
Abstract
Ionic liquid-modified magnetic polymeric microspheres (ILMPM) were prepared based on Fe3O4 magnetic nanoparticles (MNPs) and ionic liquids (ILs) incorporated into a polymer. The composites were characterized using scanning electron microscopy, Fourier transform infrared analysis, thermogravimetric analysis, X-ray diffraction, and vibrating magnetometer, which indicated that ILMPM had a regularly spherical shape and strong magnetic property. The obtained ILMPM were successfully applied as a special adsorbent of magnetic dispersive solid phase extraction (MDSPE) for the rapid extraction and isolation of sulfamonomethoxine sodium and sulfachloropyrazine sodium in urine. The factors that affected extraction efficiency, such as adsorption conditions, desorption conditions, washing and elution solvents, and pH of the sample solution, were optimized. Under the optimum condition, good linearity in the range of 0.005–2.0 μg g−1 (r ≥ 0.9996) was obtained for the two sulfonamides (SAs); the average recoveries at three spiked levels ranged from 86.9 to 102.1 %, with relative standard deviations of ≤4.3 %. The presented ILMPM-MDSPE method combined the advantages of ILs, MNPs, and MDSPE and therefore could be potentially applied for rapid screening of SAs in urine.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Over the past few decades, determination of pharmaceutical and contaminant residues in complex samples has received a great deal of attention [1]. Due to the low concentrations and the complexity of sample matrices, an appropriate pretreatment procedure for both enrichment and purification before analysis is of great importance in the analytical process prior to instrumental analysis [2]. In recent years, several sample pretreatment methods including solid phase extraction [3,4], solid phase microextraction [5–7], dispersive solid phase extraction [8], matrix solid phase dispersion [9–11], liquid phase microextraction [12,13], and stir bar sorption extraction [14] have been reported for the effective pretreatment of complex samples. Although these pretreatment methods are extensively applied, they often suffer disadvantages such as being time-consuming, involving tedious operations, or are relatively expensive. Therefore, considerable efforts have been exerted to develop new sample pretreatment methods to overcome these drawbacks in recent years [15]. Magnetic dispersive solid phase extraction (MDSPE) has become one of the most potential strategies for pretreatment procedure as a rapid and effective extraction method based on the use of magnetic nanoparticles (MNPs) [16]. An attractive property of MNPs is that they can be isolated from sample solutions more conveniently under an external magnetic field [17]. Consequently, the time-consuming column transfer process or centrifugation operation encountered in other methods is avoided. However, several unavoidable problems are associated with MNPs, such as their intrinsic instability over long periods due to their tendency to aggregate in order to reduce their surface energy, as well as being easily oxidized in air [18]. Therefore, it is crucial to develop new strategies to improve their stability, and surface coating of a shell layer seems to be an efficient strategy. It can endow MNPs with an easily modifiable surface, which can be used to graft various desirable functional groups for effective adsorption. Recently, various materials have been used as the protective layer for MNPs, such as surfactant, inorganic metals, and organic polymers [19–22]. Among them, organic polymers are particularly appealing as they exhibit high stability in acid or base, low toxicity, and simple preparation by using different modifier agents such as methacrylic acid [23], acrylamide [24], and ionic liquids (ILs) [25].
The great interest in ILs is motivated by some unique properties, such as negligible vapor pressure, thermal stability, and non-flammability, combined with high ionic conductivity and electrochemical stability [26]. During the past few years, ILs have been widely used as an alternative solvent for extraction and isolation [27–29], running buffer additives in capillary electrophoresis [30], and mobile phase additives [31]. Recently, ILs have received a lot of attention as a surface modifier of polymers or functional monomers to prepare polymers in analytical chemistry [32,33]. For material applications, ILs keep the main properties when they are immobilized on silica surface or organic polymer [34]. Therefore, IL-modified materials have been successfully used in separation fields due to their particular characteristics such as hydrophobicity, dispersibility in inorganic/organic solvents, π–π interactions, hydrogen bonding, dipole–dipole interactions, electrostatic attraction between analytes, and functional groups of ILs [35–37]. ILs had been widely used to prepare various functional polymers [38,39]; however, few of them combined the advantages of magnetic polymers and ILs together [40].
Sulfonamides (SAs) are a group of synthetic antibiotics which are administered orally or mixed with animal feed of meat-producing animals to prevent and control a great variety of bacterial diseases [41]. However, the widespread use of SAs in veterinary may lead to SA accumulation in foodstuffs [42,43], which probably causes a variety of untoward reactions (such as urinary tract disorders, hemopoietic disorders, and toxicity) to humans after intake of animal-derived food products [44]. Urine samples are commonly selected in clinical pharmacokinetic studies to research drug excretion, and determination of SAs in human urine can provide help for clinical drug monitoring after oral administration and reflect the influence of animal-derived food products for humans. Several methods utilizing liquid chromatography with ultraviolet, fluorescence, electrochemical detector, and mass spectrometry have been used for the determination of SAs; meanwhile, a series of sample pretreatment methods including solid phase extraction [4], solid phase microextraction [5], dispersive solid phase extraction [8], and matrix solid phase dispersion [9] have been used to remove potential interferences from the sample matrix. MDSPE has also been applied in the monitoring of SAs owing to its rapid, efficient, and separation convenience. The objective of this study was to synthesize new IL-modified magnetic polymer microspheres (ILMPM) through aqueous suspension polymerization based on Fe3O4 nanoparticles and ILs and then applying it as a magnetic adsorbent of MDSPE for the rapid screening of sulfamonomethoxine (SMM) and sulfachloropyrazine (SPZ) in urine since they are two widely used representative drugs of SAs. The obtained ILMPM could be dispersed into the sample solution directly and separated via external magnetic field, avoiding the tedious process of making packed columns as in traditional SPE. Compared with the magnetic polymer which was synthesized using 4-vinyl pyridine (4-VP), methacrylic acid (MAA), and acrylamide (AAM) as monomers, ILMPM provided higher purification ability and extraction recovery to SAs.
Experimental
Chemicals and reagents
Sulfamonomethoxine sodium (SMM) and sulfachloropyrazine sodium (SPZ) were purchased from Dingxin Pharmaceutical Co. Ltd. (Shijiazhuang, China). 1-Butyl-3-methylimidazolium hexafluorophosphate ([Bmim][PF6]) was obtained from Chengjie Chemical Co., Ltd. (Shanghai, China). Ethylene glycol dimethacrylate (EGDMA) and 4-VP were obtained from Sigma-Aldrich (St. Louis, MO, USA). 2,2-Azobisisobutyronitrile (AIBN), MAA, AAM, polyvinylpyrrolidone (PVP), FeCl2⋅4H2O, FeCl3⋅6H2O, NH4OH, oleic acid (OA), methanol (MeOH), acetonitrile (ACN), acetone, acetic acid (HAc), and trifluoroacetic acid were obtained from Kermel Chemical Reagents Center (Tianjin, China). Dichloromethane (DCM), ethyl acetate (EA), isopropanol (IPA), and chloroform were purchased from Huandong Chemical Reagent Co. Ltd. (Tianjin, China). Nd–Fe–B magnet (80 × 60 × 17 mm) was purchased from a local market (Baoding, China). All other reagents used in the experiments were of the highest grade commercially available. Double deionized water was filtered through a 0.45-μm filter membrane before use.
Instrumentation and conditions
HPLC analysis was performed using a Shimadzu HPLC system equipped with two LC-20AT Solvent Delivery Units, an SUS-20A gradient controller, and an SPD-20A detector (Shimadzu, Kyoto, Japan). An N-2000 chromatographic workstation (Zheda Zhineng Co., Ltd., Hangzhou, China) was used as a data acquisition system. The analytical column (C18, 5 μm, 150 × 4.6-mm ID) was purchased from Agilent Co., Ltd. (Santa Clara, CA, USA). The mobile phase was water (containing 0.5 ‰ trifluoroacetic acid)–methanol (7:3, v/v, pH 3.0) with a flow rate of 1.0 mL min−1. The detection wavelength was set at 270 nm. A KQ3200E ultrasonic oscillator (Kunshan Instrument Co., Jiangsu, China) was set at 40 kHz and 25 °C for Fe3O4@OA emulsification.
Synthesis of the ILMPM
Preparation of Fe3O4@OA
The Fe3O4 nanoparticles were prepared using a modified coprecipitation method as follows: FeCl2⋅4H2O (3.9 g) and FeCl3⋅6H2O (8.1 g) were dissolved in deionized (100 mL) water and OA (2 mL) was added into the solution and then sonicated 20 min under vigorous stirring of a glass bar until OA was divided into small oil droplets to form a microemulsion. Then, NH4OH (15 mL) was added drop by drop as the temperature elevated to 60 °C, and then OA (2 mL) was added when the temperature elevated to 80 °C. The reaction lasted 1 h at 80 °C by stirring at 600 rpm. The obtained black precipitates were isolated from the solution with an external magnetic field and washed with deionized water to pH 7.0, and then it was washed with MeOH three times and dried in vacuum at 60 °C for 24 h.
Preparation of ILMPM
Fe3O4@OA (1.0 g) was dispersed in chloroform (20 mL) by ultrasound and then [Bmim][PF6] (8 mmol), EGDMA (50 mmol), and AIBN (1.0 mmol) were added into the mixture and sonicated for 5.0 min to make it fully dissolved. PVP (3.0 g) as a dispersion agent was dissolved in water (120 mL) by stirring at 600 rpm and then the organic mixture admitted into the water solution under a nitrogen stream. Polymerization was performed at 60 °C for 24 h in a water bath. After polymerization, the products were separated with an external magnetic field and washed successively with MeOH-HAc (9:1, v/v) under ultrasonic vibration and MeOH to neutral and then dried in a vacuum oven at 60 °C for 24 h. The synthetic procedure was illustrated in Fig. 1.

Synthetic procedure of the ILMPM
Characterization measurements
A Fourier transform infrared (FTIR) spectrometer (Shimadzu) was employed to examine the infrared spectra of the obtained particles. Morphological evaluation of the particles was carried out with scanning electron micrography (SEM, KYKY-2800B, operating at 25 kV). Magnetic properties were measured with a LakeShore 7307 (Lakeshore Cryotronic) vibration magnetometer (VSM) at 300 K. The samples were also characterized with X-ray diffractometer (XRD, D8 ADVANCE) and thermogravimetric analysis (TGA, SDTA851E).
ILMPM-MDSPE-HPLC procedures
Urine samples were collected from healthy volunteers and adjusted to pH 5.0 using acetic acid. Then, they were centrifuged at 4,000 rpmin for 10 min and the upper solutions filtered through a 0.22-μm membrane and stored at 4 °C before use.
ILMPM (100 mg) were immersed in urine (10 mL) or spiked urine (0.005–2.0 μg g−1). After vortex shaking for 5 min, ILMPM were magnetically separated and then washed with water (5 mL) and eluted with 5 % NH4OH–MeOH (4.0 mL). The eluted solution was dried under nitrogen and then redissolved with mobile phase (0.5 mL) for further HPLC analysis.
Adsorption capacity experiment
The maximum adsorption capacity of ILMPM to SMM and SPZ was calculated using static absorption experiment. Of the ILMPM, 20 mg was suspended in 5.0 mL of water solution with various concentrations (1–50 μg mL−1) of SMM and SPZ. After 4 h, the saturated ILMPM were separated by a magnet and the residual concentration of SAs was determined with HPLC-UV. The adsorption amounts (Q) of SAs were calculated as Q = (W 0 − W 1)/W, where W 0 and W 1 (in milligrams) are the initial and residual amounts of SMM and SPZ at different concentrations, respectively, and W (in grams) is the amount of ILMPM.
Results and discussion
Characterization of the ILMPM
The SEM image of ILMPM shown in Fig. 2 illustrated that the diameter of the microspheres was about 30–80 μm. Majority of the ILMPM had a regular spherical shape with a rough surface, which was attributed to the interaction of the microspheres and target molecules. Meanwhile, the uniform core–shell structure will be effective in the mass transport between the solution and the shell surface of ILMPM. However, it was worth noticing that ILMPM showed no aggregation, which may result from the weaker magnetic force after shelling with organic polymers. Additionally, the results of the static absorption experiment showed that the maximum adsorption capacities of ILMPM to SMM and SPZ were 77 and 61 μmol g−1, respectively.

SEM images of ILMPM with different magnifications
The synthesized Fe3O4 (a) and ILMPM (b) were investigated using FTIR spectroscopy (Fig. 3a). The peak at 580 cm−1 was from the characteristic adsorption of Fe–O–Fe vibrations. The typical adsorption peak at 2,930 cm−1 was attributed to the C–H vibration of CH3, and the peaks at 2,850 and 1,470 cm−1 were from the C–H vibration of CH2. Absorption peaks between 1,500 and 1,600 cm−1 corresponded to the skeletal vibration of an aromatic ring, and a peak at 1,650 cm−1 corresponded to the C–H vibration of an imidazole ring. All these adsorption peaks indicated that Fe3O4 nanoparticles and ILs were successfully incorporated into the polymer under the polymerization procedure.

Characterization of the adsorbent
The amount of Fe3O4 encapsulated in the particles was measured through TGA. Figure 3b shows the TGA curves of Fe3O4 (a), Fe3O4@OA (b), and ILMPM (c), respectively. There was no dramatic weight loss within the initial temperature range (<230 °C), except for a small weight loss of free water. For Fe3O4, the weight loss of 9.85 % above 230 °C was attributed to chemical dehydration. The weight loss of Fe3O4@OA (15.7 %) was more than that of Fe3O4 because of the modification by OA. Significant mass loss (80.4 %) of ILMPM from 270 to 340 °C might be attributed to the existence of ILs and organic compounds. The remaining mass was assigned to the residue of Fe3O4, and the quantity of Fe3O4 in the microspheres was 3.98 %.
The XRD patterns for Fe3O4 (a), Fe3O4@OA (b), and ILMPM (c) were displayed in Fig. 3c. In the 2θ range of 10–90°, six characteristic peaks for Fe3O4 (2θ = 30.24°, 36.05°, 43.59°, 53.41°, 57.48°, 62.54°) were observed for the three samples, respectively, which matched well with the database of magnetite in the JCPDS–International Center for Diffraction Data file (JCPDS card no. 19-629) [45]. The presence of the specific diffraction peaks of the synthesized particles indicated that ILMPM were crystalline core materials. However, it was insufficient to exclude the possibility of γ-Fe2O3 due to the proximity of the peaks, which probably formed during the synthesis process [46]. Additionally, the characteristic peaks have been weakened due to the surface coating of shell layers.
The magnetic properties of the obtained materials were analyzed using a VSM at room temperature; the results were illustrated in Fig. 3d. It is obvious that there was no hysteresis; both remanence and coercivity were zero, suggesting that the samples exhibit superparamagnetism. The saturation magnetizations (M s) of Fe3O4 (a), Fe3O4@OA (b), and ILMPM (c) were about 50.98, 43.59, and 25.83 emu g−1, respectively. The decrease of the M s value was due to the coating of the nonmagnetic shell layer. A shell layer of OA and organic polymers were grafted onto the surface of Fe3O4, so the M s value decreased in sequence. Although the M s value of ILMPM was reduced to 25.83 emu g−1, they were very susceptible to magnetic fields and could easily separate from a suspension. The picture inserted in the right corner of Fig. 3d shows the separation and redispersion process of ILMPM. In the absence of an external magnetic field, a homogeneous dispersion was demonstrated; the ILMPM particles were attracted to the wall of the vial in a few seconds when an external magnetic field (Nd–Fe–B magnet) was applied.
Optimization of the ILMPM-MDSPE-HPLC procedures
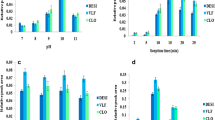
In order to optimize the process of ILMPM-MDSPE-HPLC for isolation of trace levels of SAs in urine, general parameters of MDSPE (the amount of ILMPM, pH, kind and volume of washing and elution solvent, adsorption and desorption time, etc.) were investigated to achieve rapid isolation and good selectivity and avoid interference. All the experiments were performed in triplicate and the concentration of SAs in spiked samples was 0.05 μg g−1. As shown in Fig. 4a, the recovery of SMM and SPZ was increased with the amount of ILMPM. When the amount of ILMPM was up to 100 mg, their recoveries were intended to remain stable. Thus, 100 mg of ILMPM was employed in further experiments. For SMM and SPZ are amphoteric compounds with a wide pK a range, therefore, their extraction efficiency was pH-dependent (Fig. 4b). With the decrease of pH, the recoveries of the target analytes were increased; however, the impurities of the sample matrix would increase accordingly. This was due to the fact that under pH 3.0–5.0, SMM and SPZ existed in neutral forms and a few in protonated form, which could interact with the polymer and its acidic pendant groups via hydrophobic and ion exchange interactions, respectively. To obtain comfortable extraction efficiency, the sample matrix was adjusted to pH 5.0 for the following experiments. Additionally, the adsorption and desorption times of ILMPM were also optimized at 1.0, 3.0, 5.0, 10, 15, 30, and 60 min. The results in Fig. 4c indicated that 5.0 min was enough to achieve adsorption and desorption equilibrium between the two SAs and the ILMPM sorbent.

Optimization of the ILMPM-MDSPE procedures
The washing solvent should eliminate more interference as soon as possible with the least loss of analytes. Different washing solvents were investigated; the results (Fig. 4d) indicated that using water as the washing solvent could obtain satisfactory recoveries and the chromatogram was obviously clean. This is mainly due to SMM and SPZ being insoluble when the pH of urine was adjusted to 5.0, and most of the water-soluble impurities in urine could be washed by water. Further investigation (Fig. 4e) showed that the volume of water had no significant influence on the recovery of the two SAs, but the impurities could decrease with increasing volume of water. Considering purification efficiency and occupation time, 5.0 mL of water was employed as the washing solvent.
The elution solvent should ensure all the analytes are eluted out and help obtain a clean chromatogram. Various elution systems were studied; the results (Fig. 4f) showed that 15 % HAc–acetone and 5 % NH4OH–MeOH provide a better recovery. Compared with the chromatogram using 15 % HAc–acetone as elution solvent, the chromatogram of 5 % NH4OH–MeOH eliminated most of the interference, so it was employed as the elution solvent. The main elution mechanism was the addition of alkali in favor of ionization of SMM and SPZ. Furthermore, the effect of elution volume on the recovery (Fig. 4g) was also investigated; after optimization, 5 % NH4OH–MeOH (4.0 mL) as an eluting solvent was applied for further work. Additionally, as ILMPM are stable, these could be repeatedly used six times without obvious effect on the recovery of SMM and SPZ, which further proved the practicality of the ILMPM sorbent.
Comparison of different adsorbents
To compare the extraction efficiency of ILMPM with different adsorbents which were synthesized using various traditional monomers, a series of magnetic polymers were synthesized using 4-VP, MAA, and AAM as monomers, respectively. The results (Fig. 5) showed that the recoveries of SAs on ILMPM were much higher than that of 4-VP and AAM as monomers. In addition, the purification effect of ILMPM was much better than the other adsorbents. The results were mainly attributed to the π–π, dipole–dipole, and electrostatic attraction interactions between SAs and ILs since there was less interaction between SAs and traditional monomers.

Chromatograms of spiked urine by adsorbents based on different monomers: 4-VP (a); MAA (b); AAM (c); Bmim[PF6] (d)
Validation of the ILMPM-MDSPE-HPLC method
In order to validate the ILMPM-DSPE-HPLC method, linearity, detection limits, precision, and inter-day and intra-day deviations were tested using spiked urine samples under the optimized condition. Calibration curves were constructed using the areas of chromatographic peaks measured at seven increasing concentrations of spiked urine samples, in the range of 0.005–2.0 μg g−1. Good linearity was obtained for all analytes, with regression equations of Y = 1.16 × 103 X + 2.70 × 104 and Y = 1.03 × 103 X + 1.76 × 104 for SMM and SPZ and with correlation coefficient (r) ≥ 0.9996. The limit of detection and limit of quantification calculated at signal-to-noise ratios of 3 and 10 were 0.0012 and 0.004 μg g−1 for SMM and 0.0019 and 0.005 μg g−1 for SPZ, respectively. Precision was assessed by performing replicate analyses of the spiked samples in five replicates in the same day and consecutive 3 days at a concentration of 0.05 μg g−1. The intra-day precision of the method was RSD ≤ 2.8 % and the inter-day reproducibility was below 5.7 % in all cases. The accuracy of the method was evaluated with a recovery experiment at three different spiking levels (0.01, 0.1, and 1.0 μg g−1) of SAs in urine samples. The results in Table 1 showed that the recoveries ranged from 86.9 to 102.1 %, which indicated that the method was reliable and could be applied to the determination of trace levels of SAs in urine samples. Moreover, there were no apparent influences on extraction efficiency after the ILMPM were used several times.
Analysis of real urine samples
In order to evaluate the proposed ILMPM-MDSPE-HPLC method, three kinds of urine samples collected from healthy volunteers were pretreated under the optimized condition. No residuals of SAs were observed in all urine samples. Then, urine samples obtained from volunteers after oral administration of a tablet of SPZ were analyzed; 0.046 μg g−1 of SPZ was observed in the urine even after 48 h (d in Fig. 6). As shown in Fig. 6, the chromatogram of the spiked urine (a) was complex and it was difficult to separate the two SAs from other interferences due to the complicated matrix effect of the sample solution. Similarly, SAs could not be easily detected in spiked urine without a pretreatment and preconcentration procedure (c); however, the good peak pattern of the two SAs was obtained after the ILMPM-MDSPE procedure (b). It demonstrated that the ILMPM adsorbent could remove potential matrix interferences and preconcentrate SAs rapidly and effectively.

Chromatograms of spiked and real urine samples: without purification (a); after ILMPM-MDSPE (b); without preconcentration (c); real urine after oral administration (d)
Conclusion
In this work, new ILMPM were synthesized with aqueous suspension polymerization and applied as the absorbent of MDSPE to extract and concentrate two SAs from urine. The obtained ILMPM combined the advantages of ILs and MNPs, showed good recognition and affinity to SAs, and could effectively remove potential matrix interferences and preconcentrate SAs from urine rapidly. Under the optimum condition, the ILMPM-MDSPE-HPLC method was successfully applied to the determination of SAs in urine.
References
Chen L, Liu J, Zeng Q, Wang H, Yu A, Zhang H, Ding L (2009) Preparation of magnetic molecularly imprinted polymer for the separation of tetracycline antibiotics from egg and tissue samples. J Chromatogr A 1216:3710–3719
Xu X, Su R, Zhao X, Liu Z, Zhang Y, Li D, Li X, Zhang H, Wang Z (2011) Ionic liquid-based microwave-assisted dispersive liquid–liquid microextraction and derivatization of sulfonamides in river water, honey, milk, and animal plasma. Anal Chim Acta 707:92–99
Gehring TA, Griffin B, Williams R, Geiseker C, Rushing LG, Siitonen PH (2006) Multiresidue determination of sulfonamides in edible catfish, shrimp and salmon tissues by high-performance liquid chromatography with post column derivatization and fluorescence detection. J Chromatogr B 840:132–138
Fuh MRS, Chu SY (2003) Quantitative determination of sulfonamide in meat by solid-phase extraction and capillary electrophoresis. Anal Chim Acta 499:215–221
Wen Y, Zhang M, Zhao Q, Feng YQ (2005) Monitoring of five sulfonamide antibacterial residues in milk by in-tube solid-phase microextraction coupled to high-performance liquid chromatography. J Agric Food Chem 53:8468–8473
Rezazadeh M, Yamini Y, Seidi S, Ebrahimpour B (2013) Electromembrane surrounded solid phase microextraction: a novel approach for efficient extraction from complicated matrices. J Chromatogr A 1280:16–22
Song XY, Shi YP, Chen J (2013) Carbon nanotubes reinforced hollow fiber solid phase microextraction for the determination of strychnine and brucine in urine. Talanta 116:188–194
Posyniak A, Zmudzki J, Mitrowska K (2005) Dispersive solid-phase extraction for the determination of sulfonamides in chicken muscle by liquid chromatography. J Chromatogr A 1087:259–264
Kishida K, Furusawa N (2001) Matrix solid-phase dispersion extraction and high-performance liquid chromatographic determination of residual sulfonamides in chicken. J Chromatogr A 937:49–55
Barfi B, Asghari A, Rajabi M, Barfi A, Saeidi I (2013) Simplified miniaturized ultrasound-assisted matrix solid phase dispersion extraction and high performance liquid chromatographic determination of seven flavonoids in citrus fruit juice and human fluid samples: hesperetin and naringenin as biomarkers. J Chromatogr A 1311:30–40
Rallis GN, Sakkas VA, Boumba VA, Vougiouklakis T, Albanis TA (2012) Determination of organochlorine pesticides and polychlorinated biphenyls in post-mortem human lung by matrix solid-phase dispersion with the aid of response surface methodology and desirability function. J Chromatogr A 1227:1–9
Wen YY, Li JH, Zhang WW, Chen LX (2011) Dispersive liquid–liquid microextraction coupled with capillary electrophoresis for simultaneous determination of sulfonamides with the aid of experimental design. Electrophoresis 32:2131–2138
Lin CY, Huang SD (2008) Application of liquid–liquid–liquid microextraction and high-performance liquid-chromatography for the determination of sulfonamides in water. Anal Chim Acta 612:37–43
Zhu X, Cai J, Yang J, Su Q, Gao Y (2006) Films coated with molecular imprinted polymers for the selective stir bar sorption extraction of monocrotophos. J Chromatogr A 1131:37–44
Yan H, Qiao F, Row KH (2007) Molecularly imprinted-matrix solid-phase dispersion for selective extraction of five fluoroquinolones in eggs and tissue. Anal Chem 79:8242–8248
He Z, Liu D, Li R, Zhou Z, Wang P (2012) Magnetic solid-phase extraction of sulfonylurea herbicides in environmental water samples by Fe3O4@dioctadecy ldimethyl ammonium chloride@silica magnetic particles. Anal Chim Acta 747:29–35
Wang X, Wang L, He X, Zhang Y, Chen L (2009) A molecularly imprinted polymer-coated nanocomposite of magnetic nanoparticles for estrone recognition. Talanta 78:327–332
Liu J, Qiao SZ, Hu QH, Lu GQ (2011) Magnetic nanocomposites with mesoporous structures: synthesis and applications. Small 7:425–443
Hu Y, Liu R, Zhang Y, Li G (2009) Improvement of extraction capability of magnetic molecularly imprinted polymer beads in aqueous media via dual-phase solvent system. Talanta 79:576–582
Hu Y, Li Y, Liu R, Tan W, Li G (2011) Magnetic molecularly imprinted polymer beads prepared by microwave heating for selective enrichment of β-agonists in pork and pig liver samples. Talanta 84:462–470
Sun L, Chen LG, Sun X, Du XB, Yue YS, He DQ, Xu HY, Zeng QL, Wang H, Ding L (2009) Analysis of sulfonamides in environmental water samples based on magnetic mixed hemimicelles solid-phase extraction coupled with HPLC-UV detection. Chemosphere 77:1306–1312
Sun L, Sun X, Du XB, Yue YS, Chen LG, Xu HY, Zeng QL, Wang H, Ding L (2010) Determination of sulfonamides in soil samples based on alumina-coated magnetite nanoparticles as adsorbents. Anal Chim Acta 665:185–192
Zhang Y, Liu R, Hu Y, Li G (2009) Microwave heating in preparation of magnetic molecularly imprinted polymer beads for trace triazines analysis in complicated samples. Anal Chem 81:967–976
Jing T, Xia H, Guan Q, Lu W, Dai Q, Niu J, Lim JM, Hao Q, Lee YI, Zhou Y, Mei S (2011) Rapid and selective determination of urinary lysozyme based on magnetic molecularly imprinted polymers extraction followed by chemiluminescence detection. Anal Chim Acta 692:73–79
Guo L, Deng Q, Fang G, Gao W, Shuo W (2011) Preparation and evaluation of molecularly imprinted ionic liquids polymer as sorbent for on-line solid-phase extraction of chlorsulfuron in environmental water samples. J Chromatogr A 1218:6271–6277
Bideau JL, Viau L, Vioux A (2011) Ionogels, ionic liquid based hybrid materials. Chem Soc Rev 40:907–925
Zhou X, Li ZJ, Yuan R, Liu HZ (2006) A novel room temperature ionic liquid extraction spectrophotometric determination of trace germanium in natural water with methybenzeneazosalicylfluorone. Anal Lett 39:863–877
Meindersma GW, Podt AJG, de Haan AB (2005) Selection of ionic liquids for the extraction of aromatic hydrocarbons from aromatic/aliphatic mixtures. Fuel Process Technol 87:59–70
Ye CL, Zhou QX, Wang XM (2006) Headspace liquid-phase microextraction using ionic liquid as extractant for the preconcentration of dichlorodiphenyltrichloroethane and its metabolites at trace levels in water samples. Anal Chim Acta 572:165–171
Marszałł MP, Markuszewski MJ, Kaliszan RJ (2006) Separation of nicotinic acid and its structural isomers using 1-ethyl-3-methylimidazolium ionic liquid as a buffer additive by capillary electrophoresis. Pharm Biomed Anal 41:329–332
Herrera-Herrera AV, Hernández-Borges J, Rodríguez-Delgado MÁ (2009) Fluoroquinolone antibiotic determination in bovine, ovine and caprine milk using solid-phase extraction and high-performance liquid chromatography-fluorescence detection with ionic liquids as mobile phase additives. J Chromatogr A 1216:7281–7287
Ho TD, Yu HL, Cole WTS, Anderson JL (2012) Ultraviolet photoinitiated on-fiber copolymerization of ionic liquid sorbent coatings for headspace and direct immersion solid-phase microextraction. Anal Chem 84:9520–9528
Ho TD, Joshi MD, Silver MA, Anderson JL (2012) Selective extraction of genotoxic impurities and structurally alerting compounds using polymeric ionic liquid sorbent coatings in solid-phase microextraction: alkyl halides and aromatics. J Chromatogr A 1240:29–44
Vidal L, Riekkola ML, Canals A (2012) Ionic liquid-modified materials for solid-phase extraction and separation: a review. Anal Chim Acta 715:19–41
Tian M, Bi W, Row KH (2011) Molecular imprinting in ionic liquid-modified porous polymer for recognitive separation of three tanshinones from Salvia miltiorrhiza Bunge. Anal Bioanal Chem 399:2495–2502
Zhang R, Li N, Wang C, Bai Y, Ren R, Gao S, Yu W, Zhao T, Zhang H (2011) Ionic liquid foam floatation coupled with solid phase extraction for separation and determination of hormones by high-performance liquid chromatography. Anal Chim Acta 704:98–109
Jiang HL, Ying LY, Zhou SC, Ying M, Shen W, Qiu DH (2011) Chromatographic determination of biogenic amines in wines after treatment with ionic liquids as novel media. J Sep Sci 34:1055–1062
Tao Y, Liu JF, Hu XL, Li HC, Wang T, Jiang GB (2009) Hollow fiber supported ionic liquid membrane microextraction for determination of sulfonamides in environmental water samples by high-performance liquid chromatography. J Chromatogr A 1216:6259–6266
Han J, Wang Y, Liu Y, Li YF, Lu Y, Yan YS, Ni L (2013) Ionic liquid-salt aqueous two-phase extraction based on salting-out coupled with high-performance liquid chromatography for the determination of sulfonamides in water and food. Anal Bioanal Chem 405:1245–1255
Galán-Cano F, Alcudia-León MC, Lucena R, Cárdenas S, Valcárcel M (2013) Ionic liquid coated magnetic nanoparticles for the gas chromatography/mass spectrometric determination of polycyclic aromatic hydrocarbons in waters. J Chromatogr A 1300:134–140
Shi X, Meng Y, Liu J, Sun A, Li D, Yao C, Lu Y, Chen J (2011) Group-selective molecularly imprinted polymer solid-phase extraction for the simultaneous determination of six sulfonamides in aquaculture products. J Chromatogr B 879:1071–1076
Arvand M, Alirezanejad F (2011) Sulfamethoxazole-imprinted polymeric receptor as ionophore for potentiometric transduction. Electroanalysis 23:1948–1957
Evgenév MI, Garmonov SY, Shakirova LS, Levinson FS (2000) Spectrophotometric and chromatographic determination of sulfanilamides in biological fluids and pharmaceuticals. J Anal Chem 55:799–805
Iammarino M, Palermo C, Nardiello D, Muscarella M (2011) Optimization and validation of a confirmatory method for determination of ten sulfonamides in feeds by LC and UV-diode array detection. Chromatographia 73:75–82
Kong X, Gao R, He X, Chen L, Zhang Y (2012) Synthesis and characterization of the core–shell magnetic molecularly imprinted polymers (Fe3O4@MIPs) adsorbents for effective extraction and determination of sulfonamides in the poultry feed. J Chromatogr A 1245:8–16
Maciel JC, Andrad PL, Neri DFM, Carvalho LB Jr, Cardoso CA, Calazans GMT, Aguiar JA, Silva MPC (2012) Preparation and characterization of magnetic levan particles as matrix for trypsin immobilization. J Magn Magn Mater 324:1312–1316
Acknowledgments
The project was sponsored by the National Natural Science Foundation of China (21175031, 31301464) and the Natural Science Foundation of Hebei (B2012201052).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yan, H., Gao, M., Yang, C. et al. Ionic liquid-modified magnetic polymeric microspheres as dispersive solid phase extraction adsorbent: a separation strategy applied to the screening of sulfamonomethoxine and sulfachloropyrazine from urine. Anal Bioanal Chem 406, 2669–2677 (2014). https://doi.org/10.1007/s00216-014-7665-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-014-7665-3