
Abstract
In the present study, a new, simple, rapid, and environmentally friendly headspace-liquid phase microextraction method followed by gas chromatography–flame ionization detection has been developed for the extraction/preconcentration and determination of 1,4-dioxane from shampoo. The developed procedure is performed in a home-made extraction vessel, connected to a glass vial containing sample and extraction solvent. In this method, an aliquot weight of shampoo is mixed with a binary mixture of n-hexane and dichloromethane (50:50, v/v) as the extractant and the target analyte is extracted during a liquid–liquid extraction procedure. Then a home-made extraction vessel containing a few microliters of a collection/extraction solvent is contacted to a glass vial containing the organic phase obtained from the previous step. By heating 1,4-dioxane is vaporized and enriched in a μL volume of the collection/extraction solvent. Then an aliquot volume of the collected phase is injected into the separation system. The effect of several factors which may influence performance of the method, including kind and volume of the extraction solvents used in both steps, extraction temperature, extraction time, and salt addition were evaluated. Under the optimum extraction conditions, limits of detection and quantification for the target analyte were obtained 0.52 and 1.73 μg kg−1, respectively. Enrichment factor and extraction recovery were 333 and 89 %, respectively. The method precision was evaluated at a concentration of 25 μg kg−1 and relative standard deviation was less than 6.9 % for intra-day (n = 6) and inter-day (n = 4) precisions. Finally, the proposed method has been successfully applied in analysis of 1,4-dioxane in different shampoo samples.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Sodium lauryl ether sulfate (SLES) is an anionic surfactant which is used in production of cosmetics, dish washing and autocare [1]. During the production of polyethoxylated detergents by the reaction of fatty alcohols with ethylene oxide, a portion of ethylene oxide is polymerized to form 1,4-dioxane [2]. 1,4-Dioxane is a cyclic ether which is used as a solvent in chemical reactions, a fluid for scintillation and a dehydrating agent in the preparation of tissue section for histology. Also, it is used as a component of paint and varnish removers, and a wetting and dispersion agent in textile industry [3]. In many consumer products, e.g., cleaning products, cosmetics, shampoos, and lotion formulations 1,4-dioxane is present as a byproduct. 1,4-Dioxane is a moderately volatile, colorless and water miscible solvent, with a mild and ethereal odour. Exposure from occupational as well as environmental sources may occur by all routes; inhalation, ingestion and dermal contact, that absorption is rapid following inhalation and oral administration, whereas penetration through skin is slow [4–6]. Several reports have described adverse health effects due to chronic dioxane exposure [7]. It is toxic for liver, lungs, kidneys and central nervous system [8–13]. Due to adverse effects of 1,4-dioxane, US Department of Health and Human Services has determined less than 20 mg L−1 as its maximum residue limit (MRL) in detergents and hygienes [14]. Hence level of 1,4-dioxane in commercial cosmetic products is of direct concern to consumers. The assay of this substance in cosmetics products has been carried out by several analytical methods such as gas chromatography–mass spectrometry (GC–MS) [15–22], reversed phase-high performance liquid chromatography (RP-HPLC) [23], and headspace-gas chromatography–mass spectrometry (HS-GC/MS) [24]. Although these methods are selective and sensitive, sample preparation is essential due to trace level of 1,4-dioxane present in the samples and potential interferences of the matrices. Liquid–liquid extraction (LLE) [25] and solid phase extraction (SPE) [26–28] have been used for sample preparation prior to 1,4-dioxane analysis. However, these conventional pretreatment methods need large amounts of sample and organic solvents, are time-consuming and expensive, and the materials used are not reusable. Recent researches have been oriented toward the development of efficient, economical, and miniaturized sample preparation methods. As a result, analysis of 1,4-dioxane in cosmetics was performed using headspace-solid phase micro-extraction (HS-SPME) coupled with GC–MS analysis [29, 30]. This technique has advantages of high purity of the extract, avoiding organic solvents consumption and simplicity. Its disadvantages are the memory effect, chromatographic peak broadening and the high price of the fiber used [31–34]. Unfortunately only one report (the above mentioned paper) has been published in the literature regarding application of microextraction techniques in sample preparation of 1,4-dioxane in different samples [29].
In the present study, a new sample preparation technique is proposed based on μL level of a safe solvent. In fact, this technique is a dynamic headspace-liquid phase microextraction (DHS-LPME) method which is reported for the first time and used in the extraction and preconcentration of 1,4-dioxane from different shampoo samples. Determination of the analyte is carried out using gas chromatography–flame ionization detection (GC–FID). Effect of experimental parameters including type of extraction solvent and its volume, extraction time, salt addition, and temperature are studied and optimized in details. Analytical performance of the proposed method in real samples is explored.
Experimental
Chemicals and reagents
1,4-Dioxane (99.5 %) was purchased from Merck (Darmsadt, Germany). Solvents including n -hexane, dichloromethane, dimethyl sulfoxide (DMSO), n -octanol, n -hexanol, and n -pentadecane tested as the extraction solvents were supplied from Merck. A stock solution of 1,4-dioxane in de-ionized water was prepared at a concentration of 500 mg L−1. Working standard solutions were prepared daily by appropriate dilutions of the stock solution with de-ionized water. A standard solution of 1,4-dioxane and toluene (internal standard) (IS) was prepared in n -octanol (500 mg L−1 of each) and injected into GC–FID system directly (three times in a day) for quality control of the separation system and the obtained peak areas were used in calculation of enrichment factor (EF) and extraction recovery (ER).
Samples
Eight shampoo samples including Baby (Johnson, Milvaki, USA), Sehat (Cedr, Tehran, Iran), Darugar (Super, Tehran, Iran), Parzhak (Garlic, Kerman, Iran), Fulica (Antidandruff, Tehran, Iran), Ave (Pro-Vitamin, Alborz, Iran), Clear (Cool Sport, Alborz, Iran), and Corea (Antidandruff and Scalp Care, Seoul, Korea) from various producers were purchased from local pharmacies (Tabriz, Iran). All samples were used without any pretreatment. A portion of baby shampoo was transferred into a 250-mL Erlenmeyer and nitrogen gas was passed through its headspace for 1 h for removing the analyte. Then the sample was analyzed by headspace GC–MS and no peak was observed in the retention time of the analyte. It was used as a matrix (blank shampoo) and all optimizations were performed using this shampoo spiked with known amounts of the analyte.
Instrumentation
Chromatographic analysis of the samples was performed on a Shimadzu 2014 gas chromatograph (Kyoto, Japan) equipped with a split/splitless injector used in a splitless mode (sampling time 0.5 min and split ratio 1:10) and an FID. Helium (99.999 %, Gulf Cryo, United Arabic Emirates) was used as the carrier gas at a constant linear velocity of 30 cm s−1 and make up gas (40 mL min−1). Injector and FID temperatures were maintained at 200 °C. Chromatographic separation was performed on an HP-1 capillary column (100 % poly dimethyl siloxane, 30 m × 0.25 mm ID and 0.5 µm film thickness) (Agilent Technologies, CA, USA). The column oven temperature was initially held at 40 °C for 8 min, then ramped at 20 °C min−1 to 200 °C, and held at 200 °C for 5 min. Hydrogen gas was generated with a hydrogen generator (OPGU-1500 S, Shimadzu, Japan) for FID at a flow rate of 30 mL min−1. Air flow rate for FID was 300 mL min−1. GC–MS analysis was carried out on an Agilent 7890A gas chromatograph with a 5975C mass selective detector (Agilent Technologies, CA, USA). MS operational conditions were: electron ionization (EI), 70 eV; ionic source temperature, 250 °C; transfer line temperature, 260 °C; mass range, m/z 20–100; acquisition rate, 20 Hz; and detector voltage, −1700 V. Library searching was performed using the commercial NIST library. The carrier gas was helium at a flow rate of 1.0 mL min−1. The column oven temperature programming and capillary column kind used in GC–MS were the same as used in GC–FID analysis mentioned above. The Hettich centrifuge, model D-7200 (Germany) was used for accelerating phase separation.
Extraction/preconcentration procedure
LLE procedure
To 3 g blank shampoo, 1.00 g NaCl was added and spiked with 1,4-dioxane at a concentration of 500 µg kg−1 into a 50-mL separatory funnel. Then, 5 mL n -hexane and dichloromethane mixture (50: 50, v/v) was transferred into the funnel. After manually shaking (2 min) the mixture was transferred into a 10-mL centrifuge tube. After centrifuging at 7000 rpm for 5 min, the upper phase was removed and used in the following microextraction procedure.
DHS-LPME procedure
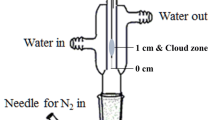
The extract (5 mL) obtained from the previous LLE step was transferred into a 10-mL glass vial and it was sealed by a PTFF-silicon septum. Then the taper end of the home-made extraction vessel (GC liner shaped) (Scheme 1) was passed through the septum and inserted in the headspace of solution. Then 2.5 μL n -octanol containing 500 mg L−1 toluene (IS) was added to wide section of the extraction vessel, and vial was placed into a water bath thermostated at 35 °C. After 8 min, the vessel was disconnected and 1 μL of the organic phase placed within the narrow section of the vessel was removed using a 1-μL microsyringe and injected into the separation system. The extraction procedure is shown schematically in Scheme 1. It is noted that during DHS-LPME procedure, the analyte was transferred to headspace of the solution placed into the vial and then passed through the extraction vessel. During passing the vessel, 1,4-dioxane was dissolved into n -octanol and concentrated. The narrow section of the vessel avoided n -octanol to pour into the vial. Indeed in the period of the extraction (8 min) many bubbles were produced in narrow section of the vessel and moved-up. When they reached to wide section of the vessel, the bubbles were cracked up and n -octanol re-collected in narrow section of the vessel. This action was repeated for many times during 8 min extraction time. The procedure is a dynamic method because the analyte is passed through the extraction vessel continuously. In spite of single drop micro-extraction in which one equilibrium is established and low ER is achievable, in this method many equilibriums are occurred and an exhaustive extraction can be obtainable (see Sect. 3.3 for ER of 89 %).

Extraction procedure
Calculation of EF and ER
Two main parameters, namely EF and ER, have been used for evaluation of the proposed procedure. The EF is defined as a ratio of the analyte concentration in the final organic phase into the extraction vessel (C f) to its initial concentration (C 0) within the sample.
C f is obtained by comparison of peak area of the analyte obtained by direct injection of a standard solution (three times in a day) prepared in the extraction solvent. The ER is defined as the percentage of the total analyte amount (n 0) which is extracted into the final organic phase (n f):
where V f, d s, and M s are volume of the final organic phase into the extraction vessel, sample density, and sample weight, respectively.
Results and discussion
In the present study 1,4-dioxane is extracted into an organic phase from shampoo during an LLE procedure. Then a new dynamic micro-extraction method is performed for the preconcentration of the analyte prior to its injection into GC–FID. In this procedure, a home-made extraction vessel is used and all parameters that can affect the extraction efficiency including type and volume of extraction solvent, extraction time, temperature, and ionic strength are investigated.
Optimization of parameters in LLE procedure
Selection of extraction solvent
As mentioned above in this study, the target analyte is extracted from shampoo sample by an LLE method and the extracted analyte is preconcentrated in the following microextraction step. In LLE, selection of an appropriate extraction solvent has an important role on performance of the developed method. The extraction solvent should have good extraction capability for the target analyte from relatively complex matrix, e.g., shampoo, is immiscible with the sample, and has sufficient volatility due to this fact that, the extracted phase will be used in the following micro-extraction method. It assists to transfer of the analyte to headspace in DHS-LPME step. According to these requirements three extraction solvents including acetone, n -hexane, and dichloromethane (individual or binary mixtures of them) were selected for this purpose. In all experiments, 3 g blank shampoo spiked with 500 µg kg−1 of 1,4-dioxane along with 5 mL of each extraction solvent or binary mixture (50:50, v/v) of them were subjected to the same LLE procedure in a 50-mL separatory funnel. After manually shaking for 2 min, transferring into a 10-mL tube and centrifuging at 7000 rpm for 5 min, 1 μL of the upper phase was injected into GC. According to the obtained results (Fig. 1) the better analytical signal is observed in the binary mixture of dichloromethane: n-hexane (50:50, v/v). Therefore, it was selected for the further experiments.

Selection of extraction solvent in LLE step. Extraction conditions: sample 3 g blank shampoo spiked with the analyte (500 µg kg−1); extraction solvent 5 mL of each solvent or binary mixture (50:50, v/v) of them; temperature 35 °C; centrifuging rate 7000 rpm; and centrifuging time 5 min. The error bars indicate the minimum and maximum of three independent determinations
Study of dichloromethane/n-hexane volume ratio
Dichloromethane and n -hexane mixtures with different volume ratios have various polarities and can affect distribution coefficient of the target analyte between the sample and extractive phase. Therefore, selection of dichloromethane and n -hexane volume ratio is important. For this purpose, a series of extractive phases was prepared by mixing different volumes of dichloromethane and n -hexane while total volume was kept constant (5 mL). The obtained results showed that the high extraction efficiency was obtained at a ratio of 50:50 (v/v) dichloromethane:n-hexane. It seems that in the case of 50:50 (v/v) dichloromethane:n-hexane distribution coefficient of the analyte between sample and the mentioned extraction solvents mixture is high due to their polarities matching. Subsequently, it was selected as an extractant for the further experiments.
Optimization of extractant volume
The effect of extractant volume in LLE step was examined using a series of experiments using different volumes of dichloromethane: n -hexane (50:50, v/v) mixture including 3, 4, 5, 6, and 7 mL according to the extraction procedure described above. The results indicated that ER increased from 3 to 5 mL and then remained constant. It is plausible that at low volumes of the extractant, extraction of 1,4-dioxane is incomplete and at volumes more than 5 mL dilution of the extracted analyte in the extractive phase can be occurred. This can be concluded from decreasing peak area of the analyte at high volumes of extractive phase used. Therefore, 5 mL was selected as the optimum volume for the extractant.
Study of salt addition
Salting-out effect is observed in most LLE methods. Addition of a salt decreases solubility of analyte in an aqueous phase and increases its partitioning into an organic phase. In the present study, addition of salt may increase extraction efficiency of 1,4-dioxane from shampoo into the extractant, and assists two-phase system formation. Therefore, effect of salt addition was investigated by adding NaCl in the range of 0–1.50 g into 3 g blank shampoo sample spiked with the target analyte at a concentration of 500 mg kg−1. The obtained results indicated that analytical signal increased up to 1.0 g and then decreased. It should be noted that volume of the collected organic phase increased from 5 to 6 mL by adding NaCl in the range of 0.0–1.50 g. Increasing the collected organic phase volume has no evil effect on ER of the analyte at 1.25 and 1.50 g with respect to 1.00 g NaCl. So, NaCl (1.00 g) was selected as a salting out agent for the further studies.
Optimization of parameters in DHS-LPME step
Selection of extraction/preconcentration solvent
Selection of extraction solvent is very important in all LPME methods as well as in this study in which a DHS-LPME is used. DHS-LPME was proposed in this work mainly to preconcentrate the analyte from the extract obtained in LLE step. Extraction/preconcentration solvent was added to the home-made extraction vessel at μL level to dissolve analyte released to headspace of the LLE extractant. In this study a dynamic mode of headspace extraction was used to have an exhaustive extraction. The analyte released gradually from the solution to headspace and passed through the extraction vessel containing extraction/preconcentration solvent. 1,4-Dioxane was dissolved into μL level of the solvent and preconcentrated. The extraction/preconcentration solvent should have a low volatility to avoid its loss during extraction period. Also, it must properly dissolve/preconcentrate the analyte and its chromatographic peak is well separated from the analyte peak in the chromatogram. Several organic solvents including n -hexanol, DMSO, n -octanol, and n -pentadecane were investigated for this purpose. It should be noted that to provide an acceptable precision the use of an internal standard in this step is necessary. For this purpose, toluene was used as an internal standard and it was added to each of the studied solvents at a concentration of 500 mg L−1. In the following experiments, 1,4-dioxane was spiked at a concentration of 0.5 mg kg−1 to blank shampoo. Figure 2 depicts the ratio of peak area of analyte to peak area of toluene versus extraction solvent kind. According to the obtained results, the best analytical signal is observed using n-octanol and it was selected as the extraction/preconcentration solvent in the subsequent studies.

Selection of extraction/preconcentration solvent volume in DHS-LPME. Extraction conditions: the same as used in Fig. 1, except n-octanol was used as the extraction/preconcentration solvent. The error bars indicate the minimum and maximum of three independent determinations
Optimization of n-octanol volume
In a micro-extraction method, the volume of extraction solvent should be optimized because this factor can affect the extraction efficiency. To evaluate the effect of extraction/preconcentration solvent volume, different volumes of n -octanol (0.5, 1.0, 1.5, 2.5, 5.0, 7.5, and 10 μL) were subjected into the same micro-extraction procedures. The obtained results showed that the peak areas ratio increased until 2.5 μL due to increase in extraction efficiency of the method and then decreased at volumes 2.5–10 μL probably due to dilution of the extracted analyte into the organic phase which in turn led to decrease in EF and analytical signal. So, 2.5 μL n-octanol was chosen as the optimum volume for the extraction/preconcentration solvent.
Influence of extraction temperature
Temperature has a significant effect on the kinetic and thermo dynamic of an extraction process. Mass transfer (rate and amount) of the analyte from the sample to headspace and then from the headspace to the extraction/preconcentration solvent depends on temperature. High temperature leads to increased vapour pressure of the analyte and hence its high concentration in headspace. On the other hand, distribution coefficient of the analyte between the headspace and extraction/preconcentration solvent decreases at high temperatures. Also at high temperatures, vaporization of the extraction/preconcentration solvent is a problem. Therefore, temperature should be optimized in this study. The effect of temperature was evaluated in the range of 25–45 °C. The obtained results are shown in Fig. 3 which reveal that extraction efficiency of the proposed method increases up to 35 °C due to increasing distribution coefficient of the analyte between solution (LLE extract) and headspace and then decreases. Decreasing in extraction efficiency at temperatures higher than 35 °C can be attributed to decrease in distribution coefficient of 1,4-dioxane between headspace and the extraction/preconcentration solvent. Therefore, 35 °C was selected as the optimum extraction temperature.

Effect of temperature on the extraction efficiency. Extraction conditions: the same as used in Fig. 2, except 2.5 μL n-octanol containing 500 mg L−1 toluene was used. The error bars indicate the minimum and maximum of three independent determinations
Heating/extraction time study
In this study, a DHS-LPME method was developed in which heating/extraction time has an important effect on performance of the developed method. This parameter was studied in the range of 0.5–10 min. The obtained data showed that the peak areas ratio and ER increase up to 8 min and then remained constant. It can be concluded that in heating/extraction time of 8 min the analyte removed quantitatively from the solution and dissolved in the extraction/preconcentration solvent placed into the extraction vessel. The ER near to 100 % at the times of 8–10 min is another reason for this subject. So, 8 min was selected as the optimum time for heating/extraction in the following experiments.
Analytical parameters
Under the optimum conditions, quantitative characteristics of the developed method including linear range (LR), coefficient of determination (R 2), limit of detection (LOD), limit of quantification (LOQ), EF, and ER were obtained. A calibration curve was obtained with LR of 1.7–10,000 μg kg−1 with a regression equation of A = 14.96C + 4.3 (A = peak area and C = concentration, μg kg−1) and coefficient of determination of 0.992. The LOD and LOQ, calculated for a signal-to-noise ratio of 3 and 10, were 0.52 and 1.7 μg kg−1, respectively. Repeatability of the developed method, expressed as relative standard deviation (RSD %), was evaluated by performing the method on six repeated experiments (for intra-day) and four repeated experiments (for inter-day) at a concentration of 25 μg kg−1 and they were obtained 5.9 and 6.9 %, respectively. The EF and ER values of the proposed method were 333 ± 7 and 89 ± 2 %, respectively, for three repeated experiments performed on the blank shampoo spiked with a concentration of 25 μg kg−1.
Real samples analysis
Applicability of the developed method to real samples was investigated by determining 1,4-dioxane in different shampoo samples. Figure 4 depicts typical GC–FID chromatograms of shampoo Sehat before and after spiking with 100 μg kg−1 of 1,4-dioxane along with chromatogram of a standard solution. Considering the chromatograms, there is a suspected peak (indicated by an asterisk) in unspiked shampoo chromatogram eluated in the retention time of 1,4-dioxane. To identify this compound, the sample was injected into GC–MS after performing the developed method. The presence of 1,4-dioxane was verified by comparison of mass data for scan 524 (retention time 5.23 min) with those of 1,4-dioxane (Fig. 5). 1,4-Dioxane content of the studied shampoos (8 samples) was determined using standard addition method at three concentration levels of 50, 250, and 500 μg kg−1 by the proposed DHS-LPME–GC–FID method and it was obtained in the range of 9.40–1030 μg kg−1 which are lower than MRL value proposed by US Department of Health and Human Services. The obtained concentrations are summarized in Table 1. To evaluate matrix effect in different samples, the added-found method was used. The samples were spiked with the target analyte at three concentrations (50, 250, and 500 μg kg−1) and the proposed method was applied to them (three times for each concentration). The relative recovery values for the target analyte are summarized in Table 1. Considering relative recoveries (>88 %), it can be concluded that the matrices of the studied samples are less effective in performance of the developed method.

GC–FID chromatograms of: a standard solution of the analyte and toluene prepared in n -octanol (500 mg L−1 of each), b shampoo Sehat, and c shampoo Sehat spiked with (50 μg kg−1 of analyte). In all cases, except chromatogram (a), the proposed method was performed on them and 1 μL of the final organic phase was injected into the separation system

a GC-total ions current-MS chromatogram of shampoo Sehat, b mass spectrum of 1,4-dioxane, and c mass spectrum of the compound eluated in retention time of 5.23 min
Comparison of the developed method with other approaches
Table 2 indicates the values of LOD, LR, and RSD of other methods and the proposed method for the extraction and determination of 1,4-dioxane from different samples. The repeatability of method is good and RSD for the proposed method is lower than those of most mentioned methods. In comparison with other microextraction methods, the presented method provides lower or comparable LOD and wider LR. These results reveal that the proposed method is a sensitive and repeatable technique that can be used for the preconcentration and determination of 1,4-dioxane in shampoo samples.
Conclusion
In this study, for the first time, a new microextraction method based on DHS-LPME has been developed for the extraction and preconcentration of 1,4-dioxane in shampoo. The method was combined with GC–FID for quantitative analysis of 1,4-dioxane in different shampoo samples. The experimental results show that this technique exhibits many merits such as high ER and EF, low LOD and LOQ, low cost, short extraction time and good repeatability. Excellent performance of the method in analysis of 1,4-dioxane in shampoo samples shows that it can be successfully applied to relatively complex matrices. In the light of these advantages, the developed method can be considered as an efficient, simple and rapid technique for the determination of 1,4-dioxane in shampoo samples.
Abbreviations
- DHS-LPME:
-
Dynamic headspace-liquid phase microextraction
- DMSO:
-
Dimethyl sulfoxide
- EF:
-
Enrichment factor
- ER:
-
Extraction recovery
- FID:
-
Flame ionization detection
- GC:
-
Gas chromatography
- LLE:
-
Liquid–liquid extraction
- LOD:
-
Limit of detection
- LOQ:
-
Limit of quantification
- RSD:
-
Relative standard deviation
- SPE:
-
Solid phase extraction
- SPME:
-
Solid phase microextraction
References
S. Scalia, F. Testoni, G. Frisina, M. Guarneri, J. Soc. Cosmet. Chem. 43, 207–213 (1992)
S.C. Rastogi, Chromatographia 29, 441–450 (1990)
T. Nishimura, S. Lizuka, N. Kibune, M. Ando, J. Health. Sci. 50, 101–107 (2004)
C.T. Derosa, S. Willbur, J. Holler, P. Richter, Y.W. Stevens, Toxicol. Ind. Health 12, 1–43 (1996)
Occupational exposure limits for 1,4-dioxane recommendations of the Scientific Committee for Occupational Exposure Limits (SCOEL) to chemical agents. Office for official publications of the European Communities (2005)
R.E. Black, F.J. Hurley, D.C. Havery, J. AOAC Int. 84, 666–670 (2001)
J.D. Young, W.H. Braun, L.W. Rampy, M.B. Chenoweth, G.E. Blau, J. Toxicol. Environ. Health 3, 507–520 (1997)
J. Matthew, R.C. Zenker, A.B. Morten, J. Environ. Eng. Sci. 20, 423–432 (2003)
G.D. Clayton, F. E. Clayton (eds.), Patty’s Industrial Hygiene and Toxicology, 3rd edn (Wiley, New York, 1982), pp 3947–3956
A. Fairly, E.C. Linton, A.H. Ford-Moore, J. Hyg. 34, 486–501 (1934)
International Agency For Research on Cancer in IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Man, International Agency for Research on Cancer (Lyons, France, vol. 11, 1976), pp. 247–256
IARC Monographs on the Evaluation of Carcinogenic Risks to Human, Re-evaluation of Some Organic Chemicals, Hydrazine and Hydrogen Peroxide (World Health Organization, Lyon, vol. 71, 1999), pp. 589–602
National Cancer Institute (NCI) Carcinogenicity Technical Report No. NCL-GC-TR-80 (Bethesda, 2015)
L.M. Sweeney, K.D. Thrall, T.S. Poet, R.A. Corley, T.J. Weber, B.J. Locey, J. Clarkson, Sh Sager, M. Gargas, Toxicol. Sci. 101, 32–50 (2008)
US Department of Health and Human Services, Agency for toxic substances and disease registry: toxicological profile for 1,4-dioxane (draft for public comment) (Atlanta, 2004)
W.M. Draper, J.S. Dhoot, J.W. Remoy, S.K. Perera, Analyst 125, 1403–1408 (2000)
R. Otson, C. Chan, Int. J. Environ. Anal. Chem. 30, 275–287 (1987)
N. Ramirez, R.M. Marce, F. Borrull, Int. J. Environ. Anal. Chem. 91, 911–928 (2011)
M. Li, P. Conlon, S. Fiorenza, S.R.J. Vitale, P.J.J. Alvarez, Ground Water Monit. Rem. 31, 70–76 (2011)
Y.P.T. Bonnie, M. Zulina, M. Halimah, J. Am. Chem. Soc. 91, 1103–1110 (2014)
A.B. Waldman, J. Soc. Cosmet. Chem. 33, 19–25 (1982)
J.J. Robinson, E.W. Ciurczak, J. Soc. Cosmet. Chem. 31, 329–337 (1980)
S. Scalia, J. Pharm. Biomed. Anal. 8, 867–870 (1990)
S.H. Hong, J.B. Lee, S.H. Lee, H.H. Lim, H.S. Shin, J. Anal. Sci. Technol. 27, 22–26 (2014)
Y.M. Park, H. Pyo, S.J. Park, S.K. Park, Anal. Chim. Acta 548, 109–115 (2005)
P.E. Grimett, J.W. Munch, J. Chromatogr. Sci. 47, 31–39 (2009)
M.A. Jochmann, M.P. Kmiecik, D.C. Schmidt, J. Chromatogr. A 1115, 208–216 (2006)
C. Isaacson, T.K.G. Mohr, J.A. Field, Environ. Sci. Technol. 40, 7305–7311 (2006)
G. Ortiz, G.M.T. Tena, J. Chromatogr. A 1101, 32–37 (2006)
C.B. Fuh, M. Lai, H.Y. Tsai, C.M. Chang, J. Chromatogr. A 1071, 141–145 (2005)
P.N. Nomngongo, J.C. Ngila, J. Iran. Chem. Soc. 12, 2141–2147 (2015)
S. Bahar, F. Karami, J. Iran. Chem. Soc. 12, 2213–2220 (2015)
G. Khayatian M. Jodan, S. Hassanpoor, S. Mohebbi, J. Iran. Chem. Soc. (2016) doi:10.1007/s13738-015-0798-2 (Article in press)
H. Sereshti, M. Karimi, J. Iran. Chem. Soc. 11, 1129–1136 (2014)
R.E. Shirey, C.M. Linton, J. Chromatogr. Sci. 44, 444–450 (2006)
W. Yufei, S. Jiawei, W. Li, Chin. J. Chromatogr. 33, 441–445 (2015)
S. Nakamura, S. Daishima, Anal. Chim. Acta 548, 79–85 (2005)
Acknowledgments
The authors thank the Research Council of University of Tabriz for financial support and scientific encouragement. The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Farajzadeh, M.A., Nassiry, P., Mogaddam, M.R.A. et al. Development of dynamic headspace-liquid phase microextraction method performed in a home-made extraction vessel for extraction and preconcentration of 1,4-dioxane from shampoo. J IRAN CHEM SOC 13, 1385–1393 (2016). https://doi.org/10.1007/s13738-016-0853-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-016-0853-7