
Abstract
Attenuated total reflectance mid-infrared spectra of serum and blood samples were obtained from 4,000 to 600 cm−1. Models for the determination of albumin, immunoglobulin, total globulin, and albumin/globulin coefficients were established for serum samples, using reference data obtained by capillary electrophoresis. Based on the use of the amide bands I and II regions, the relative root mean square error of prediction (RRMSEP) was 4.9, 14.9, 4.5, and 7.1 % for albumin, immunoglobulin, total globulin, and albumin/globulin coefficients, respectively, determined in an independent validation set of 120 samples using 200 samples for calibration. Additionally, the use of Kennard–Stone method for the selection of a representative calibration subset of samples provided comparable results using only 60 samples. For whole blood analysis, hemoglobin was determined in 40 validation samples using models built from 40 calibration independent samples with RRMSEP of 8.3, 5.5, and 4.9 % with models built from direct spectra in the first case and from sample spectra recorded after lysis by sodium dodecyl sulfate and freezing, respectively, for the last two ones. The developed methodologies offer green alternatives for patient diagnosis in a few minutes, minimizing the use of reagents and residues and being adaptable for its use as a point-of-care method.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fourier transformed infrared spectroscopy (FTIR) is a promising technique to complement or replace the current commercial methods used in clinical laboratories for the determination of biochemical parameters in clinical samples [1]. The measurement of the mid-infrared (MIR) spectrum trough attenuated total reflectance (ATR) could be carried out by using low-cost and potentially compactable equipment, which can be easily adaptable for point-of-care analysis, avoiding the weaknesses of the current clinical techniques, such as the use of costly equipment, the utilization of expensive reagents, the production of high volumes of toxic residues, and the difficulties in the compaction of the highly sophisticated instruments. Additionally, with a simple and direct measurement of the IR infrared spectrum of clinical samples, very useful information about the major and minor compounds present in the sample, which necessitate in many cases the use of chemometric tools for the correct extraction of as much information as possible from the samples [2], can be obtained.
There are precedents for the use of direct measurements of MIR spectra of serum [3] and blood [4] samples for the determination of essential parameters of the comprehensive metabolic panel, such as glucose, urea, creatinine, albumin, and total proteins, besides other important analytes, such as triglycerides, cholesterol, HDL, and LDL. The measurements of dry plasma samples [5] have been used for the determination of numerous analytes including immunoglobulin and other protein parameters providing good results. The evidenced capability of the MIR spectrum of untreated serum samples to provide information about biochemical parameters justifies the exploration of the simultaneous determination of additional parameters of clinical interest. In this sense, albumin, α1-globulin, α2-globulin, β-globulin and immunoglobulin, total globulin, and the albumin/globulin coefficient are serum protein parameters commonly determined through capillary electrophoresis which offer interesting data for the diagnosis of different diseases. Furthermore, the concentration of some of these parameters is high enough to overcome the lack of sensitivity of direct ATR measurements. For the direct analysis of the aforementioned parameters related to the proteins present in sera, fluorescence spectroscopy [6] and also near-infrared spectroscopy [7–10] have been used. However, the utilization of these techniques is not effective for the simultaneous determination of several parameters in a same sample.
ATR-FTIR measurements directly made on serum samples can provide quantitative or at least semiquantitative data of many parameters related to the presence of proteins in serum and blood. Changes on the amide bands of the MIR spectrum are very specific among the different secondary structure [11], and these differences could be used for the identification of changes in the protein composition of sera and for the prediction and determination of different secondary structure proportions [12]. Considering that serum proteins under study have different proportions of secondary structures [13, 14], the use of these bands could be useful for the modelization of those parameters, based on both intrinsic and extrinsic information on the size and shape of amide bands, suitable to be modelized by chemometrics.
Another protein susceptible to be determined by FTIR in blood is hemoglobin. Its common determination is based on transmittance measurements made with cytometers. However, there are recent attempts to determine hemoglobin by ATR-FTIR through the use of partial least squares and multiple linear regression (MLR) data treatments [15]. Although the aforementioned study provides promising results, the evaluation of the model was carried out with the same set of samples used for model optimization and thus there is no evidence on the validation of prediction capability of the method for an independent set of samples.
Additionally, in order to minimize the required number of samples for model building the models, randomly selection of samples to be employed as a calibration subset was compared with two methods of sample subset selection as Kennard–Stone [16, 17] and successive projections algorithm (SPA) [18].
In short, the aim of this work has been the evaluation of the capability of the partial least squares (PLS)-ATR-FTIR for the prediction of serum protein parameters. Moreover, a preliminary study of the determination of hemoglobin in whole blood was also made.
Materials and methods
Sample description
Three hundred twenty serum samples and 80 blood samples employed through this study were provided by the Hospital Dr. Peset Alexandre in Valencia (Spain) from the Protein Analysis Department and the Hematology Department respectively. Serum samples were characterized using a capillary electrophoresis equipment Paragon from Beckman-Coulter Inc. (Brea, CA, USA) while hemoglobin in blood samples was determined optically based on transmittance measurements after RBC lysis, in a GenS Beckman-Coulter, Inc. cytometer (Brea, CA, USA). The spectra of samples were obtained with a maximum delay of 7 days after to be taken and analyzed by the hospital through the reference procedures. Prior to ATR-FTIR analysis, samples were stored in glass tubes at 4 °C. Samples were chosen randomly and encompassed a wide range of concentrations (see Table 1).
Apparatus and reagents
Sodium dodecyl sulfate (SDS; >98 %), albumin (human serum, 97–99 %), and immunoglobulin (human serum, >95 %) were purchased form Sigma-Aldrich (St. Louis, MO, USA). SDS solution was prepared by dissolving the appropriate amount of SDS in a sodium chloride 0.9 % (w/v) solution. The homogenization of solutions and samples was carried out in all cases with a Velp Scientifica Vortex (Usmate, Italy) at 2,400 rpm for 5 s.
FTIR spectra for serum samples were measured using a Bruker (Bremen, Germany) Tensor 27 FTIR spectrometer equipped with a temperature-stabilized deuterated lanthanum tryglycine sulphate detector and for whole blood spectra measurements were made with a Bruker IFS 66/v FTIR spectrometer equipped with a liquid nitrogen-refrigerated mercury–cadmium–telluride detector, a vacuum system, and a dry air purged sample compartment. In all cases, an in-compartment ATR DuraSampleIR accessory with a nine reflection diamond/ZnSe DuraDisk from Smiths Detection Inc. (Warrington, UK) was adapted to the spectrophotometer. The OPUS software version 6.5 from Bruker was employed for the acquisition and handling of spectra and for data treatment.
Measurement of ATR-FTIR spectra
Fifty microliters of each sample were deposited in the ATR crystal cell and covered using an N-BK7 PCV lens as a dome to avoid sample evaporation. Spectra were recorded in the range between 4,000 and 600 cm−1, with a resolution of 4 cm−1, averaging 100 scans/spectrum and making ATR correction. A spectrum of the clean ATR crystal, obtained immediately before acquiring the spectrum of each sample, was used as a background. Spectra were measured in triplicate and a water spectrum, obtained in the same experimental conditions, was subtracted to the average spectrum of each sample. A spectrum of the empty cell was measured after cleaning the cell between measurement series of each three samples in order to verify the absence of memory effects on the ATR crystal. The cell was cleaned mechanically with a soft cellulose paper after each measurement using a solution of 0.9 % (w/v) sodium chloride in order to avoid protein precipitation and, finally, with Milli-Q water. Three samples could be measured in 20 min using the Tensor 27 spectrophotometer and in 3 min using the IFS 66/v instrument.
Direct spectra of untreated samples were obtained for serum sample analysis. For blood samples, three aliquots of each sample were taken for the obtention of the spectra from: i) untreated samples, ii) samples lysed by simple freezing overnight and iii) samples lysed by shaking in a vortex 175 μL of sample with 25 μL of SDS 7 % (w/v) solution.
PLS modeling of ATR-FTIR spectra
OPUS software version 6.5 from Bruker and MatLab 7.7.0 from Mathworks (Natick, MA, USA) were employed to build the PLS calibration models from the obtained ATR-FTIR spectra and to evaluate their prediction capability. Serum sample dataset were randomly divided into a calibration set with 200 samples and a validation set with 120 samples. For hemoglobin modelization, 40 samples were randomly chosen for calibration and the rest were included in the validation set. Different number of latent variables (LV; from 1 to 10), wavenumber spectrum ranges, and diverse preprocessing methods were considered for choosing the best calibration model that provides the smallest value of root mean square error of cross validation (RMSECV) for each analyte under study leaving out six samples as the selection criterion.
Study of the selection of a representative sample subset
For PLS model building to determinate proteins in serum samples, aleatory selection, Kennard–Stone, and SPA methods were assayed to select an appropriate sample calibration set. In all cases, first derivative and mean centering of data was applied before selection of representative subset. The aim was to compare the models built from a calibration set composed with m samples selected through the three methods above commented and to validate those models with the rest of 320 m samples. The number of samples in the calibration set was increased in ten samples each time from 20 to 170. The RMSECV was calculated using venetian blinds with (m/10) splits. For the validation, the number of LV which reached the minimum RMSECV from a maximum of 9 was used. All the MatLab codes employed for the calculations are available in the Electronic supplementary material.
Aleatory selection
PLS models were built using from 20 to 170 samples in the calibration set and validating with the rest of samples (from 300 to 150). The RMSECV and the RMSEP values were monitored as a function of the number of samples in the calibration set. As the evolution of those errors was strongly dependent to the nature of samples included in the calibration sets, this experiment was repeated several times modifying randomly the composition of the calibration and validation sets. The average of the prediction parameters obtained for each calibration size was determined.
Kennard–Stone
Kennard–Stone is a well-known method for the selection of representative subset of samples based only on the instrumental response matrix. The selected subset should cover the experimental region homogeneously by choosing objects which are “uniformly” distributed in the experimental space. The first sample selected is the closest to the mean of all the samples and the following objects were selected sequentially, based on the squared distances to the objects already assigned to the calibration set. Further information can be obtained in the literature [16–18].
Kennard–Stone algorithm was applied to the raw set of samples for the selection of the most representative m samples and a PLS model was built with the m selected samples and validated with the rest of 320 m objects, being m augmented again 20 to 170 increasing ten samples each time.
SPA
SPA is an iterative forward selection method that was proposed to minimize collinearity problems. The method uses a first sample data set (K 0) and searches an additional one which has the largest projection on the subspace orthogonal to K 0. Then, this new sample is included in the representative subset and it becomes K 0 for the selection of a third object. SPA was originally used for the variable selection in the instrumental response matrix (X). Araújo et al. [17] applied the algorithm to the transpose of X for the selection of a representative sample subset for calibration. Further information can be obtained in Ref. [17]
In this study, SPA was applied considering each sample as K 0 and selecting from 20 to 170 samples for calibration of different PLS models. The K 0 which raised the lowest RMSECV was chosen for each m samples and the model was validated with the rest of (320 m) samples for calculating the RMSEP.
Results and discussion
FTIR spectra of standards and samples
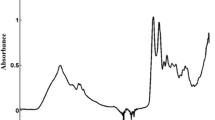
>The huge similarities found among protein and sera spectra after water subtraction (Fig. 1) evidences the high proportion of protein in the non-aqueous portion (≈90 %) of serum. Great differences between the whole spectra of those proteins standard, being the major dissimilarities in the position of the amide bands I, II, and III, especially in the peak maximum of amide band I (in albumin at 1,653 cm−1 and in immunoglobulin at 1,635 cm−1) can be appreciated. Those differences may be ascribed to the different secondary structures of globulin and albumins; while human serum albumin is mainly composed by α-helix [14], β-sheet is the most common secondary structure in immunoglobulin [12]. According to the literature [11], the position of this band is strongly affected by the secondary structure of the protein, differing between the reported interval position for α-helix (1,648–1,657 cm−1) and that for immunoglobulin (1,623–1,641 cm−1). Thus, the aforementioned differences could be very useful for the discrimination of samples and the correct selection of the interval range to be used in our predictive purpose.

ATR spectra of human serum albumin at 3 g/dL (solid line) and immunoglobulin at 1 g/dL (dotted line) (1) and raw sample spectra (2). Note: spectra were shift on the y-axis to clearly show their bands. In all cases, water blank was subtracted. Insert, regression coefficients divided by 1,000 for the PLS models of human serum albumin (solid line) and immunoglobulin (dotted line)
In the case of whole blood (Fig. 2), spectra present typical protein bands, evidencing a major presence of proteins in the samples. Spectra of untreated and lysed samples evidence problems of hemoglobin detection in untreated blood samples being similar the absorbance of the amide bands in untreated samples to that in serum spectra. In fact, the absorbance of protein bands in untreated samples is significantly lower than for lysed ones, being the spectra very similar for both lysis preprocesses employed through this study. This fact could be interpreted considering the low penetration depth of the infrared light in the ATR cell. In the case of the direct measurements, the light cannot reach the hemoglobin confined inside the erythrocytes. When the erythrocytes are lysed, free hemoglobin in dissolution is efficiently detected as well as all other blood proteins. Consequently, the difficulties expected for the correct modelization of hemoglobin in direct measurements were solved by means of a previous cell lysis.

Comparison between spectra of untreated blood sample and the same sample after SDS and freezing treatment. Note: spectrum obtained with SDS was multiplied by 1.125 in order to correct the dilution. In all cases, water blank was subtracted
PLS models of serum parameters
For α-1 globulins, α-2 globulins, and β-globulin, prediction errors equal or higher than 30 % were found thus evidencing that PLS-ATR-FTIR has a low prediction capability for these parameters. This fact can be explained considering that the aforementioned parameters are composed by a series of different proteins whit nonspecific spectra for each group.
The parameters and results obtained for albumin, globulin, immunoglobulin, and albumin/globulin coefficient are reported in Table 2. Regions used for modelization were different among the analytes, however the region of the amide bands I and II (1,400–1,700 cm−1) was included in all the cases. The importance of those bands is confirmed by the regression coefficients values obtained for albumin and immunoglobulin (see the inset of Fig. 1).
Final models provide correlation values higher than 85 % for the calibration of all analytes and a number of latent variables between 4 and 6. The prediction of the independent set of samples showed residual prediction deviation (RPD) values upper than 2.2 for all parameters considered. Data in Table 2 show that the prediction capability of models in the case of albumin and globulin was better than for immunoglobulin and the coefficient albumin/globulin. Taking into account the relative root mean square error of prediction (RRMSEP) values obtained for albumin and total globulin, this technique can be used as a routine assay, with errors lower than 5 % while relative errors for immunoglobulin and A/G coefficient were between 7 and 14 %. Nevertheless, in all the cases, results obtained evidenced that PLS-ATR-FTIR provides a good tool for protein determination in serum samples, at least as a screening level. As compared with results previously reported for the direct analysis of protein serum parameters, it can be seen that the method proposed provides, comparable or lower errors for immunoglobulin and albumin than those obtained by NIR spectrometry for real [10] or artificial samples [9]. The use of fluorescence spectroscopy in artificial samples improved the sensitivity [6] but the capability ATR-FTIR for the simultaneous determination of serum parameters is a key factor. A slight enhancement of sensitivity can be also achieved when plasma is dried before measurement [5]. However, the pre-processing step reduces the precision of the method and increases the time of analysis.
Studies on different alternatives for the selection of a representative sample subset for calibration
The evolution of the RMSEP with the size of the calibration set was different among the different strategies used for selection of the calibration set. As it can be seen in Fig. 3, the prediction error decreases slightly when increasing the number of samples employed for calibration, being this evolution slower for the use of an aleatory selection. Models constructed were not improved by using SPA regarding the RMSEP in spite of the fact that this method provided the best RMSECV (see Fig. 4).

Evolution of the RMSEP as a function of the size of the calibration set for different strategies used for its selection: randomly selection (green asterisks), Kennard–Stone (red cross), and SPA (blue circles)

Evolution of the RMSECV for the modelization of albumin as a function of the size of the calibration set for the different strategies used in this study: mean of the aleatory selection (green asterisks), Kennard–Stone (red cross), and SPA (blue circles). Insert, green pictogram of the methods developed
The best models were achieved with the Kennard–Stone method, which improves in most of the cases the prediction capability also reducing the number of required calibration samples. In short, with only 60 samples and using the Kennard–Stone algorithm, prediction errors of 0.19, 0.16, 0.18, and 0.12 g/dL were obtained for albumin, immunoglobulin, globulin, and albumin/globulin coefficient, respectively.
PLS models for determination of hemoglobin in blood samples
The parameters chosen for the best modelization of hemoglobin in blood and the values of the obtained errors are reported in Table 3. In this table, the expected differences between models based on ATR-FTIR measurements made on untreated and lysed samples can be seen.
In all three cases considered, the region of amide I band was selected, confirming the high importance of this interval in protein determination in blood. In the case of the modelization of spectra of untreated blood samples, a good correlation was found in the calibration set as it is indicated by the values of R 2 and the calibration error obtained. Nevertheless, when the model was validated with and independent set of samples, the prediction error increased till 1 g/dL and a RPD value of 1.3 was obtained, thus evidencing a poor prediction capability. On the contrary, models built from spectra of lysed blood samples provided a low number of optimal LV and better prediction capability than the previous one. On considering the treatment of samples with SDS, good correlations and good prediction capability of PLS-ATR-FTIR were observed as it was evidenced by the low RRMSEP and the high RPD results found. It must be noticed that the interval selected in this case includes the region between 1,076 and 1,231 cm−1, while the SDS spectra provide a strong band from 1,180 till 1,280 cm−1. However, the concentration of SDS was enough low to avoid any contribution of lysis reagent to the ATR spectra. The best prediction capability was found for models realized from spectra of previously freezing blood samples, where a relative prediction error lower than 5 % and a RPD value of 2.2 were found. However an excellent repeatability was obtained after sample lysing with SDS. This fact could be explained because SDS breaks the erythrocytes membrane but, in addition, produces the denaturalization of proteins. However, it also eliminates the information associated to the secondary structure and, because of that, the prediction capability found by measurements made after sample freezing provides a better prediction capability than the SDS treatment.
In the case of the determination of hemoglobin, modelization based on direct measurement of spectra provided comparable results to those obtained by PLS modelization reported from recent studies. On the other hand, the modelization based on MLR of selected wavenumber [15] provided better results than ours. Nevertheless, it has to be considered that these results were obtained without using any independent validation set, and thus the selection of the number of latent variables and wavenumbers regions in MLR was based on the date provided by the prediction set.
Environmental side effects of method proposed
Inset in Fig. 4 evidences that the developed procedures agree well with the Green Analytical Chemistry objectives [19] being the operator risk medium, based on the intrinsic nature of blood and sera and their potential infectious character. On the other hand, the energy consumed, the absence of the use of additional reagents except for SDS solutions, and the fact that the total volume of waste, for the three replicate analysis of each sample, also including the cleaning of the cell, is less than 10 mL, justify that the present FTIR procedure is a green method.
Conclusions
In this work, PLS-ATR-FTIR methods have been successfully employed for the determination of many important protein parameters, such as total albumin, total globulin, immunoglobulin, and albumin/globulin coefficient in serum samples as well as total hemoglobin in whole blood. Considering the serum parameters, the modelization was possible based on the huge differences found in the amide I region between the FTIR spectra of the different target proteins. The determination of albumin and total globulin provided low relative errors of prediction to be considered as a good alternative to the current commercial methods for their quantitative determination while for the determination of immunoglobulin and the albumin/globulin coefficient the method can be used only as a screening tool. On the other hand, our study has evidenced that the hemoglobin determination by PLS-ATR-FTIR based on direct measurements provide a bad prediction capability and a previous sample lysis was required to improve this methodology. Modelization of samples defrosted or pre-treated with SDS raised relative prediction errors lower than 5.5 %. Nevertheless, whereas the calibration and validation sets were integrated by only 50 samples, additional studies are required for a correct evaluation of this technique as an alternative to the existing methods.
In short, methods proposed offer green alternatives to the traditional ones used in hospitals because they do not involve the use of reagents, not a chemical sample pretreatment and provide clinical data in a fast and safe way and from measurements made by a relatively simple and low-cost instrumentation. The weakness of the method being basically the need of a previous signal modelization from a series of samples analyzed by a reference procedure and, as it has been indicated through this study, the higher is the number of samples used for calibration the powerful is the prediction capability of the method. However, it has been also evidenced that with the use of Kennard–Stone strategy for calibration sample subset selection it can be obtained a representative calibration set with a reduced number of samples. So, it is our own opinion that the opportunities offered by this kind of methods concern the possibility to develop diagnostic analytical tools for the point-of-care determination of clinical parameters without using any additional reagent [20].
References
Wang LQ, Mizaikoff B (2008) Application of multivariate data-analysis techniques to biomedical diagnostics based on mid-infrared spectroscopy. Anal Bioanal Chem 391:1641–1654
Moros J, Garrigues S, de la Guardia M (2010) Vibrational spectroscopy provides a green tool for multi-component analysis. TraC Trends Anal Chem 29:578–591
Perez-Guaita D, Ventura-Gayete J, Pérez-Rambla C, Sancho-Andreu M, Garrigues S, de la Guardia M (2011) A green tool for serum analysis based on Fourier transform infrared spectrometry. Abstract of papers, International conference on materials and technologies for green chemistry, Tallin
Hosafci G, Klein O, Oremek G, Mantele W (2007) Clinical chemistry without reagents? An infrared spectroscopic technique for determination of clinically relevant constituents of body fluids. Anal Bioanal Chem 387:1815–1822
Petibois C, Cazorla G, Cassaigne A, Deleris G (2001) Plasma protein contents determined by Fourier-transform infrared spectrometry. Clin Chem 47:730–738
Wiberg K, Sterner-Molin A, Jacobsson SP (2004) Simultaneous determination of albumin and immunoglobulin G with fluorescence spectroscopy and multivariate calibration. Talanta 62:567–574
Kasemsumran S, Du YP, Murayama K, Huehne M, Ozaki Y (2004) Near-infrared spectroscopic determination of human serum albumin, gamma-globulin, and glucose in a control serum solution with searching combination moving window partial least squares. Anal Chim Acta 512:223–230
Baddini ALQ, da Cunha LER, de Oliveira AMC, Cassella RJ (2010) Determination of total protein in hyperimmune serum samples by near-infrared spectrometry and multivariate calibration. Anal Biochem 397:175–180
Murayama K, Yamada K, Tsenkova R, Wang Y, Ozaki Y (1998) Near-infrared spectra of serum albumin and gamma-globulin and determination of their concentrations in phosphate buffer solutions by partial least squares regression. Vib Spectrosc 18:33–40
Hazen KH, Arnold MA, Small GW (1998) Measurement of glucose and other analytes in undiluted human serum with near-infrared transmission spectroscopy. Anal Chim Acta 371:255–267
Barth A (2007) Infrared spectroscopy of proteins. Biochim Biophys Acta Bioenerg 1767:1073–1101
Wang YQ, Boysen RI, Wood BR, Kansiz M, McNaughton D, Hearn MTW (2008) Determination of the secondary structure of proteins in different environments by FTIR-ATR spectroscopy and PLS regression. Biopolymers 89:895–905
Kong J, Yu S (2007) Fourier transform infrared spectroscopic analysis of protein secondary structures. Acta Biochim Biophys Sin 39:549–559
Sugio S, Kashima A, Mochizuki S, Noda M, Kobayashi K (1999) Crystal structure of human serum albumin at 2.5 angstrom resolution. Prot Eng 12:439–446
Yin H, Pan T, Xie J, Chen J, Chen H, Wei X, Tian P, Wang B (2011) Wavenumbers selection for FTIR/ATR spectroscopy analysis of hemoglobin in human whole blood. Abstracts of papers, 4th International conference of bioinforma and biomedical engineering, Chengdu (China).
Daszykowski M, Walczak B, Massart DL (2002) Representative subset selection. Anal Chim Acta 468:91–103
Dantas HA Jr, Galvão RKH, Araújo MCU et al (2004) A strategy for selecting calibration samples for multivariate modeling. Chemom Intell Lab Syst 72:83–91
Kennard RW, Stone LA (1969) Computer aided design of experiments. Technometrics 11:137–148
de la Guardia M, Armenta S (2011) Green analytical chemistry: theory and practice. Elsevier, Amsterdam
de la Guardia M, Garrigues S (eds) (2011) Challenges in green analytical chemistry. RSC, London
Acknowledgments
The authors gratefully acknowledge the financial support of the Ministerio de Educación y Ciencia (Projects CTQ2008-05719/BQU and CTQ2011-25743) and the Generalitat Valenciana project PROMETEO 2010-055 and the kindly support of the Fundación para la Investigación from the University Hospital Dr. Peset. D.P.G. acknowledges the “V segles” grant from the University of Valencia.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the special paper collection on Green Analytical Methods with guest editors Miguel de la Guardia and Sergio Armenta.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 435 kb)
Rights and permissions
About this article
Cite this article
Perez-Guaita, D., Ventura-Gayete, J., Pérez-Rambla, C. et al. Protein determination in serum and whole blood by attenuated total reflectance infrared spectroscopy. Anal Bioanal Chem 404, 649–656 (2012). https://doi.org/10.1007/s00216-012-6030-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-012-6030-7