
Abstract
Specific legislation in the EU and several other countries requires that foods containing genetically modified organisms (GMOs) should be approved and labelled. This has necessitated the development of methods for detection of such materials. For screening purposes these methods should preferably enable detection of several different GMOs. Here we present a simple, robust, qualitative, nineplex PCR method for event-specific detection of maize T25, GA21, TC1507, MON863, MON810, NK603, construct specific detection of BT176, BT11 and detection of the endogenous hmga maize reference gene. PCR is carried out with primers labelled with fluorescent groups and the amplicons are detected using fluorescence capillary electrophoresis. Using mixtures of DNA from different certified reference materials, the detection limit was determined to approximately 0.1% for each GMO. Good agreement was observed in 85 of 88 determinations when eleven food and feed samples were analysed using the multiplex PCR assay and compared to results from quantitative real-time 5′-nuclease PCR. Discrepancies were only observed for one GMO at or close to the detection limit. The presented method is therefore suitable for screening purposes for food and feed containing the most common maize GMOs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The employment of genetically modified plants (GMPs) is the fastest growing trend in the history of agriculture, with an annual growth exceeding 10% every year for the last decade. The global planted area has increased from 1.66 million ha in 1996 to 102 million ha in 2006 [1]. The main crops used for GM traits are soybean, maize, canola and cotton, and the main traits introduced into GMPs are tolerance to pesticides and resistance to insects. There is great public concern regarding the ecological effects of GM agriculture and the safety of foods derived from GMO plants among consumers in parts of the world. This has led several countries including the EU, Brazil, Canada, and Russia to establish threshold levels for GMO events [2]. The threshold levels range from 0.9 to 5%. The EU has the most stringent legislation, where the threshold for technologically unavoidable presence of approved GMOs is set to 0.9%. Foods that contain the threshold must be labelled as “this product contains GMO” (http://europa.eu.int/eur-lex/pri/en/oj/dat/2003/l_268/l_26820031018en00240028.pdf). For unapproved GMOs that have received a favourable opinion by the EU’s Community Scientific Committee, the tolerance threshold is 0.5%. For all other unapproved GMOs the tolerance threshold is 0.0%. In addition, the EU also requires traceability, meaning ability to trace GMOs and products made from GMOs at all stages of their placing on the market through the production and distribution chains.
The legislation has necessitated development of detection methods for GMO. Protein-based GMO detection methods have been described, and are reviewed by Grothaus et al. [3]. DNA-based methods often offer increased specificity, sensitivity and robustness. A frequently used method for GMO detection is real-time PCR. DNA based methods have been reviewed previously [4, 5]. The level of multiplexing is a key feature of DNA methods. As the number of GMO events on the market increases, it is clear that methods that can detect several targets in a single reaction, i.e. multiplex detection, is preferable when enforcing the legislation ruling the occurrence of GMOs in food. For large-scale screening, where most samples are expected to be negative for the presence of GMOs, qualitative analysis is a well-suited approach. To ensure specific detection, amplicons should span event-specific regions, either in the overlap between insert and plant DNA or event-specific rearrangements in the insert or plant DNA. Several reports on multiplex PCR-based techniques for the detection of GMO events have been published, most reports regard combinations of only a few amplicons. Nadal et al. [6] reported on the detection of four events of GM maize using PCR and capillary electrophoresis detection. Onishi et al. [7] were able to monitor eight different GM maize events in a single reaction detected by agarose gel electrophoresis and microchip electrophoresis. The same eight maize GMOs were also amplified and detected by agarose gel electrophoresis [8]. We have previously developed a two-step, array-based quantitative multiplex method for seven GMO constructs, MON810, BT176, BT11, T25, GA21, DBT418 and CBH351 [9].
Here we present a simple, robust and sensitive method for the multiplex, qualitative detection and identification of eight GMO events of maize, of which six are event-specific (MON810, MON863, GA21, T25, TC1507 and NK603) and two (BT11 and BT176) are construct-specific.
Materials and methods
Materials
Certified reference materials (CRMs) of GM maize lines of Bt11, Bt176, Mon810, Mon863, Ga21, NK603, TC1507 were purchased from Fluka. These weight-based materials are made by dry mixing after milling of seeds of defined cultivars of GMO and a non-GMO variety with the same genetic background. T25 material was kindly provided by H. Broll. Commercial food samples were bought in supermarkets in California, USA, 2002. Samples with known combinations of GMO content were made by mixing DNA isolated from individual reference materials (Table 1).
DNA isolation
An amount of 0.12–0.15 g of standard material or grinded food samples was used for DNA extraction with Qiagen Plant Mini Kit (Qiagen, Hilden, Germany) as described by the manufacturer with the following modifications. The initial buffer volume was doubled and lysis was carried out for 30 min at 65 °C in a shaking incubator. When eluting DNA bound to the column, 50 μL of buffer was used. Chromosomal DNA was analysed by agarose gel electrophoresis. The isolated DNA was analysed for quality and quantity with a Nano-Drop ND-1000 spectrophotometer (Saveen Werner, USA) and stored at −20 °C.
Oligonucleotide primers
A total of nine primer pairs, specific for eight GMO events and one maize reference gene, were designed and tested for suitability for multiplex PCR using FastPCR version 3.6.95
(Institute of Biotechnology, University of Helsinki, Finland, 2005) and Primer Express version 2.0 (Applied Biosystems) or taken from the literature (Table 2). Primers and probes for 5′-nuclease PCR were from the literature as listed in Table 3. The unlabelled primers were purchased from MWG-Biotech AG (Ebensburg, Germany), and the labelled primers from Applied Biosystems (Foster City, Ca, USA).
PCR conditions
The multiplex PCR was performed with Qiagen Multiplex PCR Kit (Qiagen, Hilden, Germany), in a total volume of 25 μL. Approximately 100 ng of template DNA was used in each reaction. The amplification protocol was slightly modified from the manufacturer’s instructions, and was as follows; an initial activation of the enzyme at 95 °C for 15 min; six cycles of 94 °C for 30 s, 60 °C for 90 s, 72 °C for 90 s, followed by 30 cycles of 94 °C for 30 s, 70 °C for 90 s, 72 °C for 90 s. The PCRs were carried out on a GeneAmp® PCR System 9700 (Applied Biosystems). Samples were analysed in triplicate.
Capillary electrophoresis
Nine microlitres of Hi-Di formamide and 0.5 μL Rox-500 standard (Applied Biosystems,) was added to 1–2 μL of the PCR product, and the sample was incubated at 95 °C for 5 min, and immediately put on ice. The sample was then loaded onto a 36 cm capillary in the ABI GA3100 sequencer (Applied Biosystems), containing the performance optimised polymer 4 (POP-4) (Applied Biosystems). The electrophoretic conditions were 5 seconds injection, run time 1,500 s at 15,000 Volts, run current 100 μAmps and 60 °C run temperature. Filter set D was used to detect the following fluorophores NED, VIC, 6-FAM and ROX. GeneScan 3.7 software was used to determine the size of the fragments. The observed sizes of the fragments were determined using singleplex reactions and the ROX-500 standard. The sizes of the fragments in the nineplex reaction did not deviate from the singleplex values.
5′-Nuclease PCR
Separate PCRs were carried out for each event under the same condition in a total reaction volume of 25 μL. Amplification reactions contained 2.5 μL of sample (approximately 100 ng), 2.5 μL TaqMan 10× Buffer A, 5 mmol/L MgCl2, 200 μmol/L dATP, dCTP and dGTP and 400 mmol/L dUTP, 0.2 μmol/L of each specific primer, 0.1 μmol/L of specific probes, 0.004 U of AmpErase Uracil N-glycolsylase (UNG) and 1.25 U AmpliTaq Gold DNA polymerase (Applied Biosystems). Before amplification, the mixture was heated to 50 °C for 2 min to let the UNG destroy possibly contaminating PCR products, and 95 °C for 10 min to inactivate UNG, to denature the DNA template and to activate the polymerase. Amplification profile; 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The reactions were performed on a 7900 HT Sequence Detection System (Applied Biosystems). The probes consisted of oligonucleotides labelled with 5′-FAM reporter dye and a 3′-TAMRA quencher dye. Real-time PCR primer and probe sequences are listed in Table 3. All samples were analysed in triplicate, and for a sample to be determined as positive, at least two parallels needed to be positive. Standard curves for each GMO were constructed by plotting differences in C t values between the GMO event and the endogenous maize reference gene (hmga) against log of the GMO content in percent of known certified reference materials. Unknown samples were determined by comparing to the standard curves. All calculations were carried out using Excel 2003.
Results
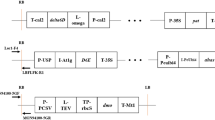
Many aspects of the multiplex reaction needed thorough optimisation. We tested several DNA polymerases and PCR kits, and the choice of DNA polymerase proved to be essential to the performance of the system. We found that the HotStart DNA polymerase, coupled with a pre-optimised mastermix for multiplex reactions, gave the best results in terms of reproducibility and robustness (results not shown). No further optimalisation of the mastermix components was necessary. The primers were tested in silico for suitability in the multiplex set-up, i.e. tested for primer-dimer formation. Primers that showed poor performance in the PCR were redesigned, or primers from other regions were selected. The positions of the amplicons are given in Fig. 1. Preliminary experiments using single templates in combination with single primer pairs and primer mixes gave single peaks and showed that all primer pairs were specific for their corresponding template (results not shown). The optimal primer concentration was determined empirically by selecting levels that gave approximately even heights of the signals in the capillary electrophoresis for a mix containing 0.4–0.6% of all GMO events. The final primer concentrations varied 16-fold, from 0.05 μM for the ZM reference gene to 0.8 μM for T25. The addition of a common 5′-sequence in all primers resulted in more similar amplification efficiencies for the different amplicons than the same primers without the common sequence did (results not shown). Further, the PCR temperature profile was optimised from the supplier’s protocol by varying number of cycles and annealing temperature. In addition, it was shown that raising the annealing temperature from 60 to 70 °C after the initial six cycles, added specificity to the method (results not shown). In order to obtain reliable results and minimise variables in the PCR, the amount of DNA used as template in the multiplex PCR was standardised to 100 ng. However, object of the standardisation was the total amount of DNA, not maize DNA. For mixed products containing other DNA materials than maize, the maize DNA concentration will deviate from this value. In the capillary electrophoresis, the only optimisation of the supplied protocol was the choice of injection time for the samples, in our case a 5-s injection time was the optimal. The lengths of the individual PCR products were determined in singleplex reactions. A single peak was detected at approximately the expected lengths for all amplicons, except for GA21, which gave two peaks, one at the expected 135 bp and an additional peak at 276 bp. The T25 peak had a neighbouring, unspecific peak at 125.3 bp while the T25 specific peak was located at 126.4 bp.

Positions of amplicons in the different GM maize constructs. Amplicons are shown as horizontal bars. P-35S, sequence derived from cauliflower mosaic virus promoter; hsp70, sequence containing intron no. 1 of the 70 kDa heat-shock protein of maize; cry1A(b), synthetic delta endotoxin gene derived from Bacillus thuringiensis; IVS2, intron from maize alcohol dehydrogenase; pat, phosphinotricin N-acetyl transferase gene from Streptomyces viridochromogenes; T-nos, transcription terminator from Agrobacter tumefaciens nopalin synthase gene; pepC, phosphoenolpyruvate carboxylase promoter; T-35S, terminator from cauliflower mosaic virus; R-act, rice actin promoter, OTP optimised transit peptide sequence; m-EPSPS, point mutated epsps gene encoding 5-enolpuruvylshikimate-3-phosphate synthase from maize; PHI8999A, DNA from transformation vector insert containing the cry1F and pat genes; IR-cry1F, inverted repeat region of the cry1F gene; nptII, neomycin phosphotransferase from Escherichia coli; EPSPS, the epsps gene from Agrobacter tumefaciens, vDNA, construct vector DNA; chlDNA, Zea maize plastid DNA. The TC1507 detection is event specific since it encompasses an event specific integration rearrangement. Maize DNA is shown in grey
Mixed GMO samples prepared from purified DNA from certified reference materials (CRMs) with known contents of the individual GMOs were further used to evaluate the specificity and the detection levels of the multiplex PCR (Table 1; Fig. 2). The method was optimised using a mixture containing 0.4–0.6% of all GMOs to ensure a reasonable sensitivity of the assay (Fig. 2a) and it was shown that in both this mix and a mixture of 0.1% of all the eight GMOs (Fig. 2b), all GMOs were detectable. To test the effect of high total GMO contents on events present at lower concentrations, mixtures of four GMOs present in high concentration (0.6–2.2% of the individual GMOs) and four GMOs present in low concentration (0.1–0.3%), yielding 5.7–5.9% total GMO concentration were analyzed together (Fig. 2c, d). All GMOs present in low concentrations were detectable. Similar results were obtained when low concentrations of GMO were detected in presence of high concentrations of NK603 or MON863 (Fig. 2e, f). In a mixture containing 50.7% T25 and 0.3–0.5% of the seven other GMOs (Fig. 2g), all the GMOs could be detected. To further test the specificity, we also examined samples containing non-GMO maize and samples not containing maize DNA. The non-GMO samples produced a peak for the ZM reference gene, but no other specific peaks (Fig. 2h). The non-maize DNA sample did not produce any specific peaks (Fig. 2i).

Nineplex assay of eight maize GMO events and one maize endogenous control (ZM). The panels show the mixtures listed in Table 1. a Contains 0.4–0.6% of all GMO events, b contains 0.1% of all events, c contains 0.1% T25, 1.2% GA21, 0.3% TC1507, 0.1% BT176, 1.6% MON863, 1.0% BT11 1.5% MON810 and 0.1% NK603 and d contains 1.4% T25, 0.1% GA21, 2.2% TC1507, 0.6% Bt176, 0.1% MON863, 0.1% BT11, 0.1% MON810 and 1.1% NK603, and, e contains 2.5% NK603 and 0.1–0.2% of all other GMOs, f contains 2.5% MON863 and 0.1–0.2% of all other events, g contains 50.7% T25 and 0.3–0.5% of all other events, h contains non-genetically modified maize and i contains no maize DNA
Fifteen commercial food and feed samples were tested by the multiplex PCR approach (Fig. 3) and by 5′-nuclease PCR. Four of the samples, mostly pet food products, contained insufficient amounts of amplifiable maize DNA, according to the 5′-nuclease PCR, to give reliable results. The results from the multiplex PCR and 5′-nuclease PCR for the remaining eleven commercial samples were compared and presented in Table 4, and examples of panels are given in Fig. 3. Data from the multiplex qualitative PCR agreed well with the quantitative data from the 5′-nuclease PCR. For samples 12 and 14 containing 0.1% BT11, no signal was obtained with the multiplex method. Likewise for sample 13 where 1 out of 3 parallels gave a positive 5′-nuclease PCR signal, the multiplex method gave a weak BT11 signal (Fig. 3c). Also, in some cases 5′-nuclease PCR gave weak positive signals for MON863 and MON810, while the multiplex method gave negative results. In all these cases the amount of GMO was less than 0.1%.

Nineplex assay of selected commercial food and feed samples. a Sample 1, contains GA21, BT176 and MON810, b sample 9 contains none (<0.1%) of the maize GMO events in the assay, c sample 13 contains T25, BT176 and MON810 and d sample 19 contains T25, GA21, BT176, BT11 and MON810
Discussion
The legislation on the occurrence of GMP in foods calls for qualitative screening methods that are specific, sensitive, robust, cost effective and easy to perform. Our approach was based on the use of PCR with forward primers labelled with FAM, NED or VIC fluorophores and unlabelled reverse primers, yielding labelled amplicons of specific lengths that were detectable by capillary electrophoresis. This is a simple, robust and high throughput method, which easily lends it self to automatisation.
Generally, development of multiplex PCR is not as straight-forward as conventional PCR. As the number of primers increases, so does the number of possible sequence dependent interactions between primers of different primer pairs, yielding primer–dimers. In addition, small differences in amplification efficiencies for the different primer pairs might result in preferential amplification of some of the PCR products, leaving other PCR products at sub-detectable levels. Thus, designing a multiplex assay is a laborious and complicated task, where primer design, cycling conditions (including extension time, extension temperature, annealing time, annealing temperature and number of PCR cycles), amount of reaction components (including primers, dNTPs, MgCl2, PCR buffer, Taq polymerase and DNA template) and the use of adjuvants (such as DMSO and BSA) should be carefully optimised in order to detect all DNA targets present in the sample [10]. In this study, we used a master mix containing pre-optimised concentrations of HotStart Taq DNA polymerase, MgCl2, dNTPs and a PCR buffer that contains a commercial factor “MP”. The synthetic factor MP increases the local concentration of primers at the DNA template and stabilises specifically bound primers, and thus ensures efficient primer annealing and extension irrespective of primer sequence. The use of HotStart DNA polymerase prevents the formation of misprimed products and reduces primer–dimer formation.
In order to further ensure that all primers anneal and extend with a similar efficiency, the primers were extended with a common 5′-sequence. The rationale behind the use of the common 5′-sequence is that primers that have similar sequences should have more similar amplification efficiencies than primers that have very different sequences. In relation to this principle, the annealing temperature was raised after the initial six cycles, since the whole primer sequence anneals to the PCR products formed in the early cycles of the PCR. As an added benefit, the elevated annealing temperature further decreases the chance of mispriming.
Also important is the choice of detection method. The most basic approach used to detect GMO events is agarose gel electrophoresis (AGE) [11]. Although AGE is regarded as a simple and cost effective standard technique, it has obvious limitations, such as low resolution and sensitivity, hampering the analysis of fragments with similar lengths or fragments that are present in low concentration. AGE also lacks automatisation capabilities, which is preferable for a technique used in screening purposes. This has led developers of multiplex PCR methods to look for alternative detection methods. Among these alternative detection methods the array approach has found a central place. In array-based GMO detection, labelled ligation or PCR products are hybridised to capture probes immobilised on a solid support [9, 12, 13] and later detected by scanners. Array-based detection offers potential for detection of a vast number of target sequences, high throughput capabilities, sensitivity and miniaturisation. However, array detection requires specialised detection equipment, and multiple steps need to be optimised in array production, printing and post-hybridisation steps to ensure sensitivity and reproducibility. Capillary electrophoresis (CE) offers many of the same advantages as arrays, such as simultaneous detection of many target sequences, high sensitivity and high throughput capabilities. CE demands fewer optimalisation steps than arrays, and it is thus easier to ensure reproducibility and is much easier to perform. Several previous papers describe different methods of CE detection using LabChips [14, 15] or different intercalating dyes and laser-induced fluorescence [16, 17].
Our approach is based on labelled primers, a pre-optimised PCR mix containing a HotStart DNA polymerase and CE. It allows for the simultaneous amplification and detection of the eight most common maize GMO events. The CE revealed very little background. Most unspecific peaks were random and very weak, only a few peaks showed to persist. The green T25 peak at the observed position 126.4 bp had an unspecific neighbouring peak at 125.3 bp of the same colour. This demonstrates the high-resolution capabilities of CE, which readily distinguishes peaks that differ only one nucleotide in size. The height of the unspecific peak did not show any relation to the percentage of T25 present in the sample. The GA21 signal had an unspecific signal at the same length, but because the unspecific peak was green, the black GA21 signal could be detected without being confused with the unspecific peak. Both the unspecific peaks could have been mistakenly regarded as false positives in a system without the high resolution and multicolour capabilities of our CE system.
We have demonstrated that the technique allows detection of GMO contents down to 0.1% for all events in reference materials. For food and feed samples, positive signals were obtained for T25 and BT176 at GMO content less than 0.1%. For other events such as MON863 and MON810 trace amounts less than 0.1% were not detected. For BT11 some samples containing 0.1% GMO appeared negative in the multiplex assay. It therefore appears reasonable to regard 0.1% as appropriate detection limit although no formal determination of LOD (Limit of detection) for each event has been performed (http://gmo-crl.jrc.it). A thorough determination of LOD is complex and will vary with many factors such as the amount of DNA used, the quality of the DNA and the composition of the sample [18]. Some authors have suggested to use DNA copy numbers as LOD instead of relative amounts [18]. In our case, using pure maize samples, 100 ng template and an estimated value of 2.6 × 109 bp/haploid maize genome [19], 0.1% would correspond to approximately 40 DNA copies.
A total of 11 commercial samples were tested for presence of eight GMO events in the multiplex approach. Of the total 88 outcomes, three deviated from the data from the 5′-nuclease PCR, all of which regarded BT11 content at or close to the detection limit of the method.
Albeit the system is qualitative, there is a certain quantitative aspect of the results. High levels of GMO generally produced signals of greater strength than low levels of GMO. This was clearly evident for the reference samples containing mixtures of GMO with four GMOs at high levels and four at low levels (Fig. 2c, d) and for the sample containing 50% T25 (Fig. 2g). Although some indications as to the quantitative content may be observed, such indications must be interpreted with extreme caution since the signal strength depends on the presence of other GMO events, e.g. the MON810 signal is much lower in Fig. 2g than in a, even though both samples contain 0.5% MON810. Many other factors may also influence the signal strength.
The nineplex PCR with CE detection described here represents an easy-to-perform and robust qualitative method for screening of samples for presence of eight different maize GMOs in food and feed samples at concentraton levels down to 0.1% GMO.
References
James C (2006) ISAAA Brief No. 35
Demeke T, Perry DJ, Scowcroft WR (2006) Can J Plant Sci 86:1–23
Grothaus GD, Bandla M, Currier T, Giroux R, Jenkins GR, Lipp M, Shan GM, Stave JW, Pantella V (2006) J Aoac Int 89:913–928
Holst-Jensen A (2007) Food toxicants analysis. Techniques, strategies and developments. Elsevier, The Netherlands 231–268
Griffiths K, Partis L, Croan D, Wang N, Elmslie KR (2002) Review of technologies for detecting genetically modified materials in commodities and food. Department of Agriculture, Fisheries and Forestry, Australia, pp 1–118
Nadal A, Coll A, La Paz JL, Esteve T, Pla M (2006) Electrophoresis 27:3879–3888
Onishi M, Matsuoka T, Kodama T, Kashiwaba K, Futo S, Akiyama H, Maitani T, Furui S, Oguchi T, Hino A (2005) J Agric Food Chem 53:9713–9721
Kim JH, Song NS, Heo MS, Lee WY, Lee SH, Park SH, Park HK, Kim MC, Kim HY (2006) Food Sci Biotech 15:148–151
Rudi K, Rud I, Holck A (2003) Nucleic Acids Res 31:e62
Henegariu O, Heerema NA, Dlouhy SR, Vance GH, Vogt PH (1997) Biotechn 23:504–511
Germini A, Zanetti A, Salati C, Rossi S, Forre C, Schmid S, Marchelli R (2004) J Agric Food Chem 52:3275–3280
Leimanis S, Hernandez M, Fernandez S, Boyer F, Burns M, Bruderer S, Glouden T, Harris N, Kaeppeli O, Philipp P, Pla M, Puigdomenech P, Vaitilingom M, Bertheau Y, Remacle J (2006) Plant Mol Biol 61:123–139
Xu J, Miao HZ, Wu HF, Huang WS, Tang R, Qiu MY, Wen JG, Zhu SF, Li Y (2006) Biosens Bioelectron 22:71–77
Birch L, Archard CL, Parkes HC, McDowell DG (2001) Food Control 12:535–540
Burns M, Shanahan D, Valdivia H, Harris N (2003) Eur Food Res Technol 216:428–433
Garcia-Canas V, Gonzalez R, Cifuentes A (2002) J Agric Food Chem 50:1016–1021
Garcia-Canas V, Gonzalez R, Cifuentes A (2002) J Agric Food Chem 50:4497–4502
Holst-Jensen A, Ronning SB, Lovseth A, Berdal KG (2003) Anal Bioanal Chem 375:985–993
Delseny M, Glaszmann JC (1995) Biofutur 146:52–56
Hernandez M, Duplan MN, Berthier G, Vaitilingom M, Hauser W, Freyer R, Pla M, Bertheau Y (2004) J Agric Food Chem 52:4632–4637
Vaitilingom M, Pijnenburg H, Gendre F, Brignon P (1999) J Agric Food Chem 47:5261–5266
Holck A, Vaitilingom M, Didierjean L, Rudi K (2002) Eur Food Res Technol 214:449–453
Nielsen CM, Berdal KG, Holst-Jensen A (2004) Eur Food Res Technol 219:421–427
Acknowledgments
This work was supported by the Norwegian Research Council project 154254/130 and the EU 6th Framework project “Co-Extra”. We want to thank Signe Drømtorp and Brit Oppegård Pedersen for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Heide, B.R., Heir, E. & Holck, A. Detection of eight GMO maize events by qualitative, multiplex PCR and fluorescence capillary gel electrophoresis. Eur Food Res Technol 227, 527–535 (2008). https://doi.org/10.1007/s00217-007-0751-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-007-0751-4