete the data on published real-time PCR assay (including Gottsberger 2010), summarize the use and potential of real-time PCR methods in the detection of fire blight and describe transfer and optimization of a previously developed Ams assay (Pirc et al. 2009) onto a faster SmartCycler® real-time PCR detection system.
Real-time PCR assays for detection and identification of Erwinia amylovora
Available chemistries and target DNA sequences
Real-time PCR systems for detection of Erwinia amylovora were developed with different aims and authors have thus concentrated on different characteristics to make their assays fit for their purpose (Dreo 2009). First real-time PCR for detection of E. amylovora (Salm and Geider 2004) included SYBR Green and TaqMan chemistry (hydrolysis probes). SYBR Green, an intercalating dye, is included in the reaction mixture and specifically binds to double stranded DNA and emits fluorescence upon binding. The SYBR Green chemistry was popular due to its low costs, since no labeled oligonucleotides are required. Basically any PCR assay can be run with a suitable intercalating dye, SYBR Green or other, and the accumulation of product then measured by an increase in fluorescence using real-time PCR instrument. The downside of SYBR Green chemistry and the reason for its current lesser use in phytobacteriology is that any double stranded DNA including primer-dimers will generate fluorescence. Thus, a positive signal is not necessarily specific for our target and by itself provides no information on the DNA sequence of the amplified product or its size. To derive as much information from real-time PCR employing SYBR Green chemistry as from classical PCR, the reaction should be followed by an additional step in which melting temperature (Tm) of generated product is determined i.e. melting curve analysis. The shape of the melting curve and the determined melting temperature depend on the size of the PCR product, its concentration, nucleotide base composition and to some extent on its nucleotide sequence.
Hydrolysis (TaqMan) probes rely on the 5′ > 3′ nuclease activity of Taq polymerase. When polymerase encounters annealed probe during elongation of primer, the probe is cleaved and the two labels attached to the ends of the probe dissociate in solution. Due to their physical properties their fluorescence changes when the distance between them increases, energy transfer between the two dyes can no longer take place and the fluorescence of a reporter dye (e.g. FAM) is detected and measured. In case of fluorescent quencher dyes (e.g. TAMRA) the wavelength of their fluorescence also changes during amplification due to a decrease in the energy transferred to them from reporter dyes. Using TaqMan chemistry, primers and probes are designed in such a way that the extent of target sequence not complementary to them is minimized, which is reflected in a shorter final product than in conventional PCR with final amplification products typically ranging from 70 to 110 base pairs. The signal is generated only when both primers and a probe, together often covering most of the target sequence anneal to it. The choice of the target sequence is therefore crucial for the specificity of the test. Regardless of the chemistry employed, generated fluorescence is measured by real-time PCR instrument and analyzed by accompanying software which also provides data normalization and usually some automatic options of analysis. The result is a calculated cycle in which a positive signal is obtained i.e. a cycle when the generated fluorescence significantly rises above the background fluorescence or a point of maximum second derivative of the signal amplification. This point is labeled Ct for ‘cycle threshold’ on Applied Biosystems and SmartCycler® instruments; however, we use term ‘quantification cycle’ or ‘C q’ in this manuscript as proposed in MIQE guidelines (Bustin et al. 2009).
Targets for conventional PCR methods for detection of E. amylovora have historically included plasmid pEA29 or chromosomal DNA. Due to several copies of pEA29 per cell as opposed to often one copy of chromosomal DNA targets, plasmid based system were considered more sensitive and were thus a preferred method. Plasmid pEA29 is present in vast majority of E. amylovora strains and seems to be specific to E. amylovora, while cross-reactions and lower sensitivity were sometimes reported in PCRs targeting chromosomal targets (Bereswill et al. 1995; Maes et al. 1996; Llop et al. 2000; Roselló et al. 2002). Not surprisingly therefore, the first real-time PCR developed for E. amylovora was designed to detect a target sequence on the pEA29 plasmid (Salm and Geider 2004). Similarly, De Bellis et al. (2007) have chosen pEA29 as a target in their proof-of-principle paper combining Scorpion primers with nested PCR.
Potential existence of E. amylovora strains without pEA29 plasmid has been recognized early on but they have only been found in nature fairly recently (Llop et al. 2006) and seem to be rare (Mohammadi et al. 2009). While the biological significance of strains lacking pEA29, or any plasmid, is not yet resolved, a method based on detection of chromosomal DNA rather than plasmid DNA seems more suitable when detection of all strains is desired. Real-time PCR systems developed later therefore employed chromosomal targets, either those previously used in classical PCR or newly selected DNA sequences.
The ams region of the E. amylovora chromosome that is involved in the synthesis of the capsular polysaccharide amylovoran has been used as a target sequence in several classical PCR and later in real-time PCR methods (gene amsK in Mohammadi et al. 2009; gene amsC in Pirc et al. 2009). The amylovoran is reported to be unique to E. amylovora (Bugert and Geider 1995; Bereswill et al. 1995) and is strongly associated with its multiplication in plants and its virulence (Steinberger and Beer 1988; Menggad and Laurent 1998). Sequences for the production of levan sucrose, another exopolysaccharide contributing to E. amylovora virulence, have been chosen as a target by Lehman et al. (2008). Intergenic spacer regions between 16S and 23S ribosomal DNA (ITS) have been useful in the detection of many bacterial species (Li and De Boer 1995; Pastrik et al. 2002). By analyzing ITS regions in E. amylovora several copies of rRNA operons were found, with a 139 bp sequence named ‘optional sequence’ present only in E. amylovora strains from fruit trees and not in the isolate from Rubus spp. (McGhee et al. 2002); this ‘optional sequence’ was consequently used in real-time PCR developed by Pirc et al. (2009) and the assay shown to detect strains without pEA29 plasmid as well as Erwinia strains from a Japanese island Hokkaido. Real-time PCR designed by Gottsberger (2010) targets a hypothetical protein.
As in other research fields, early papers report the use of both SYBR Green and hydrolysis probes (TaqMan) chemistries for detection purposes (Salm and Geider 2004). Also a real-time PCR system based on the use of Scorpion bi-probes was developed but has not shown advantages over other systems particularly with respect to sensitivity. The authors have overcome this shortcoming by combining the real-time PCR assay with a previous nested step (De Bellis et al. 2007). Despite successfully increasing sensitivity, the transfer of reaction products from nested to the real-time PCR step increases the possibility of contaminations, and thus this method has not been widely accepted. All other assays were developed with TaqMan probes employing technological advances such as availability of more efficient quencher molecules (Mohammadi et al. 2009; Svircev et al. 2009) that increase the signal to noise ratio (Marras et al. 2002).
Most published real time PCR assays for E. amylovora detection are tested with regards to their specificity and sensitivity however; the extent of testing does vary according to the purpose of the study. High sensitivity is crucial if a test is to be used as a screening test and specificity is of higher importance when it is to be applied to pure cultures for identification purposes. While the extent of validation certainly depends on the purpose, a need for a good and reliable method is common to research and diagnostic laboratories. Lots of effort has therefore been put into agreeing on a common strategy to validations in the last years among researchers in the EPPO region. Consequently, basic requirements for validation of diagnostic methods were published as EPPO standards (EPPO 2007) and there is a continuous effort to gather validation data on the most frequently used tests.
It is to be expected that novel potential DNA targets for real-time PCR and other tests will become available with further accumulation and critical analysis of data obtained with genome sequencing (Sebaihia et al. 2010; Smits et al. 2010a; Powney et al. 2011).
Analytical specificity
The first step in guarantying specificity is choosing appropriate DNA target sequence. Oligonucleotide primers and probes are then usually designed using suitable design software. Checking primer’s and probe’s sequences, as well as the whole PCR product against sequences deposited in nucleotide databanks such as NCBI (Genbank, National Center for Biotechnology Information, Bethseda, MD) is an easy step that can prevent many unnecessary experiments. The conclusive results however are still generated through empirical testing. Specificity has been tested for all designed real-time PCR assays although the degree of testing differs between laboratories and research projects (Dreo 2009). Commonly tested strains used by most authors include, in addition to a selection of known E. amylovora strains, Erwinia pyrifoliae and Erwinia spp. isolated in Japan. The last two bacteria are the most likely non-E. amylovora bacteria to give positive reactions in tests designed for E. amylovora due to their intermediate taxonomical status and genetic similarity (Won-Sik et al. 2001; Maxson-Stein et al. 2003; Geider et al. 2009). Indeed, their intermediate status has been supported by real-time PCR analysis targeting hrpW and ITS sequences. Real-time PCR analysis of hrpW, originally designed to detect E. pyrifoliae however, also detected Erwinia spp. from Japan while no amplification was observed with E. amylovora (Lehman et al. 2008). The ITS real-time PCR gave a positive reaction in more than 200 E. amylovora strains isolated from different hosts (Pirc et al. 2009), indicating that this sequence is widely present in the target pathogen; and although no unspecific amplifications were observed a clear positive signal was generated with Erwinia spp. strains from Japan (Hokkaido) but not with E. pyrifoliae. As in both cases the signals are generated because of specific amplification of similar/identical sequences, these tests have potential for detection of a broader range of pathogens. Several genome sequencing projects have been completed or are in progress; it is expected that a comparative genomic analysis of these species will eventually lead to clarification of the relationships among these strains (see e.g. Smits et al. 2010b).
Other closely related bacteria or other bacteria expected in the same environment have been tested at least by some authors in determining specificity: Erwinia spp. isolated in Australia, Erwinia sp. (Roselló et al. 2002; López et al. 2010) isolated from necrotic pear blossoms in Spain, Erwinia billingiae, Erwinia tasmaniensis, Erwinia persicina, Pectobacterium atrosepticum, Dickeya chrysanthemi, different Pseudomonas spp. and Pantoea agglomerans, including biocontrol agent strains (Pirc et al. 2009). In most of these cases the number of available strains was limited and the current results on specificity are only as reliable as the available isolates are representative of the species diversity. Pirc et al. (2009) focusing on development of real-time PCRs for diagnostic purposes in plant samples, have included additional uncharacterized strains isolated from necrotic tissues of fire blight hosts and DNA extracted from the same hosts and associated micro flora (24 bacterial strains isolated from necrotic apple, pear and quince samples which tested negative for E. amylovora by conventional methods), while Gottsberger (2010) included other selected bacteria e.g. Enterobacter amnigenus (ammonia reducing product) and Raoultella terrigena (tree fertilizer injector).
As with any other test, most relevant information on the specificity of real-time PCR will be gathered during its use on a wide range of samples. In the absence of other as sensitive or more sensitive test it is useful to have several assays targeting different regions of E. amylovora genome thus increasing reliability of diagnosis (Pirc et al. 2009; Gottsberger 2010).
High analytical sensitivity of real-time PCRs and DNA extraction
The approach to determine analytical sensitivity, i.e. smallest amount of target that can be detected reliably (EPPO 2010a), commonly involves spiking, that is adding target bacteria in a range of concentrations to plant samples or extracts prepared from plant tissues, and then processing them by the chosen methods of DNA extraction and real-time PCR (EPPO 2010b).
Sensitivity of real-time PCR is affected by many factors including: inherent characteristics of the real-time PCR assay, type and amount of plant material, DNA target copy number, DNA extraction and purification method itself, volume of sample analyzed in real-time PCR, total reaction volume and the number of reactions performed. The reported sensitivity encompasses all these details and changes in any of them may influence performance of real-time PCR and merit verification of method’s performance.
A reliable detection method for fire blight aims to be able to detect E. amylovora in different plant species, symptomatic and asymptomatic plant material and, ideally, in host plants of different genera. Using a detection method based on amplification of DNA usually requires its extraction and purification from samples, particularly if samples are rich in polyphenols or other inhibitors of polymerase. In case of real-time PCR assays, a critical step to address in this respect is the use of DNA extraction methods that successfully removes inhibitors.
While in most studies using real-time PCR healthy plant material is used for preparation of spiked plant samples, such an approach may seriously underestimate the influence of inhibitors present in natural samples and may be too optimistic in reporting sensitivity. Especially, comparing different methods, these may be affected by necrotic plant tissues to a different extent and thus their sensitivity and reliability in real-life situations can differ significantly from their performance in healthy tissues. The different amounts of inhibitors in individual plant material give rise to additional variation in sensitivity. Variability in the level of inhibition among different plant hosts and between individual samples of the same host was reported (Pirc et al. 2009), confirming observation by Maes et al. (1996) that the inhibition in a particular sample seems to be more affected by the amount of necrosis and physiological age of tissue than the plant species itself.
When selecting a DNA extraction and purification method, the final decision on it often depends not only on the type of samples but also on their number (requirement for a high-throughput method) and current protocols already in place in laboratories. No doubt many of the available methods are suitable and efficient in isolating E. amylovora bacteria from plant tissues and DNA from bacteria themselves as well as being able to remove inhibitors. The potential of using real-time PCR for comparison and evaluation of DNA extraction methods was fully exploited by Pirc et al. (2009) comparing methods based on different principles of DNA extraction (isopropanol precipitation—modified Llop et al. 1999, silica columns, magnetic beads). Of the tested DNA extraction methods, magnetic beads and silica column-based methods were found to be most successful in removing inhibitors leading to increased amplification efficiencies in real-time PCR (Pirc et al. 2009). However, irrespective of the DNA extraction (isopropanol, silica-columns, magnetic beads) and real-time PCR system used (targets amsC, ITS and protocol originally developed by Salm and Geider 2004 with modifications), concentrations at and above 103 cells/ml plant extracts (less than 4 cells per reaction) could always be detected (Pirc et al. 2009). Detection at low concentrations (samples with 100 cells/ml plant extract) was sometimes observed with probability of detection from 0.09 to 0.91 and was affected by both DNA extractions and real-time PCR, specifically target copy number. Another example of a comprehensive comparison of DNA extraction method for detection of E. amylovora, although using healthy plant material, was done in the frame of Euphresco ERA-NET project ERWINDECT (Reisenzein et al. 2010).
Sensitivity of a test can be reported in different ways e.g. 8.6 × 103 cells from leaf sample and 1.8 × 104 cells from bark sample (Salm and Geider 2004), 3.2 × 104 CFU/mL (De Bellis et al. 2007), 20 CFU/reaction (Lehman et al. 2008), <500 CFU/reaction (Mohammadi et al. 2009) and 100 CFUs/reaction (Svircev et al. 2009). Taking into account the way the samples are prepared and analyzed these values generally correspond to 103 E. amylovora cells/mL of starting plant extract.
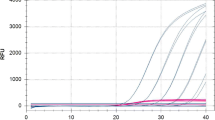
In reporting sensitivity, authors rarely define term ‘reliable detection’. There is no consensus on what is the probability of detection corresponding to reliable detection although a probability of 0.90 or 0.95 is often mentioned. To accurately determine a concentration at which probability of detection is e.g. 0.90 high numbers of replicates need to be analyzed (Navidi et al. 1992) which is rarely practical. Close enough approximation can be determined however through non-linear modeling of a limited amount of data (Burns and Valdivia 2008). The R Project for Statistical Computing (R Development Core Team 2010) allows such modeling in the frame of drc package (Ritz and Streibig 2005). Using the two-parameter Weibull model with upper limit at 1 resulted in determined analytical sensitivity of the Ams assay of 5 × 103 CFU/mL plant extract (P = 0.90) and 9 × 103 CFU/mL plant extract (P = 0.95) with calculated C q values of 34.60 and 33.23 respectively (Fig. 1).

Non-linear modeling of probability of detection of E. amylovora DNA in real-time PCR as a function of its concentration on 7900HT Sequence detection System (full line) and SmartCycler® (dotted line) using Ams assay. Data points represent average C q values obtained from three separate decimal dilutions of E. amylovora in necrotic tissue of apple with final concentrations of E. amylovora from 0 to 106 CFUs/mL of plant extract. DNA was extracted by QuickPick™ SML Plant DNA Kit (Bionobile, Turku, Finnland). Horizontal line indicates probability of detection of 0.90
Another useful information on analytical sensitivity of real-time PCR can be obtained through analysis of basic characteristics such as (i) quantification (de Kok et al. 1998), (ii) estimation of PCR efficiency calculated e.g. using formula E = (10[−1/k]) − 1 (Pfaffl 2001) and theoretical one copy detection C q (Fig. 2). The aim is to have an efficient amplification (corresponding to efficiency of 1.00) that leads to lower C q values which are easier to distinguish from background signals.

Determination of theoretical C q value from linear regression between logarithmic values of Erwinia amylovora CFUs per reaction and determined C q values. Data points represent average C q values obtained from three separate decimal dilutions of E. amylovora in necrotic tissue of apple with final concentrations of E. amylovora from 0 to 106 CFUs/mL of plant extract. DNA was extracted by QuickPick™ SML Plant DNA Kit (Bionobile, Turku, Finnland). Ams assay was performed on 7900HT Sequence Detection System. Theoretical C q value of one target copy per reaction is 37.30, while a slope of −3.47 corresponds to 94% amplification efficiency
Due to higher sensitivity of real-time PCR there is usually a range of low target concentrations that can only be detected as positive using real-time PCR and not by other, less sensitive methods. This can give rise to some difficulties in reporting results and their interpretation. Concept of zero-tolerance for quarantine pathogen presence can come in conflict with requirement of diagnostic schemes to have at least two methods with positive results to be able to report a sample as suspicious or with a requirement to isolate bacteria in pure culture (EPPO 2004). To some extent this can be resolved by using a combination of real-time PCRs with different target sequences to increase the reliability of detection (Pirc et al. 2009; Gottsberger 2010). Recently, several approaches were described on determining a so called “cut-off value” C q above which signals are no longer interpreted as positive based on statistical analysis of detection probability and analysis of naturally infected samples (Chandelier et al. 2010; European Patent Application EP1770172). It is important to note that this cut-off C q value enables us to distinguish between samples with low-level target organisms from samples where its presence is not conclusive due to stochastic effects and Poisson distribution of positive results.
In contrast with detection of E. amylovora in low concentrations, DNA extraction is not as important in analysis of symptomatic material. Especially when the target plant material is expected to contain low levels of inhibitors, simple DNA extraction procedures may be sufficient and there is a lesser need for extensive purification of DNA. Analyzing pure bacterial cultures or ooze, colony PCR is a quick and valuable option as most DNA polymerases in use today require an incubation step at 95°C for 5–15 min for enzymatic activation during which bacterial cells added directly to the reaction are disrupted and release DNA. Such approach has been used for analysis of pure cultures (Svircev et al. 2009) and is indeed a practical approach for identification of E. amylovora isolates. In a study conducted on pure bacterial cultures, Mohammadi et al. (2009), as well as previously Salm and Geider (2004), have used Tween 20 detergent (0.1–1%, with or without heating) to disrupt cells prior to real-time PCR reaction. However, this may not be necessary and in fact, too high concentrations of the detergent may have an adverse effect on DNA polymerase stability and PCR amplification.
Lehman et al. (2008) reported using a simple Direct Plant Extraction Buffer (DiPEB) for DNA extraction from artificially inoculated pear blossoms from which petals and peduncles were removed since it is known that they oxidize quickly and can be a source of potent PCR inhibitors. The same buffer was used by Svircev et al. (2009) to isolate E. amylovora from bark, stems, leaves, blossoms, anthers and pollen. However, little data is available on its performance in these different samples. The buffer DiPEB is expected to become commercially available from Agdia Inc. (Elkhart, IN). Stöger et al. (2006) have described a rather straightforward modified protocol of the REDExtract-N-AmpTM Plant polymerase chain reaction kit (Sigma) for detection of E. amylovora that has been extensively tested in combination with classical PCR and has good potential to perform well also in combination with real-time PCR.
Because of high amount of bacteria usually present in symptomatic fire blight samples positive results can often be obtained through direct addition of plant extract to real-time PCR reaction even when it is prepared from necrotic tissues, provided it is diluted at least 1:10 in water and buffer (Salm and Geider 2004; Pirc et al. 2009).
Interestingly, at least in some cases of enriched asymptomatic samples, colony real-time PCR was shown to be at least as or even more efficient than DNA extraction (Pirc et al. 2008).
Since the inhibition seems to depend on a particular symptomatic sample and cannot be generalized over plant species or accurately predicted, omitting DNA extraction and purification procedures in official diagnosis of symptomatic and asymptomatic plant samples is discouraged unless supported by suitable controls.
Testing for latent infection
Enrichment of E. amylovora from plant extracts in non-selective and semi-selective media is advised when low concentrations of bacteria are expected, e.g. in symptomatic samples with abundant tissue necrosis, samples treated with pesticides or bactericidal compounds and symptomless samples (EPPO 2004). Enrichment is expected to increase sensitivity of further screening tests (Llop et al. 2000; EPPO 2004) and facilitate isolation in pure culture by activating viable but non-cultivable cells (Ordax et al. 2006).
Pirc et al. (2009) have used quantification of E. amylovora using real-time PCR to assess the efficiency of enrichment method by spiking a set of 33 asymptomatic samples from twigs, prepared and previously tested as negative according to EPPO guidelines (EPPO 1992, 2004) with low concentrations of E. amylovora and quantified them before and after enrichment. From initial concentrations at LOD level (≈103 cells/ml) E. amylovora cells reliably multiplied in all tested symptomless samples with final concentrations ranging from 105 to 109 E. amylovora cells/mL. In contrast with this promising result, isolation of E. amylovora from enriched samples in pure culture proved difficult. Despite using five tenfold dilutions for plating to avoid problems with overgrowth by other bacteria (an increase from three tenfold dilutions suggested by EPPO 2004), target bacteria could be isolated in pure culture mainly from samples with at least 107 E. amylovora cells/mL of enriched extract. Isolation of bacteria in pure culture was not possible from samples where bacteria did not multiply to that level.
During the last two years we have used real-time PCR to analyze 60 asymptomatic samples of twigs in addition to standard EPPO tests (EPPO 2004). Real-time PCR was applied after enrichment of samples in King’s B and CCT media. As expected, due to sampling in presumably E. amylovora free zones where fire blight symptoms were not previously observed, most samples (93%) were clearly negative by both, real-time PCR and standard EPPO methods. In 4 samples (7%) that were taken in or in immediate proximity of infected areas, suspicious results were obtained using real-time PCR with one, two or all three real-time PCR assays used (Ams, pEA29 and ITS; Pirc et al. 2009) (Table 1). However, despite a previous enrichment step the level of contamination was extremely low and close to the detection limit. This would indicate that there was little or no amplification of E. amylovora. Not surprisingly, attempts to isolate bacteria from these samples in pure culture were not successful. Results in natural samples are thus in contrast with findings of Pirc et al. (2009) where asymptomatic samples were spiked with physiologically active reference culture of E. amylovora. In contrast, when 134 asymptomatic samples were collected from areas where fire blight was present, 15 samples were positive using real-time PCR and isolation was successful from 7 such samples (Gottsberger 2010). Research by Braun-Kiewnick et al. (2011) shows that E. amylovora can be readily detected in naturally infested apple flowers by real-time PCR however, isolation of bacteria was not attempted.
In conclusion, difficulties are expected in proving real-time PCR positive asymptomatic samples as positive according to EPPO guidelines with isolation of bacteria in pure cultures. A consensus is needed on interpretation of such results that are based on molecular methods alone. While the main disadvantage of real-time PCR in this respect is that it detects DNA and thus both living and dead bacteria (as do classical PCR and serological methods), its reliability can be increased through combined use of several assays. In case of suspicious samples the likelihood of E. amylovora presence is higher when more assays based on different DNA sequences are positive e.g. in case of samples 1 and 2 (Table 1). Quantification of E. amylovora prior and after enrichment may be useful in proving its viability however, this can only be achieved when bacteria are multiplying.
The physiological condition of E. amylovora seems to be critical for successful isolation in pure cultures and further data on its survival in asymptomatic samples is needed to optimize testing for latent infection particularly with respect to material and time of sampling.
Compared to other available methods suitable for asymptomatic samples, real-time PCR is the most sensitive, fastest and, through combined use of several assays, the most reliable. Even when its results can not be confirmed by isolation of the pathogen in pure culture it provides relevant information to plant health authorities enabling them to intensify surveys or perform additional analyses.
Quantification and its relevance to fire blight diagnostics and research
Real-time PCR assays can be used in both qualitative as well as quantitative way as the signal generated by degradation of hydrolysis probes correlates to the starting amount of the target. Relative quantification of E. amylovora concentration in a sample in comparison to standard curve provides additional information on the likelihood of its isolation in pure culture and the possibility of confirming the result with other, less sensitive methods. When normalized to cell or CFU number, quantification can only be accurate when the target copy number is constant and does not vary among isolates. Further consideration in accuracy of quantification is the similarity of PCR amplification efficiencies between samples and standard curve (Cankar et al. 2006) however, such accuracy is seldom required in the current testing scheme for detection purposes. For E. amylovora, being a quarantine pathogen with zero tolerance levels, quantification is rarely employed in routine detection as any positive result has the same consequences and is interpreted together with other methods used (EPPO 2004). In effect, real-time PCR is used as a qualitative method with plus/minus results. This does not mean that characteristics relevant to quantification are not important. Amplification efficiency and theoretical C q of one copy are good indicators of a test’s quality and robustness and data obtained from standard curves analysis is necessary for determination of analytical sensitivity and defining a C q cut-off value.
Quantification of E. amylovora using real-time PCR however is more often applied for research purposes. Both Salm and Geider (2004) and De Bellis et al. (2007) have used plasmid based real-time PCRs for quantification of E. amylovora in artificially inoculated detached flowers and leaves. Lehman et al. (2008) have used duplex real-time PCR targeting chromosomal sequences of E. amylovora and E. pyrifoliae to study their interaction upon co-inoculation of detached pear blossoms. In this study concentrations of E. amylovora of less than 100 cells per blossom lead to fire blight symptoms, indicating that threshold population sizes smaller than 4.7 × 104 CFU/blossom can results in infection of hypanthia. This is in contrast to previous studies indicating that higher populations are required for infections (Thomson et al. 1975; Johnson et al. 1993). As Lehman et al. (2008) indicated, the observed differences could be a consequence of the different analytical methods used in related studies (real-time PCR versus plating on media) or different inoculation methods.
Pirc et al. (2009) have quantified E. amylovora concentrations in asymptomatic samples in enrichment showing that subsequent isolation in pure culture requires high concentrations of bacteria above 107 cells/mL enriched plant extracts.
An interesting example of real-time PCR use has been recently employed attempting quantification of E. amylovora in asymptomatic apple blossoms under natural conditions and early in the season using real-time PCR (Reisenzein et al. 2010; Braun-Kiewnick et al. 2011). The studies aim to benefit from high sensitivity of real-time PCR and determine concentrations of E. amylovora in apple blossoms under natural conditions in areas where fire blight has been previously observed. The final objective is to integrate results of E. amylovora testing with prognostic models and thus supporting a more informed use of control substances during flowering (Braun-Kiewnick et al. 2011).
Case study: transfer of amsC targeting real-time PCR assay to a faster, portable format on SmartCycler® instrument
Its speed is one of the advantages of real-time PCR. Improved instruments and formulations of reaction mixtures allow for ever faster amplification and temperature cycling thus shortening the whole analysis time. Protocols developed under slower amplification conditions can often be transferred to faster instruments however, for optimal performance this requires optimization (Bentley et al. 2005). As a case study we have chosen to transfer real-time PCR targeting amsC gene from ABI PRISM®Applied Biosystems 7900HT Sequence Detection System (Pirc et al. 2009) to SmartCycler® detection system (Cepheid). An extra advantage of SmartCycler® instrument is the possibility to control each of the 16 reaction sites in a block (iCores) independently, allowing for parallel testing of different amplification conditions.
Real-time PCR reactions were performed on a SmartCycler® detection system (Cepheid) in 25 μL reactions using TaqMan® Fast Universal PCR Master Mix (Applied Biosystems) with sample volumes of 5 or 10 μL. Primers and probes targeting chromosomal amsC gene were as reported in Pirc et al. (2009) except that in this study black hole quencher 1 (BHQ-1™) was used instead of TAMRA. Primers and probe were synthesized and cleaned to high purity salt free standard and HPLC respectively by Eurofins MWG Operon. Transfer of real-time PCR between instruments included adaptation of amplification protocol and optimization of (i) primers and probe concentrations, tested in final concentrations in reactions from 0.1 to 0.5 μM and from 0.1 to 0.4 μM respectively (Tables 2, 3), (ii) annealing temperature (from 60 to 66°C) and time (from 10 to 40 s) (Table 4), (iii) various concentration of MgCl2 (from 2 to 6 mM) (Table 5), (iv) temperature ramping rate and (v) sample volume.
High annealing temperatures of oligonucleotides above 60, (64.3, 61.6 and 71.5°C for Ams116F, Ams189R and Ams141T respectively) as determined by in silico analysis allowed for the use of a two step protocol where annealing and extension of PCR products is performed in a single step. The starting amplification protocol was chosen based on this and master mix’s manufacturer’s guidelines and was as follows: 95°C, 20 s (initial denaturation) followed by 45 cycles of 95°C, 1 s (denaturation) and 60°C, 20 s (annealing and extension). Temperature ramping, e.g. the speed at which the temperature is changed, was set based on manufacturer’s recommendations at ±3°C/s. Several preliminary runs were performed to determine optimal analysis settings which were then re-checked at every individual optimization step.
Primer optimization revealed that their influence on determined quantification cycle can be significant, with almost 7 C q values difference between minimum and maximum C q determined at constant concentration of target DNA (Table 2). Based on these results, three combinations of forward and reverse primer concentrations were further tested in a step optimizing probe concentration; confirming previous results lower probe concentrations resulted in lower quantification cycle values (Table 3).
Both variations in temperature and time of annealing and extension step influenced quantification cycle (C q) values. As the Ams assay was designed for the universal 7900HT amplification conditions where annealing and extension is performed at 60°C, it is not surprising that at temperatures of 66°C, it resulted in very low signal (high C q) or even absent signal at 66°C, 10 s. With the aim of a faster protocol, annealing time of 20 s at 62°C was chosen for further testing as the determined quantification cycle did not differ much from the best C q obtained with longer annealing times (Table 4). Optimization of MgCl2 concentrations indicated that amplification may benefit from increased concentration of MgCl2, in reaction however the influence was not significant under tested conditions (Table 5).
In both, primer and probe optimization, as well as in further optimization steps, it could be seen that accuracy in preparation of reaction mixtures is vital and experimental set-up that avoids pipetting of small volumes is necessary.
Optimized SmartCycler® protocol for Ams assay was finalized as follows: (i) reaction mixture composition as described in Table 6; (ii) a two-step amplification protocol of initial denaturation (20 s, 95°C) followed by 45 cycles of denaturation (1 s, 95°C) and annealing and extension step (20 s, 62°C) with temperature ramping set to 3°C/s.
Optimized SmartCycler® Ams assay for E. amylovora detection was applied to three samples of apple green tissues exhibiting necrosis that previously tested negative for fire blight according to EPPO protocol and real-time PCR. The aim was to determine whether changed reaction conditions influence analytical sensitivity (EPPO 2010b). Final concentrations of E. amylovora in spiked plant extract ranged from 0 to 106 CFUs/mL. DNA was extracted by QuickPick™ SML Plant DNA Kit (Bionobile, Turku, Finland) as described previously (Pirc et al. 2009) and each sample analyzed in SmartCycler® Ams assay in single reactions. Results on analytical sensitivity and other real-time PCR characteristics were compared to real-time PCR results on 7900HT instrument (Applied Biosystems; Pirc et al. 2009). Samples were analyzed in 7900HT Sequence Detection System in triplicates.
Comparison showed that performance of real-time PCR Ams assay on SmartCycler® instrument is equivalent to its performance on 7900HT Sequence Detection System. Despite a different set up, particularly a much faster reaction and lower concentrations of oligonucleotides on SmartCycler® instrument, there were no big differences in most important characteristics, reaction efficiency and analytical sensitivity (Fig. 1; Table 6) with similar level of 103 E. amylovora CFU/mL of plant extracts detected in both cases (P = 0.90).
The main difference that could be observed was in the dynamics of probability of detecting lower target concentrations. Using 7900HT, a decline of probability of detection started already at 103 CFU/mL (0.78 vs. 1.00 probability of detection at 104 CFU/mL) warranting use of several parallel reactions in analysis. Probability of detection declined to 0.22 at 102 CFU/mL. In contrast, using the SmartCycler® protocol, probability of a positive result at 103 CFU/mL level was 1.00 and zero at 102 CFU/mL. With increased number of reactions and increased volume of sample DNA (10 μL) per reaction, positive signals were observed at 102 CFU/mL level of E. amylovora also in SmartCycler® (P = 0.56). A more accurate information on dynamics of analytical sensitivity at various target concentration levels will be obtained through further analysis of samples.
An increased sample volume lead to an increased sensitivity and extended linear range in one sample (out of 3) in SmartCycler® protocol; linear range in this case included log 2 concentration level which is an advantage when quantification of low levels of E. amylovora is desired.
Further data on a variety of samples is needed to confirm observed differences between the 7900HT and SmartCycler® protocol and future plans include application of both protocols to a variety of samples of different host plants including naturally infected samples. In conclusion, the Ams real-time PCR assay was successfully transferred to SmartCycler® instrument, decreasing time needed for amplification from 1 h 40 min to 30 min, with no impairment of analytical sensitivity.
Conclusions
Real-time PCR assays have proven to be the most sensitive reliable method so far available for detection and diagnosis of bacterial and other plant pathogens (López et al. 2009). Several real-time PCR assay have been developed for detection of Erwinia amylovora and some have been extensively tested with regards to their specificity and sensitivity (Dreo 2009). Most studies have indicated their higher sensitivity and reliability compared to classical PCR (Pirc et al. 2009; Gottsberger 2010). Their advantages warrant their use in detection and diagnosis of E. amylovora, particularly in (i) detection of low concentrations of target bacteria e.g. in testing for latent infections and in (ii) samples where other methods may be disadvantaged by the sample characteristics e.g. necrotic samples, samples recently treated with biological control agents and/or when co-infection with other harmful organisms is expected. As described, major issue that needs to be addressed with respect to diagnostic work is directly related to high sensitivity of the assays. As with PCR, interpretation is difficult in case of dubious or low level E. amylovora presence that cannot be confirmed by any other method much less by isolation in pure culture. Despite discouraging results from a limited number of plant samples, providing a proof of viability of E. amylovora by its quantification at the start and end of enrichment culture remains an option worth exploring (Pirc et al. 2009). Combined use of various real-time PCR assays greatly enhances already high reliability of testing for E. amylovora infection (Pirc et al. 2009; Gottsberger 2010).
With the advent of sensitive and reliable methods for detection of E. amylovora in low concentrations, when we are able to detect bacteria if present in a sample, focus is again returning to sampling. When and what to sample to be able to detect fire blight in a new area as early as possible is still a relevant question (Kuflik et al. 2008). It is to be expected that real-time PCR will play a major role in assessing various approaches to sampling both because of its sensitivity and high-throughput. Ams assay was successfully transferred and optimized for a faster amplification on portable SmartCycler® instrument that allows on-field analysis when relevant. In addition to real-time PCR, novel approaches for on-site detection are being developed e.g. LAMP (Notomi et al. 2000; Temple and Johnson 2010). These are faster than real-time PCR assay however, at the moment they do not enable quantification.
In research of E. amylovora, the era of real-time PCR is on the rise, with researchers beginning to exploit quantification provided by real-time PCR in biological contexts (Lehman et al. 2008; Braun-Kiewnick et al. 2011). The benefit of real-time PCR to study gene expression in E. amylovora has yet to be fully exploited and reports on such use (Pester 2011) are expected to be followed by many more.
Abbreviations
- CFU:
-
Colony forming units
- C q :
-
Quantification cycle
- LOD:
-
Limit of detection
References
Bentley HA, Belloni DR, Tsongalis GJ (2005) Parameters involved in the conversion of real-time PCR assays from the ABI prism 7700 to the Cepheid SmartCycler II. Clin Biochem 38(2):183–186
Bereswill S, Bugert P, Bruchmüller I, Geider K (1995) Identification of the fire blight pathogen, Erwinia amylovora, by PCR assays with chromosomal DNA. Appl Environ Microb 61(7):2636–2642
Bonn WG, van der Zwet T (2000) Distribution and economic importance of fire blight. In: Vanneste JL (ed) Fire blight the disease and its causative agent, Erwinia amylovora. UK CAB International, Wallingford, pp 37–53
Braun-Kiewnick A, Lehmann A, Duffy B, Dreo T, Ravnikar M, Stockwell VO (2011) Detection of Erwinia amylovora in orchards using quantitative PCR and lateral-flow immunography. Acta Hortic 896:73–76
Bugert P, Geider K (1995) Molecular analysis of the ams operon required for exopolysaccharide synthesis of Erwinia amylovora. Mol Microbiol 15:917–933
Burns M, Valdivia H (2007) Modelling the limit of detection in real-time quantitative PCR”. Eur Food Res Technol 226(6):1513–1524
Burrill TJ (1882) The Bacteria: an account of their nature and effects, together with a systematic description of the species. Illinois Industrial University 11th Report, 1882, pp 93–157
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Reinhold M, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55(4):611–622
Cankar K, Štebih D, Dreo T, Žel J, Gruden K (2006) Critical points of DNA quantification by real-time PCR—effects of DNA extraction method and sample matrix on quantification of genetically modified organisms. BMC Biotechnol 6(1):37
Chandelier A, Planchon V, Oger R (2010) Determination of cycle cut off in real‐time PCR for the detection of regulated plant pathogens. EPPO Bull 40(1):52–58
De Bellis P, Schena L, Cariddi C (2007) Real-time Scorpion-PCR detection and quantification of Erwinia amylovora on pear leaves and flowers. Eur J Plant Pathol 118(1):11–22
de Kok JB, Hendriks JCM, van Solinge WW, Willems HL, Mensink EJ, Swinkels DW (1998) Use of real-time quantitative PCR to compare DNA isolation methods. Clin Chem 44(10):2201–2204
Dreo T (2009) Is there a need and a place for Real-Time PCR in detection of Erwinia amylovora? Zaštita bilja 60(270):209–226
Duffy B, Schärer H, Bünter M, Klay A, Holliger E (2005) Regulatory measures against Erwinia amylovora in Switzerland. EPPO Bull 35:239–244
European Patent Application EP1770172, Determination of the cycle threshold (Ct) value in a PCR amplification curve by cluster analysis with variable cluster endpoint, http://www.freepatentsonline.com/EP1770172.html
Geider K, Auling G, Jakovljević V, Völksch B (2009) A polyphasic approach assigns the pathogenic Erwinia strains from diseased pear trees in Japan to Erwinia pyrifoliae. Lett Appl Microbiol 48(3):324–330
Gianetti G, Garofalo MC, Morone C, Scortichini M (2004) First report of Erwinia amylovora in Piedmont. Informatore Agrario 60(5):62–63
Gottsberger RA (2010) Development and evaluation of a real‐time PCR assay targeting chromosomal DNA of Erwinia amylovora. Lett Appl Microbiol 51(3):285–292
Johnson K, Stockwell VO, McLaughlin R, Sugar D, Loper JE, Roberts RG (1993) Effect of antagonistic bacteria on establishment of honey bee-dispersed Erwinia amylovora in pear blossoms and on fire blight control. Phytopathology 83:995–1002
Kuflik T, Pertot I, Moskovitch R, Zasso R, Pellegrini E, Gessler C (2008) Optimization of fire blight scouting with a decision support system based on infection risk. Comput Electron Agric 62(2):118–127
Lehman SM, Kim W, Castle AJ, Svircev AM (2008) Duplex real-time polymerase chain reaction reveals competition between Erwinia amylovora and E. pyrifoliae on pear blossoms. Phytopathology 98(6):673–679
Li X, De Boer SH (1995) Selection of polymerase chain reaction primers from an RNA intergenic spacer region for specific detection of Clavibacter michiganensis subsp. sepedonicus. Phytopathology 85:837–842
Llop P, Caruso P, Cubero J, Morente C, López MM (1999) A simple extraction procedure for efficient routine detection of pathogenic bacteria in plant material by polymerase chain reaction. J Microbiol Meth 37(1):23–31
Llop P, Bonaterra A, Peñalver J, López MM (2000) Development of a highly sensitive nested-PCR procedure using a single closed tube for detection of Erwinia amylovora in asymptomatic plant material. App Environ Microb 66(5):2071–2078
Llop P, Donat V, Rodríguez M, Cabrefiga J, Ruz L, Palomo JL, Montesinos E, López MM (2006) An indigenous virulent strain of Erwinia amylovora lacking the ubiquitous plasmid pEA29. Phytopathology 96(8):900–907
López MM, Llop P, Olmos A, Marco-Noales E, Cambra M, Bertolini E (2009) Are molecular tools solving the challenges posed by detection of plant pathogenic bacteria and viruses? Curr Issues Mol Biol 11(1):13–46
Maes M, Garbeva P, Crepel C (1996) Identification and sensitive endophytic detection of the fire blight pathogen Erwinia amylovora with 23S ribosomal DNA sequences and the polymerase chain reaction. Plant Pathol 45:1139–1149
Marras SAE, Kramer FR, Tyagi S (2002) Efficiencies of fluorescence resonance energy transfer and contact-mediated quenching in oligonucleotide probes. Nucleic Acids Res 30(21):e122
Maxson-Stein K, McGhee GC, Smith JJ, Jones AL, Sundin GW (2003) Genetic analysis of a pathogenic Erwinia sp. isolated from pear in Japan. Phytopathology 93:1393–1399
McGhee GC, Schnabel EL, Maxson-Stein K, Jones B, Stromberg VK, Lacy GH, Jones AL (2002) Relatedness of chromosomal and plasmid DNAs of Erwinia pyrifoliae and Erwinia amylovora. App Environ Microb 68:6182–6192
Menggad M, Laurent J (1998) Mutations in ams genes of Erwinia amylovora affect the interactions with host plants. Eur J Plant Pathol 104(3):313–322
Mohammadi M, Moltmann E, Zeller W, Geider K (2009) Characterisation of naturally occurring Erwinia amylovora strains lacking the common plasmid pEA29 and their detection with real-time PCR. Eur J Plant Pathol 124(2):293–302
Navidi W, Arnheim N, Waterman MS (1992) A multiple-tubes approach for accurate genotyping of very small DNA samples by using PCR: statistical considerations. Am J Hum Genet 50(2):347–359
Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T (2000) Loop-mediated isothermal amplification of DNA. Nucleic Acids Res 28(12):E63
Ordax M, Marco-Noales E, Lopez MM, Biosca EG (2006) Survival strategy of Erwinia amylovora against copper: induction of the viable-but-nonculturable state. App Environ Microb 72(5):3482–3488
Pastrik KH, Elphinstone JG, Pukall R (2002) Sequence analysis and detection of Ralstonia solanacearum by multiplex PCR amplification of 16S–23S ribosomal intergenic spacer region with internal positive control. Eur J Plant Pathol 108:831–842
Pester E (2011) Gene expression in the quarantine pest Erwinia amylovora during apple flower-infection. Acta Hortic 896:173–176
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29(9):e45
Pirc M, Dreo T, Ravnikar M (2008) Real-time PCR for testing of symptomless fire blight samples. Acta Hortic 793:533–538
Pirc M, Ravnikar M, Tomlinson J, Dreo T (2009) Improved fireblight diagnostics using quantitative real-time PCR detection of Erwinia amylovora chromosomal DNA. Plant Pathol 58(5):872–881
PO EP (1992) EPPO Standards PM 3/40 Erwinia amylovora. Sampling and test methods. EPPO Bull 22:225–231
PO EP (2004) Diagnostic protocols for regulated pests PM 7/20, Erwinia amylovora. EPPO Bull 34:159–171
PO EP (2007) Basic requirements for quality management in plant pest diagnosis laboratories. EPPO Bull 37(3):580–588
PO EP (2010a) PM 7/76 (2): Use of EPPO diagnostic protocols. EPPO Bull 40:350–352
PO EP (2010b) PM 7/98 (1): specific requirements for laboratories preparing accreditation for a plant pest diagnostic activity. EPPO Bull 40(1):5–22
Powney R, Smits THM, Sawbridge T, Frey B, Blom J, Frey JE, Plummer KM, Beer SV, Luck J, Duffy B, Rodoni B (2011) Genome sequence of an Erwinia amylovora strain with pathogenicity restricted to rubus plants. J Bacteriol 193(3):785–786
R Development Core Team (2010) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, URL http://www.R-project.org/
Reisenzein H, López MM, Duffy B, Dreo T, Paulin JP, Poliakoff F (2010) Development and validation of innovative diagnostics tools for the detection of fire blight (Erwinia amylovora) (ERWINDECT): EUPHRESCO final report, 2010. 65 f
Ritz C, Streibig JC (2005) Bioassay Analysis using R. J Stat Softw 12(5):22
Roselló M, Garcia Vidal S, Tarin A, Llop P, Gorris MT, Donat V, Chartier R, Paulin JP, Gardan L, Peñalver J, López MM (2002) Characterization of an Erwinia sp. isolated from necrotic pear blossoms in Valencia, Spain. Acta Hortic 590:139–142
Salm H, Geider K (2004) Real-time PCR for detection and quantification of Erwinia amylovora, the causal agent of fireblight. Plant Pathol 53(5):602–610
Sebaihia M, Bocsanczy AM, Biehl BS, Quail MA, Perna NT, Glasner JD, DeClerck GA, Cartinhour S, Schneider DJ, Bentley SD, Parkhill J, Beer SV (2010) Complete genome sequence of the plant pathogen Erwinia amylovora strain ATCC 49946. J Bacteriol 192(7):2020–2021
Smits THM, Jaenicke S, Rezzonico F, Kamber T, Goesmann A, Frey JE, Duffy B (2010a) Complete genome sequence of the fire blight pathogen Erwinia pyrifoliae DSM 12163T and comparative genomic insights into plant pathogenicity. BMC Genomics 11(1):2
Smits THM, Rezzonico F, Kamber T, Blom J, Goesmann A, Frey JE, Duffy B (2010b) Complete genome sequence of the fire blight pathogen Erwinia amylovora CFBP 1430 and comparison to other Erwinia spp. Mol Plant Microbe In 23(4):384–393
Steinberger EM, Beer SV (1988) Creation and complementation of pathogenicity mutants of Erwinia amylovora. Mol Plant Microbe In 1:135–144
Stöger A, Schaffer J, Ruppitsch W (2006) A rapid and sensitive method for direct detection of Erwinia amylovora in symptomatic and asymptomatic plant tissues by polymerase chain reaction. J Phytopathol 154(7–8):469–473
Svircev AM, Kim W, Lehman SM, Castle AJ (2009) Erwinia amylovora: modern methods for detection and differentiation. Methods Mol Biol 508:115–129
Temple TN, Johnson KB (2010) Evaluation of loop-mediated isothermal amplification for rapid detection of Erwinia amylovora on pear and apple fruit flowers. Plant Dis 12:423 –430
Thomson S (2000) Epidemiology of fire blight. In: Vanneste JL (ed) Fire blight the disease and its causative agent, Erwinia amylovora. UK CAB International, Wallingford, pp 9–36
Thomson S, Schroth M, Moller W, Reil W (1975) Occurrence of fire blight of pears in relation to weather and epiphytic populations of Erwinia amylovora. Phytopathology 65:353–358
van der Zwet T (2006) Present worldwide distribution of fire blight and closely related diseases. Acta Hortic 704:35–36
van der Zwet T, Beer SV (1995) Fire blight: its nature, prevention, and control: a practical guide to integrated disease management, vol 631. United States Department of Agriculture (Washington, DC), 91 pp
Winslow CEA, Broadhurst J, Buchanan RE, Krumwiede JRC, Rogers LA, Smith GH (1920) The families and the genera of the bacteria. Final report of the Committee of the Society of American Bacteriologists on characterization and classification of bacterial types. J Bacteriol 5(S):191–229
Won-Sik K, Jock S, Paulin JP, Rhim SL, Geider K (2001) Molecular detection and differentiation of Erwinia pyrifoliae and host range analysis of the Asian pear pathogen. Plant Dis 85:1183–1188
Acknowledgments
This work was supported by the following projects: COST-Action 864: PomeFruitHealth, Combining traditional and advanced strategies for plant protection in pome fruit growing, MORS 4300-102/2007-1 (Ministry of Defence of the Republic of Slovenia), ERWINDECT of Euphresco Phytosanitary ERA-NET. Activities were also financially supported by the Phytosanitary Administration of Slovenia (PARS, Ministry of Agriculture, Forestry and Food) and ARRS Program P165. Samples of fire blight symptomatic plant material were obtained in the frame of the official fire blight surveys coordinated by PARS.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by W. Osswald.
A contribution to the Special Issue: Pome Fruit Health.
Rights and permissions
About this article
Cite this article
Dreo, T., Pirc, M. & Ravnikar, M. Real-time PCR, a method fit for detection and quantification of Erwinia amylovora . Trees 26, 165–178 (2012). https://doi.org/10.1007/s00468-011-0654-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00468-011-0654-7