
Abstract
Cultivation and marketing of genetically modified organisms (GMOs) have been unevenly adopted worldwide. To facilitate international trade and to provide information to consumers, labelling requirements have been set up in many countries. Quantitative real-time polymerase chain reaction (qPCR) is currently the method of choice for detection, identification and quantification of GMOs. This has been critically assessed and the requirements for the method performance have been set. Nevertheless, there are challenges that should still be highlighted, such as measuring the quantity and quality of DNA, and determining the qPCR efficiency, possible sequence mismatches, characteristics of taxon-specific genes and appropriate units of measurement, as these remain potential sources of measurement uncertainty. To overcome these problems and to cope with the continuous increase in the number and variety of GMOs, new approaches are needed. Statistical strategies of quantification have already been proposed and expanded with the development of digital PCR. The first attempts have been made to use new generation sequencing also for quantitative purposes, although accurate quantification of the contents of GMOs using this technology is still a challenge for the future, and especially for mixed samples. New approaches are needed also for the quantification of stacks, and for potential quantification of organisms produced by new plant breeding techniques.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Genetically modified organisms (GMOs) are organisms in which the genetic material has been altered through the application of gene technology in a way that does not occur naturally through mating and/or natural recombination. A GMO is formed by the insertion of one or more functional genes (e.g. the association of two or more DNA sequences arising from different species) into the genome of an organism. This technique is used to produce new genetic combinations (events) that are of value to science, medicine, agriculture and industry, and it has been used with several species of plants, animals, fungi and microorganisms. At present, GM plants are the most important GMOs globally. According to a recent International Service for the Acquisition of Agri-biotech Applications report [1], since 1994, 35 countries and the European Union (i.e. a further 27 countries) have granted regulatory approval for GMOs for food and/or feed use, and for environmental release or planting. A total of 2,833 regulatory approvals involving 27 GM crops and 336 GM events have been issued by the competent authorities [1].
Nevertheless there are considerable differences between countries in the adoption of this technology. The precautionary approach that follows the Cartagena Protocol on Biosafety to the Convention on Biological Diversity [2] was agreed internationally to ensure an adequate level of protection in the transfer, handling and use of GMOs that might have adverse effects on the conservation and sustainability of biological diversity, also taking into account the risks to human health, and specifically focusing on trans-boundary movements. Each country that is party to the protocol has agreed to take the necessary and appropriate legal, administrative and other measures to implement its obligations under this protocol. Several countries have implemented, or are in the process of adopting, legislation to ensure the safe use of GMOs for cultivation, food and feed, and for the traceability of GMOs. In some countries, the legislation demands mandatory labelling, which is commonly associated with specific thresholds [3]. Tolerance thresholds or thresholds for labelling vary among countries and can also be only voluntary. As a consequence, the specific needs for quantification vary significantly [4].
Legislation across the European Union (EU) states that the GMO labelling requirements do not apply to food and feed that contain in EU approved GM events at less than 0.9 % of the food/feed ingredients when considered individually, if this presence is adventitious or technically not avoidable [5]. However, when this legislation was implemented, the measurement unit was not explicitly specified. In 2004, as technical guidance for the sampling and detection of GMOs, EU Recommendation 2004/787/EC proposed that this should be expressed as the percentage of event-specific DNA copy numbers in relation to the target taxon-specific DNA copy numbers, calculated in terms of haploid genomes [6]. However, many laboratories still express their data as mass fractions, as the majority of certified reference materials (CRMs) are certified for mass fraction. Recent EU regulation on the presence of GMOs in feed for which an authorisation procedure is pending or the authorisation of which has expired is setting the non-compliance limit to 0.1 %, as related to the mass fraction of a GMO [7]. The European Union Reference Laboratory for GM Food and Feed (EU-RL GMFF) has issued technical guidance for the implementation of this regulation, which includes also the proposal for how to convert the result of analysis from GMO % in the DNA copy number ratio to GMO % in the mass fraction for crops that are homozygous or hemizygous for the specific event [8].
As reviewed by Gruère and Rao [3], other countries or regions have other labelling policies that differ in nature, scope, coverage, exceptions and degree of enforcement (Table 1). In Korea, labelling is mandatory for agricultural products if the product contains more than 3 % of a GMO [9] and a GM food [10]. In Japan, a list of agricultural products and their processed foods have been designated as mandatory labelling items, including soy, maize, potatoes, canola, cotton, alfalfa and sugar beet [11]. If the non-GM food is handled correctly according to the identity preservation, the adventitious presence of GM crops can be accepted up to 5 % (w/w) [12]. In Brazil, the Consumer Protection Code [13] and Decree No. 4680 [14] mandate the labelling of food products for animal and human consumption that contain over 1 % GMOs. However, many countries have not implemented labelling requirements, for different reasons, while some countries, like the USA and Canada, have voluntary labelling guidelines for GM and non-GM food. Also, numerous countries still have to implement or enforce their regulations [3].
In many countries and regions, consumers have expressed their concerns over gene technology, and have demanded appropriate information and labelling for foods derived from GMOs. To provide information to consumers and to facilitate international trade, reliable GMO analysis that can comparably measure the GMO contents of products is required, especially at the respective thresholds [15]. To guarantee the reliability of such analytical data, there is the need for accurate methods that should be validated, verified in testing laboratories and used together with the appropriate controls.
The appropriate enforcement and compliance for the labelling of GMOs are constantly challenged by the increasing numbers of GMOs and their diversity, and by the different regulations in different countries, which necessitate additional efforts towards harmonisation on the one hand, and cost-efficiency analyses on the other. Several reviews are available on the analysis of GMOs [4, 15–20]. With the present review, we would like to present the state-of-the-art and currently used technologies, as well as the most recent advances and technical possibilities for the quantification of GMOs, and to highlight the existing challenges, as well as the future perspectives.
Current approaches and challenges
Quantitative real-time polymerase chain reaction
At present, quantitative real-time polymerase chain reaction (qPCR) is the most commonly accepted and used method for detection, identification and quantification of GMOs. While it is true that the technology can have exquisite sensitivity and specificity that can be coupled to high reproducibility and accuracy, it is essential to understand that qPCR analysis consists of numerous, often divergent, protocols that use different instruments, enzymes, buffers and non-identical targets [21]. To produce reliable and comparable data, laboratories need to harmonise their methods for quantification of GMOs, or to demonstrate that different methods used for the quantification of GMOs are commutable. Quantitative analysis of GMOs is usually carried out by determination of the amount of event-specific target with respect to a taxon-specific target (i.e. the reference gene). Methods for detection and quantification of many event-specific targets of commercially available GMOs and reference genes are available. Public databases, like the GMO detection database (GMDD; http://gmdd.shgmo.org/index/search) and GMOMETHODS (http://gmo-crl.jrc.ec.europa.eu/gmomethods/), have been established for the collection and exchange of developed and validated methods. There are also many publications that report on the development of methods, and sometimes on their in-house validation. However, it is important to critically evaluate any validation status before implementing a method in the laboratory for routine analyses of samples.
In this sense, a recent study from Kodama and collaborators [22] is useful, as they extensively studied the data from a large number of collaborative trials for the testing of GMO methods. To harmonise method validation and verification within Europe the European Network of Genetically Modified Organisms Laboratories (ENGL) have provided a document that details how methods for GMO analysis should be evaluated and validated by the EU-RL GMFF [23]. This document, which is in the process of revision, is in the context of Commission Regulation (EC) No. 1829/2003 [24], and in synergy with the recommendations of Codex Alimentarius Commission [25]. In addition, ENGL have prepared a guidance document for laboratories that are implementing validated methods [26]. Also, in some other countries (e.g. China), it is legally binding that the adopted methods are fully validated [27].
For accurate and comparable measurements, as well as validated methods, calibration of the measurements is needed to establish an anchor point for the measurement value and measurement units [28]. Different types of reference materials (RMs) are available, including whole seeds, seed-derived powder, genomic DNA from leaves, and plasmids. According to Commission Regulation (EC) No. 641/2004 [29], RMs with a precisely known content of the specific targets are needed for the calibration of qPCR measurements and for quality control, e.g. to check for any bias in the measurements [30].
There are two qPCR quantification approaches in use: the standard curve approach and the comparative Cq approach (ΔΔCq, Cq being the crossing point, i.e. the cycle number that corresponds to the first detectable signal above threshold). In the ΔΔCq approach, direct comparisons of the Cq of the two measurements are made, e.g. event-specific and taxon-specific targets in the sample. With the ΔΔCq method, it is assumed that reactions for event-specific targets and taxon-specific targets have similar efficiencies of amplification. However, the ΔΔCq method is only acceptable when working with well-established samples (e.g. with raw materials) and if the qPCR methods are validated in combination with the DNA extraction method (i.e. the non-modular approach). In the standard curve approach, the quantification data for each target are related to the standard curve, which is obtained with a reference material that contains the same target of known quantity. The precondition for this approach is that there are similar amplification efficiencies of target sequences in the reference material and investigated sample. This can be examined by comparisons of event-specific and taxon-specific calibration curves, and event-specific and taxon-specific curves generated from dilutions of DNA from test samples. To determine whether a quantification is feasible, the acceptance criterion has been set to a maximal difference of 0.3 in the slopes of the linear regression lines of the test sample DNA and the reference material DNA [31]. The determination of the GMO content is finally carried out by relating the measured content of the event-specific target to the content of the taxon-specific target. This approach is applicable to all samples, from raw materials to matrixes of complex materials.
For reliable quantification of GMOs, laboratories need appropriate organisation and quality management systems, and several critical points should be considered in the analytical procedures, such as sampling, sample preparation and DNA extraction, which have been described recently [18] and which will not be discussed in this review. In the EU and some other countries, GMO testing laboratories are obliged to operate under International Organization for Standardisation/Internal Electrotechnical Commission (ISO/IEC) 17025 accreditation [32]. The number of new GMOs is increasing rapidly and laboratories need to introduce new methods within the scope of their accreditation in a timely manner, which is possible if the scope of accreditation is flexible. To facilitate harmonised flexible scope accreditation within Europe, a technical guidance document has been prepared that is currently limited to laboratories that are quantifying GMOs, and that addresses different levels of flexibility concerning the products, events and analytical procedures [33]. Quality controls described in EN ISO 24276:2006 [34] and EN ISO 24276:2006/A1:2013 [35] are needed to monitor the performance of a method, and for the interpretation of the data. An additional independent sample with known GMO content (i.e. the quantification control) can be analysed in parallel with the unknowns to confirm the appropriate analytical procedure and the calculation of results, and to construct control charts.
Each step of an analysis can introduce a certain degree of uncertainty in any process and in the final data and their interpretation. The way in which such measurement uncertainty can be estimated using data from collaborative trials in combination with in-house quality control data is well described in the guidance document on the measurement of uncertainty for GMO testing laboratories [36]. The appropriate estimation of the measurement uncertainty associated with an analytical result is crucial for decisions on the compliance of the sample tested. It has been proposed that the value obtained by subtracting the expanded uncertainty from the reported GMO content is used to assess compliance. To consider a tested sample as non-compliant (i.e. beyond what is permissible), the concentration of the analyte should be above the legal threshold without any reasonable doubt.
Quantity and quality of DNA
The prerequisite for accurate quantification is a sufficient amount of DNA that is of appropriate quality. Accurate estimation of total DNA concentrations is an important component of the molecular analysis. Different methods for the measurement of DNA concentrations are available; however, there is great variability between different methods [37]. As reviewed by Bhat et al. [38], the measurement of DNA concentrations according to UV absorbance at 260 nm can lead to overestimation of the DNA levels as a result of the presence of nucleotides, RNA, single-stranded (ss)DNA and impurities, such as proteins and phenols. Quantification of double-stranded (ds)DNA with intercalating fluorescent dyes is more sensitive, and this is advantageous owing to the limited binding of these dyes to RNA and ssDNA, although this might rely on external standards that were quantified using UV absorbance at 260 nm. A study by Folloni et al. [39] indicates that methods using fluorescent dyes are more sensitive. The disadvantage of fluorescent dyes is underestimation of DNA concentrations in highly processed samples, which is believed to be due to the low binding capacity of small DNA molecules [36]. Such samples are common in GMO testing.
In everyday analysis of GMOs, the testing laboratories can avoid measuring DNA concentrations extracted from known matrixes (e.g. maize seed, soybean flour, oilseed rape leaves) and only assess the quantity and quality of DNA by measuring taxon-specific genes using qPCR and comparing these data to expected values. Nevertheless, when the presence of a GMO is determined in unknown or highly processed matrixes, or when new extraction methods are introduced, the measurement of the DNA concentration is recommended.
The GMO content has been measured across a wide range of sample matrixes. DNA extracted from processed or very complex matrixes can be of poor quality (e.g. degraded, or with co-extracted impurities), and this can influence the amplification efficiency. The influence of qPCR efficiency on the outcome of quantitative analysis was well described by Cankar et al. [40] and Demeke and Jenkins [41]. The amplification efficiency is not necessarily influenced in the same way for all amplicons, and this should be taken into account in GMO quantification, because for the determination of GMO content, two targets are quantified: those that are event specific and taxon specific. Consequently, qPCR efficiency is one of the main method performance parameters, which is monitored during qPCR analysis. A 100 % efficiency of amplification in each cycle corresponds to the theoretical doubling of the PCR product during each cycle. For GMO quantification it has been proposed that the average value of the slope of the standard curve should be in the range of −3.6≤ slope ≤−3.1, which corresponds to an amplification efficiency of 90–110 % [23, 26].
Sequence mismatches
Different varieties of the same GM crop are available on the market. For example, there are a few hundred different varieties of the insect-resistant GM maize (event MON810) worldwide. All of these varieties include the same event-specific target, which is unique, and which matches only one GMO. However, mismatches in inserted sequences that have arisen during crossing have already been reported [42]. A mismatch between a primer and the DNA template can cause partial to complete failure of the amplification of the initial DNA template, which will depend on the type and location of the nucleotide mismatch, and which affects the estimated target copy number to a varying degree. Variations are even more likely in taxon-specific sequences and can lead to overestimation of the GMO content. The maize genome, for example, is highly diverse. As reviewed by Ghedira et al. [43] and Papazova et al. [44], single base-pair substitutions or small insertions or deletions of bases are very frequent, and these can lead to the inaccurate determination of the GMO content.
Taxon-specific genes
The taxon-specific target should be specific to the taxon of interest, and it should have a stable, known and low copy number (preferably, 1 copy per haploid genome) with no allelic variations among cultivars of the same species [45–47]. Many reports on the development and evaluation of methods for taxon-specific genes have been published [44, 46, 48]. The selection of a taxon-specific target is especially challenging if differentiation of a closely related taxon and detection of introgression events are needed, also due to a lack of the genome sequences. Such an example can be seen for sugar beet, where species-specific genes should differentiate four genomic sections. The assay for glutamine synthetase [49] has been widely used as the taxon-specific target, but this does not discriminate between sugar beet (Beta vulgaris L.) and autumn beet (Brassica rapa L.), and so a new assay for a taxon-specific target, the adenylate transporter, was developed [50].
Units of measurement
The measurement units of sample quantification are determined by the use of RMs and the quantification methodology. The vast majority of commercially available RMs are certified for mass fraction. Over the last few years, some of these have been additionally certified for copy number ratios. However, the different units of measurement are not directly comparable, and consequently the data from different quantification procedures for GMOs cannot be compared. The production of CRMs is an extremely complex, time-consuming and expensive process [15], and worldwide accessible CRMs are limited to a few major producers, while some countries are developing their own. Many studies have confirmed the reliability of plasmids as an alternative calibrant for the calculation of the GMO copy number, and these might provide a cheaper and more flexible alternative to conventional reference materials [39, 51–53]. Moreover, when correctly established and expressed, the measurement data for copy number can provide a metrologically sound reference system [15].
In plants, zygosity, maturity status and endosperm tissue ploidy (parental origin of a GM trait and endosperm DNA content) are known biological factors that can significantly influence the GMO content when expressed in terms of a haploid genome, as compared to the GMO content expressed as a mass/mass ratio estimate using qPCR [54–56]. For some CRMs, data on the zygosity and the origin of the GMO are available, and this can help laboratories to improve their accuracy and to reduce measurement uncertainty. However, uncertainty connected to the biological factors of the test sample still remains [4], and a sample containing 50 % of a homozygous GMO cannot be distinguished from a sample containing a 100 % hemizygous GMO.
To harmonise the data on the expression of measurements stated in EU Regulation No. 619/2011 [7], the technical guidance sets out the conversion factors for crops [8]. A conversion factor “GM % in DNA copy number ratio = 50 % [GM % in mass fraction]” is used for crops hemizygous for an event-specific insert (e.g. hemizygous GM maize), while the conversion factor “GM % in DNA copy number ratio = 100 % [GM % in mass fraction]” is used for crops homozygous for an event-specific insert (e.g. homozygous GM soya). Even if not totally precise scientifically for each sample, this is a pragmatic approach for harmonisation among laboratories in the EU.
Statistical approaches to quantification
Statistical approaches in combination with qPCR can provide additional information for GMO detection and quantification. Differential qPCR, for the detection of non-authorised GMOs, is based on the presence of several common elements in different GMOs (e.g. promoter, genes of interest). A statistical model was developed to study the difference between the number of targets of such a common sequence and the number of event-specific targets that can identify the approved GMO and the donor organism of the common sequence. When this difference differs statistically from zero, the presence of a non-authorised GMO can be inferred [57]. However, this approach has low sensitivity and it is reliable only if the presence of an unknown GMO exceeds 30 %.
There have also been two reports on the application of statistics-based methods for GMO quantification for the determination of the absolute number of molecules. These do not require CRMs as calibrants and they enable lower limits of quantification. With this approach, quantification of GMOs is possible in samples with very low DNA concentrations. SIMQUANT (i.e. single molecule quantification) is based on the ratio of discrete volumes without or with one or more PCR-amplifiable event-specific target copies, and this results in a statistical estimate of the relative GMO concentration that is based on the probability that one or more amplifiable event-specific template copies are present in the discrete volume [58]. A very similar approach is seen with QUIZ (i.e. quantitation using informative zeros), which uses larger numbers of a dilution for both GMOs and non-GMO reference targets to obtain the GMO content estimates. Values obtained independently for a GMO-specific target and a taxon-specific target give an estimation of the GMO content [59]. QUIZ was applied to estimate the contents of two GMOs in processed food containing one or both of these GMOs. The data showed good agreement between the derived values and the known GMO input and they compared favourably with quantitative real-time PCR [59]. These methodologies are not yet widely used, as they are labour intensive and expensive. However, their development indicates the need for alternative approaches in the quantification of GMOs.
To conclude, qPCR is currently the method of choice for quantification of GMOs, although there remain some challenges that compromise the comparability of data among laboratories, such as sensitivity to inhibitors that are often present in complex matrixes; potential bias with degraded DNA and low concentrations of targets, especially in processed matrixes; lack of harmonisation in the expression of the measurement data; and diversity of the available RMs. This might be improved by the availability of the appropriate RMs, which will preferably be certified for copy number, and along with this harmonisation, in the unit of measurement. Another important challenge is the increasing number of GMOs on the market worldwide, which leads to time-consuming and more costly GMO quantification in cases where there are multiple GMOs in a sample. The currently used methodology is thus approaching its limits, and new approaches for the quantification of GMOs are needed that have improved performance and cost efficiency of these analyses.
New approaches
The development of new approaches that are suitable for the quantification of specific sequences is a promising solution that can overcome the drawbacks of the currently used methods. In the past, several alternative methods for the detection of GMOs were developed that explored different aspects of amplification, detection and identification approaches (reviewed by Holst-Jensen [4] and Žel et al. [18]). Recently, additional methods have been developed [53, 60–63]. However, not all of the newly developed methods that allow high multi-targeting and/or multiplexing enable quantification. Those addressing the quantitative aspect are described below, where more focus is given to the latest and currently most promising, the digital PCR.
Microarrays
Two of the developed multiplex approaches that enable quantification and where the detection was carried out on microarrays are multiplex quantitative DNA array-based PCR (MQDA-PCR) [64] and nucleic-acid-sequence-based amplification (NASBA)-implemented microarray analysis (NAIMA) [65, 66].
MQDA-PCR consists of two PCR steps, labelling and microarray hybridisation. In the first PCR step, tailed primers are used, harbouring universal sequences and target-specific sequences. In the second PCR step, only the universal primers complementary to universal sequences introduced by the tailed primers in the first PCR step are used. After the PCR amplification sequence-specific labelling of DNA probes by linear amplification is included. Products are hybridised overnight and detected on a DNA array. The method showed good specificity on tested samples, including food and feed samples (Table 2). Some signal deviations were observed between samples of mixed GMOs and single GMO, probably owing to non-specific side reactions. The method offers the possibility of semi-quantification by characterizing the sample as containing more than 2 %, between 1 and 2 %, between 0.1 and 1 %, or less than 0.1 % of GMO.
Similarly, NAIMA consists of different steps including a multiplex template synthesis step followed by a universal NASBA amplification, labelling and microarray detection [65]. DNA templates synthesized during the first step are directly used in the second, universal NASBA amplification step. Universal primers, binding to both universal regions created by the forward and reverse tailed primers during the first step, allow more uniform amplification of different fragments and conserving the initial ratio between them. After amplification, NAIMA products are ligated directly to fluorescently labelled 3DNA dendrimers. Products are then hybridised to a custom designed microarray. NAIMA has shown good specificity, sensitivity and wide linear range of amplification [65]. NAIMA is fully quantitative in the range of 0.1–25 % of transgenic content (Table 2).
Interestingly, the future perspectives of nucleic-acid-based methods as predicted by Holst-Jensen in 2009 [4] included non-PCR methods and microarray detection methods. As things appear today, at least from the quantification point of view, these methods are still not implemented in routine diagnostics. The reasons for this might be the additional equipment needed for the microarray hybridisation and analysis, and the more demanding validation and standardisation.
Capillary electrophoresis
Another way to perform the detection of several targets in one reaction was proposed by Heide et al. [73]. They developed a 9-plex in combination with detection on capillary gel electrophoresis (PCR-CGE). The idea of the method was to perform multiplex PCR reaction and differentiate between amplicons of the same size with the use of fluorescently labelled forward primers by performing CGE. The method was developed for the maize taxon-specific gene and 8 GM maize events (Bt11, GA21, MON810, NK603, T25, TC1507, MON863, BT176). As this method was not quantitative, Heide and co-workers went a step further and presented a two-step PCR in combination with capillary gel electrophoresis (2S-PCR-CGE) [67]. This improved method implemented the same idea of combining multiplex PCR with bipartite primers in the first step and universal PCR amplification in the second step, as presented above for MQDA-PCR and NAIMA. The method was tested on seeds and plant material and results have shown good specificity with no false positive results. Quantification was possible in the range of 0–2 % of GMO content (Table 2) [67]. A limitation of this method is its laborious optimization and primer design, when developing assays for new targets, and the availability of specific equipment. As capillary gel electrophoresis in not routinely used in GMO quantification, extensive validations and verifications would have to be performed.
Loop-mediated isothermal amplification
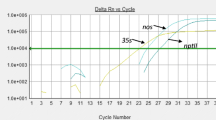
On the other hand methods that allow a faster workflow and that do not require sophisticated equipment have also been recently developed for GMO detection, such as loop-mediated isothermal amplification (LAMP)-based GMO detection methods [74–77]. They were developed for detection of cry1Ab gene from rice [76], KMD1, TT51-1 and KF6 GM rice [75] and for commonly used promoters (P-35S, P-FMV) and marker genes (aadA, nptII, uidA) [77]. LAMP employs a set of four specially designed primers that make the primer design one of the most important steps. These primers then recognize six distinct regions on target DNA template. One primer pair initiates the reaction, whereas the second pair helps to form a loop structure that increases the speed of amplification. The reaction is run using simple equipment under isothermal conditions, with amplification and detection coupled in single step (tube). LAMP methods showed high specificity, sensitivity and tolerance to inhibitors; however, they are currently used only for qualitative purposes in different areas of diagnostics. Nevertheless the first attempts to use LAMP for quantitative purposes have also been reported [78, 79]. If they become developed, quantitative LAMP methods for GMO detection could pose great competition to other new and already established methods.
Digital PCR
One of the most promising recent technical improvements in the field of quantitative PCR was the development of digital (d)PCR. The idea behind dPCR is to quantify the absolute number of targets that are present in a sample using PCR, limiting dilutions and Poisson statistics [80]. This is achieved by partitioning the PCR mix into a large number of separate reactions that contain zero or one or a few copies of the target nucleic acid. By counting the positive and negative partitions after the PCR reaction, and with the use of binomial Poisson statistics, the absolute copy number of the targets in an initial sample can be calculated [81].
Currently, there are two dPCR systems [69, 81, 82]. Chamber (c)dPCR uses microfluid chambers with partitioning into up to a few thousand individual reactions. The other dPCR system is known as droplet (d)dPCR, and this uses water–oil emulsion-based partitioning into several thousand, or even millions, of individual droplets that are counted using flow cytometry. The absolute copy number estimations of these two systems do not differ. However, the expanded measurement uncertainty is higher for cdPCR [81]. Also, ddPCR relies on end-point detection of fluorescence/amplified products, whereas cdPCR follows the amplification in real time in the same way as in qPCR (Table 2).
Although dPCR has already been used for a number of different applications, and mostly in studies of absolute copy number determination [82, 83], it has been shown to be particularly useful for the detection of rare and low copy number targets [72], to determine copy number variations (e.g. ratios of 1.25, or even below 1.2, can be distinguished) [84, 85].
In comparison to qPCR, dPCR has several advantages when it comes to quantification of GMOs. dPCR enables the determination of absolute target copy numbers present in a reaction, which thus avoids the bias of amplification efficiency between samples and reference material when performing quantification with qPCR [68, 71, 72]. The data produced by dPCR are very precise, and they provide confident results, which are necessary for metrological use [71, 72]. Additionally, measurements with dPCR also provide more accurate estimations of targets at low copy numbers [86].
As already mentioned, inhibitory substances present in a sample matrix background are one of the reasons for possible different amplification efficiencies between samples and/or the reference material. However, for dPCR, when it is considered as an end-point measurement, its tolerance to inhibitors reduces the bias of the sample matrix that is frequently observed with qPCR [70]. As has been shown experimentally for ddPCR, inhibitors can affect individual microreactions, but after analysing amplitude plots and setting the threshold, all of these reactions are counted as positive, and thus do not affect the final quantification [87].
Another very important advantage of dPCR is the easy transfer of the assays from qPCR platforms to dPCR platforms, which makes the implementation of the latter platforms in laboratories easier compared with other approaches.
The importance of reference materials detailed in the previous section diminishes with the use of dPCR, owing to the absolute quantification in which the analyses are not dependent on the availability of a reference material [70]. For example, conflicting data from quantification with qPCR when the samples and the CRMs are not of equal zygosity can be solved easily by the use of dPCR. At present, while dPCR is not yet widely used, this technology can already improve the comparability of data through the determination of the copy/copy of CRMs. The use of cdPCR for assessment of the copy number ratios in CRMs has already been demonstrated, showing that this has a suitable metrological performance [71, 72, 88].
All of the advantages mentioned above contribute to the cost-effectiveness of the dPCR methods, especially ddPCR. The experimental set-up for qPCR can be quite extensive when performing relative quantification, as it needs to include several dilutions of the reference material and at least two dilutions of each sample. When it comes to the comparison with a 96-well reaction plate, the number of samples processed with ddPCR in an equal time frame is threefold higher, and the price per sample is 30 % lower with ddPCR than with qPCR.
ddPCR has also been shown to be appropriate for routine use in control laboratories, especially when they handle high numbers of samples [68]. When comparing the ddPCR and cdPCR price performances, the machine for cdPCR is about twice the price of that for ddPCR, and the arrays used in cdPCR are relatively expensive. To increase the price performance of dPCR, multiplexing can be used. dPCR systems enable multiplexing from two to up to 10 targets in one reaction. The multiplex amplification relies on the use of differently labelled probes, with up to five different fluorophores used in cdPCR, and two different fluorophores in ddPCR. In ddPCR, additional multiplexing for up to 10 targets in the same reaction is possible through using different primer/probe concentrations [69]. Recent advances in ddPCR have included the possibility to use DNA-binding dye chemistry, which also allows multiplexing [89].
To sum up this discussion of the dPCR systems, ddPCR is at present more suitable for absolute GMO quantification, owing to its wider linearity range for quantification and its better cost efficiency.
Next generation sequencing
In GMO analysis, the development and application of accurate, comprehensive and high-throughput techniques are the core steps for molecular characterisation of GMOs and for the implementation of labelling regulations [90]. Next generation sequencing (NGS) has been widely used for mutant-site identification [91], NA expression profile analysis [92], and copy number variations in humans, animals, plants and bacteria, with the advantage of high throughput and good resolution and accuracy at the whole genome level [93, 94]. Novel analytical methods based on NGS techniques might provide a good solution for GMO analysis in the future. Recently, several studies have investigated the possibility of detection and characterisation of known and unknown GMOs using NGS techniques on different NGS platforms (e.g. FLX, Solexa, SOLiD). NGS has also been successfully used for molecular characterisation (e.g. insertion site, flanking sequence, unintended insertion) and transgene copy number evaluation, among others.
As early as 2009, Tengs et al. [95] described the possibility for characterisation and identification of unknown genetic modifications at the transcription level using the FLX sequencing platform. A total of 147 reads were observed with high similarity to the transgenic plasmid DNA sequence in GM Arabidopsis thaliana. Also, 15 and 27 sequences were found in the EST library of the GM rice ABF3 line and the GM papaya SunUp, respectively. These data have shown the potential of the use of high-throughput sequencing in GMO analysis, even though the data were only generated from the transcription profile with low coverage.
As a development of these high-throughput sequencing techniques, the Solexa and SOLiD platforms have both been used in transgenic plant and animal analyses, including for the identification of a T-DNA insertion and a flanking sequence, and for the estimation of trangene copy number and the detection of unknown elements. DuBose et al. [96] established a combined system for the identification of the transgene anatomy in transgenic mice by using the target capture array and high-throughput sequencing techniques. They successfully identified the transgenic integration, and supplied the basic junction sequence for the development of a genotyping assay to distinguish heterozygous and homozygous transgenic mouse. However, the hybridised efficiency of the target capture array is a key step for this system, as well as the known information of the transgene and its cassette.
Kovalic et al. [97] developed a method based on high-throughput sequencing and junction sequence analysis bioinformatics to fully characterise a GMO. In the typical GM soybean event, the transgene site insertion and transgene copy numbers were obtained. Additionally, in vivo DNA rearrangement of DNA inserts was observed. Yang et al. [90] reported an integrated approach that combined whole genome high-throughput sequencing and computational reassembly to comprehensively uncover transgene inserts and rearrangements in GM rice (T1c-19 and TT51-1). Three data analysis modules were established to target the molecular characterisation, the contents of known GMOs and the identification of unknown GM rice. Module 1 is intended for use when the DNA sequence of the transformation vector is known. Module 2 is intended for use when a DNA sequence database of genetic elements and transgene constructs from known GMOs is available and can be used as a reference library. Module 3 is intended for use when no a priori knowledge of the DNA sequence of the vector and insert is available (i.e. an unknown GMO).
The above studies have demonstrated well the applicability of NGS techniques in the analysis of GMOs (i.e. for transgene integration and copy number evaluation). However, the application of NGS in accurate quantification of the content of GMOs remains a challenge for the future, especially for samples of mixed GMOs, because of the massive sequence data analysis, the high similarity of sequences among different plant species and more than 10 % noise sequences from the high-throughput sequencing process. It is expected that the technology will become more sensitive and reliable, and that as well as the already known whole genomes of a few plant species (including important crops like rice, soybean and maize), the whole genomes of other plant species will also become available. It is also likely that the costs associated with NGS will continue to decrease over time. On the basis of these concepts, new pipelines for specific analysis need to be developed to lead to more successful use of NGS in the detection and quantification of GMOs.
Open issues
Quantification of stacks
An important feature of GMOs currently on the market is stacked events. In 2013, 13 countries were already growing GMOs with two or more traits. The trend of the increasing hectarage of stacked events is expected to continue, and in 2013 this had already reached almost a third of the total of 175 million hectares [1]. Stacked GMOs can be constructed in different ways (reviewed in [98]). The most challenging for quantification are stacks that are obtained by conventional crossing of individual parental GMOs. The resulting GMO contains a combination of insertion sites that are identical to the parental GMOs and thus cannot be distinguished from a mixture of individual GMOs. As no specific methods exist here, recent quantification has relied on summing up the quantities obtained for individual events, and thus the concentration of a pure double-stack GMO can be overestimated by 100 %. This limitation in distinguishing stacks from single-event GMOs might pose a regulatory problem, as GMOs that contain single events might already be approved when the stacks containing these single events are not approved. The only possibility to definitively determine the existence of stacks today is by performing an analysis of the DNA isolated from a single organism [99–101].
All of these proposed approaches can only be used on kernels or on individual plants in the field, and they are not applicable to processed samples. As these are costly and time-consuming procedures, Mano et al. [102] proposed an alternative approach: a group testing strategy was designed and assessed in which the GMO content was statistically evaluated on the basis of qualitative analyses of multiple small pools that consisted of 20 maize kernels each. The method described by Xu et al. [103] offered a mathematical solution and the combination of the qPCR data, but it was tested on a limited sample that contained only the three organisms (maize Bt11, maize GA21, Bt11xGA21 stacked GMO). Even though the technical possibilities exist (e.g. NGS), there are limited short-term perspectives for routine and large-scale implementation of GMO stack quantification, mostly because additional research and additional adaptations need to be made for these techniques. Recently, the ENGL working group was established to discuss the current state of play, to explore the feasibility of novel approaches under routine analytical conditions and to propose research strategies for the identification of stacked GM events.
New plant breeding techniques
New types of genetic modifications are under development through new plant breeding techniques. Overviews of these techniques have been published by Lusser et al. [104, 105]. These techniques might also fall under existing or new legislation, and should new legislation be needed, this would require the sort of control and traceability systems that are in place for conventional GMOs. This will pose challenges to testing laboratories, as the current detection and quantification methodologies might well be insufficient. For plants produced with ZFN-3 technology (i.e. targeted insertion of larger sequences, and even whole genes) and for cisgenic/intragenic plants, detection and quantification would be possible if information on the flanking sequences is available. For plants produced with the ZFN-1 and ZFN-2 techniques (i.e. targeted modifications of a single or a few nucleotides) or with oligonucleotide-directed mutagenesis, detection and quantification would also be possible with prior information of the nucleotide sequences flanking the introduced modification. However, the same genome modifications might be generated by other mutagenesis techniques or by natural genetic variation. Without prior knowledge, detection and quantification of such small modifications, this will require more complex technologies (e.g. full genome sequencing) and extensive background sequence information. As a further and greater challenge, there is the detection of organisms in which gene silencing is obtained through DNA and/or histone methylation (RNA-dependent DNA methylation), where the DNA sequence itself is not modified. In this case, it is not possible to differentiate analytically between the naturally induced methylation patterns and those induced by the deliberate use of RNA-dependent DNA methylation.
Conclusions
In the field of GMO quantification, qPCR is still the method of choice. For comparability of results among laboratories across the world, it is crucial that the methods used are correctly validated and that in the everyday analysis, all possible sources of bias are taken into account and controlled for, such as the quantity of extracted DNA, the presence of inhibitors and the quality of the reference materials. Considerable efforts have been invested in the understanding and critical evaluation of this technology.
However, as a result of the steadily increasing number of GMOs developed and approved worldwide, the present qPCR methodology is no longer fully suited to purpose. It is expected that in the near future new approaches, like dPCR with the ability of absolute quantification, and better sequencing technologies with the generation of large amounts of data in single experiments, will find their appropriate place in the world of GMO detection and quantification. Ideally, the combination of multitarget screening systems for the detection of several GMOs in single samples would be the method of choice for the constantly increasing number of GMOs [63, 106–108], along with the possibility for simultaneous quantification.
A recently started EU FP7 project “Development of Cost efficient Advanced DNA-based methods for specific Traceability issues and High Level On-site applicatioNs” (DECATHLON) will bring a new dimension to this situation. This investigation is horizontally linking three areas using molecular methods for detection, identification and quantification of food pathogens, GMOs, and other customs-related issues of plants and endangered animal species. In this way, new knowledge on relevant molecular biological and bioinformatics methods will be developed, and also minimum performance requirements will be set for these new methodologies.
References
James C (2013) Global status of commercialized biotech/GM crops: 2013. ISAAA briefs 46
Secretariat of the Convention on Biological Diversity (2000) Cartagena protocol on biosafety to the convention on biological diversity: text and annexes. Secretariat of the Convention on Biological Diversity, Montreal
Gruère GP, Rao SR (2007) A review of international labeling policies of genetically modified food to evaluate India’s proposed rule. AgBioforum 10:51–64
Holst-Jensen A (2009) Testing for genetically modified organisms (GMOs): past, present and future perspectives. Biotechnol Adv 27:1071–1082
European Commission (2003) Regulation (EC) No 1830/2003 of the European Parliament and of the Council of 22 September 2003 concerning the traceability and labelling of genetically modified organisms and the traceability of food and feed products produced from genetically modified organisms and amending Directive 2001/18/EC. Off J Eur Union 268:24–28
European Commission (2004) Commission recommendation of 4 October 2004 on technical guidance for sampling and detection of genetically modified organisms and material produced from genetically modified organisms as or in products in the context of Regulation (EC) No 1830/2003. Off J Eur Union 348:18–26
European Commission (2011) Regulation (EU) No 619/2011 of 24 June 2011 laying down the methods of sampling and analysis for the official control of feed as regards presence of genetically modified material for which an authorisation procedure is pending or the authorisation of which has expired. Off J Eur Union 166:9–15
European Union Reference Laboratory for Genetically Modified Food and Feed (2011) Technical guidance document from the European Union Reference Laboratory for Genetically Modified Food and Feed on the implementation of Commission Regulation (EU) No 619/2011
Ministry of Agriculture and Forestry (2000) Guidelines for labeling genetically modified agricultural products. MAF Notification No. 2000-31
Korea Food and Drug Administration (2000) Labeling standards for genetically modified foods. KFDA Notification No. 2000-43
Ministry of Agriculture, Forestry and Fisheries (2007) Notification No. 1173 (October 1, 2007) for the amendment of Notification No. 517
Ministry of Agriculture, Forestry and Fisheries (2000) Notification No. 1775
Brazil (1990) Consumer protection code. Law No. 8078 of September 11, 1990. Provides for consumer protection and other measures
Brazil (2003) Decree No. 4680 of April 24, 2003. Regulates the right to information, provided by Law No. 8078 of September 11, 1990, regarding food and food ingredients for human consumption or animal feed containing or produced from GMOs, without prejudice to compliance with other applicable rules
Trapmann S, Corbisier P, Schimmel H, Emons H (2010) Towards future reference systems for GM analysis. Anal Bioanal Chem 396:1969–1975
Holst-Jensen A (2013) Real-time PCR analysis of genetically modified organisms. In: Rodriguez-Lazaro D (ed) Real-time PCR in food science. Caister Academic, Norfolk
Broeders SRM, De Keersmaecker SCJ, Roosens NHC (2012) How to deal with the upcoming challenges in GMO detection in food and feed. J Biomed Biotechnol 2012:402418–402429
Žel J, Milavec M, Morisset D, Plan D, Van den Eede G, Gruden K (2012) How to reliably test for GMOs. Springer, New York
Lipp M, Shillito R, Giroux R, Spiegelhalter F, Charlton S, Pinero D, Song P (2005) Polymerase chain reaction technology as analytical tool in agricultural biotechnology. J AOAC Int 88:136–155
Hernandez M, Rodriguez-Lazaro D, Ferrando A (2005) Current methodology for detection, identification and quantification of genetically modified organisms. Curr Anal Chem 1:203–221
Huggett J, Bustin SA (2011) Standardisation and reporting for nucleic acid quantification. Accred Qual Assur 16:399–405
Kodama T, Kurosawa Y, Kitta K (2010) Tendency for interlaboratory precision in the GMO analysis method based on real-time PCR. J AOAC Int 93:734–749
European Network of Genetically Modified Organisms Laboratories (2008) Definition of minimum performance requirements for analytical methods of GMO testing. Publications Office of the European. Union, Luxembourg
European Commision (2003) Regulation (EC) No 1829/2003 of the European Parliament and of the Council of 22 September 2003 on genetically modified food and feed. Off J Eur Union 268:1–23
Codex Committee on Methods of Analysis and Sampling (2010) Guidelines on performance criteria and validation of methods for detection, identification and quantification of specific DNA sequences and specific proteins in foods. CAC/GL 74-2010. Codex Alimentarius Commision – WHO, Rome
European Network of GMO Laboratories (2011) Verification of real time PCR methods for GMO testing when implementing interlaboratory validated methods - guidance document from the European Network of GMO laboratories (ENGL). Publications Office of the European Union, Luxembourg
Technical Committee of National Agricultural Genetically Modified Organisms safety Management and Standardisation. China. Notification on the issuance of the “validation protocol of GMO detection methods”. Notification 2012-1
Caprioara-Buda M, Meyer W, Jenyov B, Corbisier P, Trapmann S, Emons H (2012) Evaluation of plasmid and genomic DNA calibrants used for the quantification of genetically modified organisms. Anal Bioanal Chem 404:29–42
European Commision (2004) Commission Regulation (EC) No. 641/2004 of 6 April 2004 on detailed rules for the implementation of Regulation (EC) No. 1829/2003 of the European Parliament and of the Council as regards the application for the authorisation of new genetically modified food and feed, the notification of existing products and adventitious or technically unavoidable presence of genetically modified material which has benefited from a favourable risk evaluation. Off J Eur Union 102:14–25
Gruden K, Allnutt TR, Ayadi M, Baeumler S, Bahrdt C, Berben G, Berdal KG, Bertheau Y, Bøydler Andersen C, Brodmann P, Buh Gašparič M, Burns MJ, Burrel AM, Cankar K, Esteve T, Holst-Jensen A, Kristoffersen AB, La Paz J, Lee D, Løvseth A, Macarthur R, Morisset D, Pla M, Rud RB, Skjœret C, Tengs T, Valdivia H, Wulff D, Zhang D, Žel J (2012) Reliability and cost of GMO detection. In: Bertheau Y (ed) Genetically modified and non-genetically modified food supply chains: co-existence and traceability. Wiley-Blackwell, Oxford
Meyer W, Caprioara-Buda M, Jenyov B, Corbisier P, Trapmann S, Emons H (2012) The impact of analytical quality criteria and data evaluation on the quantification of genetically modified organisms. Eur Food Res Technol 235:597–610
Regulation (EC) No. 882/2004 of the European Parliament and of the Council of 29 April 2004 on official controls performed to ensure the verification of compliance with feed and food law, animal health and animal welfare rules. Off J Eur Union 165:1–141
Trapmann S, Charles Delobel C, Corbisier P, Emons H, Hougs L, Philipp P, Sandberg M, Schulze M (2013) European technical guidance document for the flexible scope accreditation of laboratories quantifying GMOs. Publications Office of the European Union, Luxembourg
International Organization for Standardization (2006) EN ISO 24276:2006. Foodstuffs – methods of analysis for the detection of genetically modified organisms and derived products – general requirements and definitions. International Organization for Standardization, Geneva
International Organization for Standardization (2013) EN ISO 24276:2006/A1:2013. Foodstuffs – methods of analysis for the detection of genetically modified organisms and derived products – general requirements and definitions. International Organization for Standardization, Geneva
Trapmann S, Burns M, Broll H, Macartur R, Wood R, Žel J (2009) Guidance document on measurement uncertainty for GMO testing laboratories. Office for Official Publications of the European Communities, Luxembourg
Van den Bulcke M, Bellocchi G, Berben G, Burns M, Cankar K, De Giacomo M, Gruden K, Holst-Jensen A, Malcewsky A, Mazzara M, Onori R, Papazova N, Parlouer E, Taverniers I, Trapmann S, Wulff D, Zhang D (2012) The modular approach in GMO quality control and enforcement support systems. In: Bertheau Y (ed) Genetically modified and non-genetically modified food supply chains: co-existence and traceability. Wiley-Blackwell, Oxford
Bhat S, Curach N, Mostyn T, Bains GS, Griffiths KR, Emslie KR (2010) Comparison of methods for accurate quantification of DNA mass concentration with traceability to the international system of units. Anal Chem 82:7185–7192
Folloni S, Bellocchi G, Prospero A, Querci M, Moens W, Ermolli M, Van den Eede G (2010) Statistical evaluation of real-time PCR protocols applied to quantify genetically modified maize. Food Anal Methods 3:304–312
Cankar K, Stebih D, Dreo T, Žel J, Gruden K (2006) Critical points of DNA quantification by real-time PCR–effects of DNA extraction method and sample matrix on quantification of genetically modified organisms. BMC Biotechnol 6:37
Demeke T, Jenkins G (2010) Influence of DNA extraction methods, PCR inhibitors and quantification methods on real-time PCR assay of biotechnology-derived traits. Anal Bioanal Chem 396:1977–1990
Morisset D, Demšar T, Gruden K, Vojvoda J, Štebih D, Žel J (2009) Detection of genetically modified organisms—closing the gaps. Nat Biotechnol 27:700–701
Ghedira R, Papazova N, Vuylsteke M, Ruttink T, Taverniers I, De Loose M (2009) Assessment of primer/template mismatch effects on real-time PCR amplification of target taxa for GMO quantification. J Agric Food Chem 57:9370–9377
Papazova N, Zhang D, Gruden K, Vojvoda J, Yang L, Buh M, Blejec A, Fouilloux S, De Loose M, Taverniers I (2010) Evaluation of the reliability of maize reference assays for GMO quantification. Anal Bioanal Chem 396:2189–2201
Hernández M, Río A, Esteve T, Prat S, Pla M (2001) A rapeseed-specific gene, acetyl-CoA carboxylase, can be used as a reference for qualitative and real-time quantitative PCR detection of transgenes from mixed food samples. J Agric Food Chem 49:3622–3627
Paternò A, Marchesi U, Gatto F, Verginelli D, Quarchioni C, Fusco C, Zepparoni A, Amaddeo D, Ciabatti I (2009) Finding the joker among the maize endogenous reference genes for genetically modified organism (GMO) detection. J Agric Food Chem 57:11086–11091
Taverniers I, Papazova N, Allnutt T, Baumler S, Bertheau Y, Esteve T, Freyer R, Gruden K, Kuznetzov B, Luis La Paz J, Nadal A, Pla M, Vojvoda J, Wulff D, Zhang D (2012) Harmonised reference genes and PCR assays for GMO quantification. In: Bertheau Y (ed) Genetically modified and non-genetically modified food supply chains: co-existence and traceability. Wiley-Blackwell, Oxford
Wei J, Li F, Guo J, Li X, Xu J, Wu G, Zhang D, Yang L (2013) Collaborative ring trial of the papaya endogenous reference gene and its polymerase chain reaction assays for genetically modified organism analysis. J Agric Food Chem 61:11363–11370
European Union Reference Laboratory for Genetically Modified Food and Feed (2006) Event-specific method for the quantitation of sugar beet line H7-1 using real-time PCR - validation report. IHCP, Ispra
Chaouachi M, Alaya A, Ali IB, Hafsa AB, Nabi N, Bérard A, Romaniuk M, Skhiri F, Saïd K (2013) Development of real-time PCR method for the detection and the quantification of a new endogenous reference gene in sugar beet “Beta vulgaris L”.: GMO application. Plant Cell Rep 32:117–128
Guan Q, Wang X, Teng D, Yang Y, Tian F, Yin Q, Wang J (2011) Construction of a standard reference plasmid for detecting GM cottonseed meal. Appl Biochem Biotechnol 165:24–34
Ballari RV, Martin A, Gowda LR (2013) A calibrator plasmid for quantitative analysis of insect resistant maize (Yieldgard MON 810). Food Chem 140:382–389
Brod FC, Dinon AZ, Kolling DJ, Faria JC, Arisi AC (2013) Development of plasmid DNA reference material for the quantification of genetically modified common bean embrapa 5.1. J Agric Food Chem 61:4921–4926
Zhang D, Corlet A, Fouilloux S (2008) Impact of genetic structures on haploid genome-based quantification of genetically modified DNA: theoretical considerations, experimental data in MON 810 maize kernels (Zea mays L.) and some practical applications. Transgenic Res 17:393–402
Holst-Jensen A, De Loose M, Van den Eede G (2006) Coherence between legal requirements and approaches for detection of genetically modified organisms (GMOs) and their derived products. J Agric Food Chem 54:2799–2809
Liu D, Shen J, Yang J, Zhang D (2010) Evaluation of the impacts of different nuclear DNA content in the hull, endosperm, and embryo of rice seeds on GM rice quantification. J Agric Food Chem 58:4582–4587
Cankar K, Chauvensy-Ancel V, Fortabat M, Gruden K, Kobilinsky A, Žel J, Bertheau Y (2008) Detection of nonauthorized genetically modified organisms using differential quantitative polymerase chain reaction: application to 35S in maize. Anal Biochem 376:189–199
Berdal KG, Bøydler C, Tengs T, Holst-Jensen A (2008) A statistical approach for evaluation of PCR results to improve the practical limit of quantification (LOQ) of GMO analyses (SIMQUANT). Eur Food Res Technol 227:1149–1157
Lee D, La Mura M, Greenland A, Mackay I (2008) Quantitation using informative zeros (QUIZ): application for GMO detection and quantification without recourse to certified reference material. Food Chem 118:974–978
Guo J, Yang L, Chen L, Morisset D, Li X, Pan L, Zhang D (2011) MPIC: a high-throughput analytical method for multiple DNA targets. Anal Chem 83:1579–1586
Guo J, Yang L, Chen L, Liu X, Gao Y, Zhang D, Yang L (2012) A multiplex degenerate PCR analytical approach targeting to eight genes for screening GMOs. Food Chem 132:1566–1573
Kim J, Zhang D, Kim H (2014) Detection of sixteen genetically modified maize events in processed foods using four event-specific pentaplex PCR systems. Food Control 35:345–353
Shao N, Jiang S, Zhang M, Wang J, Guo S, Li Y, Jiang H, Liu C, Zhang D, Yang L, Tao S (2014) MACRO: a combined microchip-PCR and microarray system for high-throughput monitoring of genetically modified organisms. Anal Chem 86:1269–1276
Rudi K, Rud I, Holck A (2003) A novel multiplex quantitative DNA array based PCR (MQDA-PCR) for quantification of transgenic maize in food and feed. Nucleic Acids Res 31:e62. doi:10.1093/nar/gng061
Morisset D, Dobnik D, Hamels S, Žel J, Gruden K (2008) NAIMA: target amplification strategy allowing quantitative on-chip detection of GMOs. Nucleic Acids Res 36:e118. doi:10.1093/nar/gkn524
Dobnik D, Morisset D, Gruden K (2009) NAIMA as a solution for future GMO diagnostics challenges. Anal Bioanal Chem 396:2229–2233
Heide B, Dromtorp S, Rudi K, Heir E, Holck A (2008) Determination of eight genetically modified maize events by quantitative, multiplex PCR and fluorescence capillary gel electrophoresis. Eur Food Res Technol 227:1125–1137
Morisset D, Štebih D, Milavec M, Gruden K, Žel J (2013) Quantitative analysis of food and feed samples with droplet digital PCR. PLoS ONE 8(5):e62583
Baker M (2012) Digital PCR hits its stride. Nat Methods 9:541–544
Burns MJ, Burrell AM, Foy C (2010) The applicability of digital PCR for the assessment of detection limits in GMO analysis. Eur Food Res Technol 231:353–362
Corbisier P, Bhat S, Partis L, Rui Dan Xie V, Emslie K (2010) Absolute quantification of genetically modified MON810 maize (Zea mays L.) by digital polymerase chain reaction. Anal Bioanal Chem 396:2143–2150
Bhat S, Herrmann J, Armishaw P, Corbisier P, Emslie KR (2009) Single molecule detection in nanofluidic digital array enables accurate measurement of DNA copy number. Anal Bioanal Chem 394:457–467
Heide B, Heir E, Holck A (2008) Detection of eight GMO maize events by qualitative, multiplex PCR and fluorescence capillary gel electrophoresis. Eur Food Res Technol 227:527–535
Kiddle G, Hardinge P, Buttigieg N, Gandelman O, Pereira C, McElgunn C, Rizzoli M, Jackson R, Appleton N, Moore C, Tisi L, Murray J (2012) GMO detection using a bioluminescent real time reporter (BART) of loop mediated isothermal amplification (LAMP) suitable for field use. BMC Biotechnol 12:15
Chen X, Wang X, Jin N, Zhou Y, Huang S, Miao Q, Zhu Q, Xu J (2012) Endpoint visual detection of three genetically modified rice events by loop-mediated isothermal amplification. Int J Mol Sci 13:14421–14433
Li Q, Fang J, Liu X, Xi X, Li M, Gong Y, Zhang M (2013) Loop-mediated isothermal amplification (LAMP) method for rapid detection of cry1Ab gene in transgenic rice (Oryza sativa L.). Eur Food Res Technol 236:589–598
Randhawa GJ, Singh M, Morisset D, Sood P, Žel J (2013) Loop-mediated isothermal amplification: rapid visual and real-time methods for detection of genetically modified crops. J Agric Food Chem 61:11338–11346
Soleimani M, Shams S, Majidzadeh-A K (2013) Developing a real-time quantitative loop-mediated isothermal amplification assay as a rapid and accurate method for detection of Brucellosis. J Appl Microbiol 115:828–834
Huang X, Chen L, Xu J, Ji H, Zhu S, Chen H (2014) Rapid visual detection of phytase gene in genetically modified maize using loop-mediated isothermal amplification method. Food Chem 156:184–189
Sykes PJ, Neoh SH, Brisco MJ, Hugues E, Condon J, Morley AA (1992) Quantitation of targets for PCR by use of limiting dilution. Biotechniques 13:444–449
Pinheiro LB, Coleman VA, Hindson CM, Herrmann J, Hindson BJ, Bhat S, Emslie KR (2012) Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal Chem 83:1003–1011
Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, Kitano TK, Hodel MR, Petersen JF, Wyatt PW, Steenblock ER, Shah PH, Bousse LJ, Troup CB, Mellen JC, Wittmann DK, Erndt NG, Cauley TH, Koehler RT, So AP, Dube S, Rose KA, Montesclaros L, Wang S, Stumbo DP, Hodges SP, Romine S, Milanovich FP, White HE, Regan JF, Karlin-Neumann GA, Hindson CM, Saxonov S, Colston BW (2011) High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem 83:8604–8610
Sanders R, Huggett JF, Bushell CA, Cowen S, Scott DJ, Foy CA (2011) Evaluation of digital PCR for absolute DNA quantification. Anal Chem 83:6474–6484
Weaver S, Dube S, Mir A, Qin J, Sun G, Ramakrishnan R, Jones RC, Livak KJ (2010) Taking qPCR to a higher level: analysis of CNV reveals the power of high throughput qPCR to enhance quantitative resolution. Methods 50:271–276
Whale AS, Huggett JF, Cowen S, Speirs V, Shaw J, Ellison S, Foy CA, Scott DJ (2012) Comparison of microfluidic digital PCR and conventional quantitative PCR for measuring copy number variation. Nucleic Acids Res 40:e82
Whale AS, Cowen S, Foy CA, Huggett JF (2013) Methods for applying accurate digital PCR analysis on Low copy DNA samples. PLoS ONE 8:e58177
Dingle TC, Sedlak RH, Cook L, Jerome KR (2013) Tolerance of droplet-digital PCR vs real-time quantitative PCR to inhibitory substances. Clin Chem 59:11
Pérez Urquiza M, Acatzi Silva AI (2014) Copy number ratios determined by two digital polymerase chain reaction systems in genetically modified grains. Metrologia 51:61–66
McDermott GP, Do D, Litterst CM, Maar D, Hindson CM, Steenblock ER, Legler TC, Jouvenot Y, Marrs SH, Bemis A, Shah P, Wong J, Wang S, Sally D, Javier L, Dinio T, Han C, Brackbill TP, Hodges SP, Ling Y, Klitgord N, Carman GJ, Berman JR, Koehler RT, Hiddessen AL, Walse P, Bousse L, Tzonev S, Hefner E, Hindson BJ, Cauly TH 3rd, Hamby K, Patel VP, Regan JF, Wyatt PW, Karlin-Neumann GA, Stumbo DP, Lowe AJ (2013) Multiplexed target detection using DNA-binding dye chemistry in droplet digital PCR. Anal Chem 85:11619–11627
Yang L, Wang C, Holst-Jensen A, Morisset D, Lin Y, Zhang D (2013) Characterization of GM events by insert knowledge adapted re-sequencing approaches. Sci Rep 3:2839
Polko JK, Temanni MR, van Zanten M, van Workum W, Iburg S, Pierik R, Voesenek LA, Peeters AJ (2012) Illumina sequencing technology as a method of identifying T-DNA insertion loci in activation-tagged Arabidopsis thaliana plants. Mol Plant 5:948–950
Fullwood MJ, Wei CL, Liu ET, Ruan Y (2009) Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses. Genome Res 19:521–532
Campbell PJ, Stephens PJ, Pleasance ED, O'Meara S, Li H, Santarius T, Stebbings LA, Leroy C, Edkins S, Hardy C, Teague JW, Menzies A, Goodhead I, Turner DJ, Clee CM, Quail MA, Cox A, Brown C, Durbin R, Hurles ME, Edwards PA, Bignell GR, Stratton MR, Futreal PA (2008) Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet 40:722–729
Hormozdiari F, Hajirasouliha I, McPherson A, Eichler EE, Sahinalp SC (2011) Simultaneous structural variation discovery among multiple paired-end sequenced genomes. Genome Res 21:2203–2212
Tengs T, Zhang H, Holst-Jensen A, Bohlin J, Butenko M, Kristoffersen A, Sorteberg H-G, Berdal K (2009) Characterization of unknown genetic modifications using high throughput sequencing and computational subtraction. BMC Biotechnol 9:87
Dubose AJ, Lichtenstein ST, Narisu N, Bonnycastle LL, Swift AJ, Chines PS, Collins FS (2013) Use of microarray hybrid capture and next-generation sequencing to identify the anatomy of a transgene. Nucleic Acids Res 41:e70
Kovalic D, Garnaat C, Guo L, Yan Y, Groat J, Silvanovich, Ralston L, Huang M, Tian Q, Christian A, Cheikh N, Hjelle J, Padgette S, Bannon G (2013) The use of next generation sequencing and junction sequence analysis bioinformatics to achieve molecular characterization of crops improved through modern biotechnology. Plant Genome 5:149–163
Taverniers I, Papazova N, Bertheau Y, De Loose M, Holst-Jensen A (2008) Gene stacking in transgenic plants: towards compliance between definitions, terminology, and detection within the EU regulatory framework. Environ Biosaf Res 7:197–218
Akiyama H, Watanabe T, Wakabayashi K, Nakade S, Yasui S, Sakata K, Chiba R, Spiegelhalter F, Hino A, Maitani T (2005) Quantitative detection system for maize sample containing combined-trait genetically modified maize. Anal Chem 77:7421–7428
Akiyama H, Minegishi Y, Makiyama D, Mano J, Sakata K, Nakamura K, Noguchi A, Takabatake R, Futo S, Kondo K, Kitta K, Kato Y, Teshima R (2012) Quantification and identification of genetically modified maize events in non-identity preserved maize samples in 2009 using an individual kernel detection system. Food Hyg Saf Sci 53:157–165
Shin K, Suh S, Lim M, Woo H, Lee J, Kim H, Cho H (2013) Event-specific detection system of stacked genetically modified maize by using the multiplex-PCR technique. Food Sci Biotechnol 22:1763–1772
Mano J, Yanaka Y, Ikezu Y, Onishi M, Futo S, Minegishi Y, Ninomiya K, Yotsuyanagi Y, Spiegelhalter F, Akiyama H, Teshima R, Hino A, Naito S, Koiwa T, Takabatake R, Furui S, Kitta K (2011) Practicable group testing method to evaluate weight/weight GMO content in maize grains. J Agric Food Chem 59:6856–6863
Xu W, Yuan Y, Luo Y, Bai W, Zhang C, Huang K (2009) Event-specific detection of stacked genetically modified maize Bt11 x GA21 by UP-M-PCR and real-time PCR. J Agric Food Chem 57:395–402
Lusser M, Parisi C, Plan D, Rodríguez-Cerezo E (2011) New plant breeding techniques. State-of-the-art and prospects for commercial development. JRC Technical Report EUR 24760 EN. European Commission Joint Research Centre, Rome
Lusser M, Parisi C, Plan D, Rodríguez-Cerezo E (2012) Deployment of new biotechnologies in plant breeding. Nat Biotechnol 30:231–239
Querci M, Foti N, Bogni A, Kluga L, Broll H, Van den Eede G (2009) Real-time PCR-based ready-to-use multi-target analytical system for GMO detection. Food Anal Methods 2:325–336
Kluga L, Folloni S, Van den Bulcke M, Van den Eede G, Querci M (2012) Applicability of the real-time PCR-based ready-to-use multi-target analytical system for GMO detection in highly processed food matrices. Eur Food Res Technol 234:109–118
Cottenet G, Blancpain C, Sonnard V, Chuah PF (2013) Development and validation of a multiplex real-time PCR method to simultaneously detect 47 targets for the identification of genetically modified organisms. Anal Bioanal Chem 405:6831–6844
Acknowledgments
We thank Dr. Christopher Berrie for reviewing the manuscript. The work was co-financed by the Slovenian Research Agency (contract no. P4-0165), the Slovenian Ministry of Agriculture and Environment (contract no. 2330-13-000072) and the Slovenian Ministry of Economic Development and Technology, Metrology Institute of the Republic of Slovenia (contract no. 640118/2008/67). The research leading to these results has received funding from the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement n° 613908, relating to the project “Development of Cost-efficient Advanced DNA-based methods for specific Traceability issues and High Level On-site Applications”.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the topical collection Nucleic Acid Quantification with guest editors Hendrik Emons and Philippe Corbisier.
Rights and permissions
About this article

Cite this article
Milavec, M., Dobnik, D., Yang, L. et al. GMO quantification: valuable experience and insights for the future. Anal Bioanal Chem 406, 6485–6497 (2014). https://doi.org/10.1007/s00216-014-8077-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-014-8077-0