
Abstract
The sequential extraction methods according to Tessier et al. [1], Borovec et al. [2], Zhang and Moore [3] and Hall et al. [4] have been tested for their suitability for arsenic fractionation in samples of artificially prepared mineral mixtures. Mixtures containing different amounts of As-containing phases were prepared so that their compositions corresponded to weathering products on As-bearing ore deposits. A comparison of different procedures on simple mineral mixtures containing calcium arsenate (CaHAsO4·H2O), As-bearing goethite (FeOOH) and arsenopyrite (FeAsS) showed that only the results of the Hall method satisfactorily correspond to the expected arsenic distribution. A detailed verification of the Hall method was subsequently carried out on most complex synthetic mineral mixtures with varying amounts of As-containing kaolinite and carbonate, calcium arsenate, As-bearing goethite and arsenopyrite. The results confirm that the Hall method cannot be fully employed for an accurate As speciation but may be applied for a route identification of As distribution between "labile", "medium-labile" and "residual" forms in heavily polluted soils.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Because of its teratogenic and mutagenic effects on living organisms, arsenic is one of the most dangerous metalloids in the environment. Regarding other trace elements in soils and sediments, determination of the total As concentration represents only the first step of the environmental study. In fact, the environmental availability of As depends on its chemical speciation, i.e. on its bonding to the individual soil/sediment mineral components. The adsorption of As on amorphous or crystalline hydrous ferric oxides (HFO) is a key scavenging process in both natural and contaminated environments [5, 6]. Metallic arsenates commonly crystallise during treatment of mine processing effluents or industrial wastes with a Ca(OH)2 solution [6, 7]. Calcium arsenates were found in the oxidation zones in the As-containing ore districts [8, 9] and as oxidation products of As-bearing mineral wastes [10].
Sequential extraction procedures based on the original work of Tessier et al. [1] represent a powerful tool for determination of chemical speciation of metal cations. Only a few studies, however, have been performed on the suitability of sequential extraction procedures for oxyanions such as As [11]. The evaluation of As forms in soil/sediment using extraction techniques is complicated by the occurrence of As in extracts in the form of anionic complexes formed as H3AsO4 dissociates. In acidic medium AsO4 3− ions can be easily adsorbed on the surface of solid phases, commonly on HFO [12]. The goal of present study is to test the efficiency of four original sequential extraction procedures of Tessier et al. [1], Borovec et al. [2], Zhang and Moore [3] and Hall et al. [4] on synthetic mineral mixtures and to evaluate their possible application to soils heavily contaminated by As, which are commonly found in the oxidation zones of ore deposits. The described extraction methods were chosen with respect to the differences in reagents used, reaction time and temperature of equilibration.
Materials and methods
As a first step, a comparison of the efficiency of the four cited extraction procedures was carried out using two artificially prepared samples (samples A and B) with defined amounts of different As-bearing phases and exactly known As concentrations. The mineral composition and amount of As in these model samples corresponded to those found in weathered zones above As-containing gold deposits. In a second step, a detailed verification of the suitability of the use of Hall method was tested on another four synthetic samples with different As bonding (samples C, D, E and F).
Reagents and samples
Chemicals of pro-analyse quality were employed for synthetic phases preparation and for extraction steps. HF, HClO4, HCl and HNO3 of Suprapur quality (Merck, Germany) were used for mineral decomposition and for preparation of all extraction solutions. All solutions were prepared using MilliQ-Plus (Millipore, USA) deionised water.
Synthetic mineral mixture preparation
Sample A represents a soil with no primary As mineral content. Arsenic is bonded to iron(III) hydroxide oxide (goethite, FeOOH) and is partially present as a "secondary" mineral, calcium arsenate (haidingerite, CaHAsO4·H2O). Synthetic goethite was prepared by the reaction of 200 mL of 2.5 M NaOH with 825 mL of a 0.15 M solution of Fe(NO3)3 [13]. Prior to addition of the hydroxide, the ferric nitrate was enriched in As so that 1 g of the goethite formed contained about 100 μg As. The As source was a 1 g L−1 calibration standard As solution (Astasol, Analytika CZ). Crystallisation of goethite (ageing of the suspension) occurred at a temperature of 80 °C. During ageing (40 h), the suspension formed was rinsed with deionised water. The sodium hydroxide-free suspension was dried at a maximum temperature of 105 °C to constant weight (approximately 2 h). Calcium arsenate was prepared by the reaction of sodium hydrogen arsenate with calcium chloride following the scheme proposed by Pierrot [8]; 102 g Na2AsO4·7H2O was mixed with 35 g CaCl2 then dissolved in 200 mL deionised water. Then 40 mL of concentrated HCl was added and NH4OH was carefully added drop-wise to the solution. The precipitate formed was separated by filtration. The overall process of calcium arsenate crystallisation proceeded for 7 days at room temperature. Powder X-ray diffraction analysis (XRD, Dron 2.1 diffractometer) revealed the presence of haidingerite (CaHAsO4·H2O). The mineral composition of the final sample was completed by weighing and adding the other constituents (crushed quartz, muscovite and feldspar) (Table 1).
Sample B contains only primary arsenopyrite (FeAsS) and corresponds to unweathered sediments. Sediments with similar As speciation occur in the mine tailings of old gold mines in the Bohemian Massif, Czech Republic [9]. The final mineral composition of the sample was prepared by addition of quartz, muscovite and feldspar (Table 1).
In sample C, arsenic is bonded to the clay mineral kaolinite (Al2Si2O5(OH)4) and synthetic calcium carbonate (CaCO3). Arsenic-enriched kaolinite was prepared from a suspension of kaolinite enriched with a 1 g L−1 standard As solution (Astasol, Analytika CZ). Following rinsing and drying, kaolinite contained 600 mg kg−1 As. Synthetic CaCO3 was prepared with 22 g CaCl2 stirred in 100 mL deionised water and enriched with a standard solution of As, so that the final concentration was 1000 μg As in 1 g CaCO3. The CaCO3 was precipitated in 200 mLof 1 M Na2CO3 solution. The suspension was aged at room temperature for 2 days, then dried at 105 °C to constant weight and ground in an agate mortar.
In sample D, arsenic is bonded to clay mineral, carbonate and synthesised goethite. Sample E represents a mixture of synthetic haidingerite, clay mineral, carbonate and synthesised goethite. Sample F has the identical composition as sample E, but also contains primary arsenopyrite.
Following weighing of the individual phases (As-bearing phases, quartz, muscovite and feldspar), the prepared mineral mixtures were homogenised and pulverised in an agate ring mill. The approximate grain size of mineral mixture was less than 60 μm.
After preparation of As-bearing and other mineral phases, the As content in each mineral was determined. Mineral powder (0.2 g) was digested in a PTFE closed beaker (Savillex, Minetonka, USA) with 10 mL HNO3 and 5 mL HF overnight on a hotplate (150 °C). After opening the vessels and evaporation to near dryness, the residue was dissolved in 2% HNO3 and transferred into a 100-mL volumetric flask. The stock solution was diluted 10–100 times before ICP MS measurement. Certified reference materials GXR 1, GXR 2 and GXR 3 (USGS) were analysed for quality control of analytical data of each prepared As-bearing mineral. Expected values of As in the different extraction steps were calculated from mineral content in the mixture and from concentration of As in each mineral.
The mineralogical composition and corresponding As concentrations are given in Table 1.
Sequential extractions
For a comparison of efficiency in As extraction, the methods of Tessier et al. [1], Borovec et al. [2], Zhang and Moore [3] and Hall et al. [4] were selected. The successive chemical extraction steps of each sequential procedure are summarised in Table 2. To facilitate the comparison of different extraction techniques, the extraction steps were combined in four groups: (i) exchangeable ions, carbonates and calcium arsenates (AEC fraction—adsorbed/exchangeable/carbonate), (ii) Fe- and Mn-oxyhydroxides, (iii) organic matter and sulphides, and (iv) residual minerals. Calcium arsenates are relatively soluble (from 4.1×10−2 to 9×10−3 mol L−1 [10]) and will probably dissolve during the first step of the extraction procedures. For this reason, results concerning the dissolution of calcium arsenate were ranged into the AEC fraction.
The Tessier method was carried out according to the original work, with only a change in the volume of the extraction agent from 8 mL to 20 mL. The Borovec extraction method was carried out exactly according to the original work. The original definition of individual fractions (soluble, reducible, oxidisable, organic, residual) given by Zhang and Moore [3] were readjusted to agree with the terminology used in the Tessier, Borovec and Hall methods. The Zhang and Moore method was carried out according to the original work. The Hall extraction technique was also carried out according to the original work, except that the KClO3 recommended for oxidation of the organic matter and sulphides was replaced by H2O2 in acidic medium, i.e. according to the Tessier method to maintain approximately the same solid/liquid ratio for oxidation of arsenopyrite.
The initial mass of synthetic mineral mixture was 1.000±0.001 g for every extraction procedure. After each extraction step, the solution was separated from the solid phase by centrifugation (4000 rpm, time 20 min). The solution was transferred into a volumetric flask and the undissolved residue was rinsed with 20 mL of deionised water. Following centrifugation, the rinse solution was added to the original solution. Blanks were run simultaneously at all stages of the procedure, and all the extraction methods were carried out in triplicate. In addition, the concentrations in the individual extracts were also determined in triplicate. The results are given as the arithmetic mean. The total concentration of As in the "residual fraction" and in individual synthetic mixtures was determined by dissolution of solid samples in a mixture of HF and HClO4 in a ratio of 5:1. It was found that residual mineral components (quartz, muscovite, feldspar) also contained small amounts of As (Table 1).
Analysis of solutions
All the solutions were stabilised with concentrated hydrochloric acid (final HCl concentration 2% v/v). The element concentrations were determined using flame atomic-absorption spectrometry (FAAS, Varian SpectrAA 200HT) in an acetylene–nitrous oxide flame under the conditions recommended by the manufacturer with matrix-matched calibration. The detection limit of As was 10 µg g−1.
Solutions with low As concentrations were prepared in 2% v/v HNO3 and analysed by inductively coupled plasma mass spectrometry (PQ 3 VG Elemental, GB) with a glass concentric nebuliser, water-cooled spray chamber and peristaltic pump. Lenses and gas flows of the ICP MS were tuned using 50 μg L−1 As and 20 μg L−1 In solutions. Arsenic external calibration standards (1, 10, 50 μg L−1) were used; the internal standard (20 μg L−1 In solution) was mixed with measured solutions using a Y junction. An RF power of 1350 W, the pulse count mode of the detector, three points per peak, the peak jump mode and three replicates of 30 s acquisition time were used for measuring the isotopes 75As and 115In. The LOD of ICP MS measurement was 1.5 µg g−1 As.
Results and discussion
Release of metals from AEC fraction
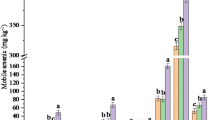
The AEC fraction comprises As adsorbed and bound in an exchangeable position, As in carbonates and easily soluble arsenates. Comparison of the different sequential extraction procedures shows that the amount of As released from the AEC fraction is significantly underestimated by the Tessier, Borovec and Zhang and Moore methods (Table 3, Fig. 1). For sample A, a good agreement between the results of the first step of the Hall method and expected values was observed (Table 3, Fig. 1). Thus, the acidity of extraction agents and especially the reaction time are the most important factors influencing As recovery from the AEC fraction. It was found that the highest amounts of As were released during the first steps of the Hall and the Borovec methods with a sufficiently long extraction interval (reaction times of 12 and 8 h, respectively) (Table 3, Fig. 1). This is in agreement with Wenzel et al. [11], who quote the importance of a sufficiently long time for the first steps of selective extraction. Less than 1.5 µg g−1 As was released using 0.25 M NaCl (Zhang and Moore method), probably due to the short reaction time and weakness of the extraction agent (Table 3, Fig. 2). The higher amount of As released from exchangeable positions by the Borovec method can possibly be linked with the preparation of goethite; the subsequent treatment of the mineral mixture (especially grinding) leads to a shift in the individual forms towards more labile bonding and part of the As enters exchangeable positions. During the first extraction steps, As was also released from sample B with arsenopyrite content; this phenomenon was observed especially in slightly acidic extraction agents at pH~5 (Table 3, Fig. 1) and confirms that sulphides are reactive even in the intermediate pH range [14].

Efficiency of different sequential extraction procedures employed for simple mineral mixtures A and B

Verification of the Hall sequential extraction method on complex mineral mixtures C, D, E and F
The Hall method seems to be the best for determining the speciation of As in synthetic soils with high amounts of calcium arsenates, which are commonly found in oxidation zones of ore districts. However, the first step of this method initiates the release of As from soils with a high amount of primary arsenopyrite.
Effect of the reducing agent (Fe- and Mn-oxyhydroxide fraction)
Hydroxylammonium hydrochloride (NH2OH·HCl) is one of the most frequently employed extraction agents with reducing properties (Table 2). The use of this reagent should lead to the reduction of synthetic goethite in sample A. If the contribution of As from the previous steps is neglected, the amount of As released by this reagent depends primarily on the reaction temperature and the acid concentration in the NH2OH·HCl solution that helps to maintain a considerable amount of released As in the extract. Comparison of the selected extraction procedures shows that in the case of sample A containing As-doped goethite, the Tessier, Borovec and Zhang and Moore methods significantly overestimate the As released (Table 3, Fig. 1). The recovery of As in this extraction step of the Tessier method is five times higher than expected values. It is probable that such an elevated release of As is caused by late dissolution of calcium arsenate, which was not dissolved in previous extraction steps due to the weakness of extraction agents or insufficient reaction time. As well as in previous fractions, the Hall method seems to give the best fit for As concentrations bound on Fe- and Mn-oxyhydroxides (Table 3, Fig. 1). Small amounts of As were released from sample B by the extraction steps of the Tessier, Borovec and Hall methods (Table 3, Fig. 1). This phenomenon is probably linked with dissolution of newly formed alteration products (hydrous ferric oxides, HFO) formed on the surface of the arsenopyrite during its exposure to extraction media in the first steps of each sequential procedure. Wang et al. [14] found that in alkaline solutions, the oxidation of arsenopyrite produces ferric hydroxide on the arsenopyrite surface. Such processes can be taken into account in the Tessier and Borovec methods using neutral and alkaline media, respectively, in the first extraction steps (Table 2). It is probable that in slightly acidic media, the oxidation of arsenopyrite and subsequent HFO precipitation will also take place especially when the extraction steps are relatively long (Table 2).
Effect of the oxidising agents (sulphide and organic fraction)
The strong oxidising action of H2O2 in acidic medium is frequently employed for oxidation of organic material and sulphides in sequential extraction (Table 2). Although the design of this extraction step in the Tessier, Borovec and Hall methods is identical, the extraction results are different as a result of the efficiency of previous extraction steps (Table 3, Fig. 1). Sample A was prepared so that it would not contain any As bound to sulphides or organic matter. However, some As from these fractions was released by allthe methods used (Table 3, Fig. 1). In particular, the Tessier method showed a significant release of As; this fact is probably related to the late dissolution of goethite and/or calcium arsenate, which were not completely dissolved in previous extraction steps. The lowest amount of As is released by the extraction step of the Hall method (Table 3, Fig. 1). A comparison of the selectivity of the extraction methods for As in sample B (arsenopyrite) shows a good fit of the Hall method with the expected values (Table 3, Fig. 2). Conversely, the Tessier, Borovec and Zhang and Moore methods significantly underestimate the As available in this extraction step (Table 3, Fig. 2). As a result, the extraction step of the Hall method was found to be the best for the speciation of As bound to sulphides and organic matter.
Metals in the residual fractions
Although the As contents in the silicate fractions are environmentally insignificant, knowledge of the As release by this extraction step is relatively important, because it indicates the efficiency of the previous extraction steps. In the case of sample A, all the mineral phases should be decomposed in the HFO fraction. However, the recoveries from the residual fraction are significantly overestimated and the amount of As released by the Tessier, Borovec and Zhang and Moore methods indicates the inadequate efficiency of the extraction agents used in previous steps (Table 3, Fig. 1). In contrast, relatively low As contents in the residual fraction in the Hall method indicate that the mineral phases were decomposed during the previous extraction steps with a sufficient efficiency of extraction agents used (Table 3, Fig. 1).
For sample B, high recoveries of As were determined in the residual fraction by the final extraction step of the Tessier, Borovec and Zhang and Moore methods (Table 3, Fig. 2). The elevated As content in this fraction in the Tessier and Borovec methods indicates that the arsenopyrite was insufficiently oxidised in sample B in the previous extraction steps. In contrast, the amount of As determined in the residual fraction by the Hall method indicates efficient action of the oxidising agents on the arsenopyrite dissolution (Table 3, Fig. 2).
Detailed study of the Hall method
The Hall method was found to be the most suitable for mineral mixtures A and B. Subsequently, a detailed study of the extraction on more complex mineral mixtures C, D, E and F was performed. The results of this verification are shown in Table 4 and Fig. 2. Besides the As concentrations in individual extraction steps, concentrations of Ca and Fe were also determined in order to confirm the dissolution of calcium arsenate and HFO as the extraction proceeded (Table 4, Fig. 2). The Ca content decreases rapidly in the AEC fraction and remains constant in the subsequent steps (approx. 100 μg g−1). The dissolution of calcium arsenate and CaCO3 in the initial steps of the extraction experiments is apparent from variations in the Ca concentration (Fig. 2). It follows from the variation in the Fe concentration that the Fe in the samples containing kaolinite is extracted in the initial steps. The dissolution of goethite also leads to the formation of the corresponding peak in the reduction of crystalline HFO in samples D, E and F (Fig. 2). Oxidation of arsenopyrite was not significantly manifested on the plot of concentration of Fe (Fig. 2); thus, the oxidation of arsenopyrite probably occurs throughout the entire experiment.
In sample C, As is only bound on kaolinite and carbonate. However, only about half (44 μg) is extracted in the AEC extraction step and the remainder is released in the following extraction steps, especially in the residual silicate fraction (Table 4, Fig. 2). In sample D, the As is present in the AEC fraction and in goethite. Similarly to the previous sample, only half of the As is extracted in the AEC fraction and the remainder is extracted together with the fraction of crystalline HFO (Table 4, Fig. 2). The low As recovery in the AEC fraction could be caused by re-adsorption of As ions by kaolinite whose pH zero point of charge (ZPC) value is close to the pH of the extraction agent employed (pH~5) [15]. In the HFO extraction step, where the reducing solutions have a sufficiently low pH, As is present as H3AsO4, which significantly limits its re-adsorption so it can be sufficiently extracted from the remaining solid phase (Table 4). Again, the fraction of As recovered from the residual minerals is slightly overestimated (Table 4, Fig. 2). In sample E, As is present in the AEC fraction, as well as in calcium arsenate and goethite. The recovery from the AEC fraction is slightly lower than the expected value; the high amount of As released from the amorphous HFO fraction corresponds to the complete dissolution of calcium arsenate (Table 4, Fig. 2). In addition to previous synthetic mineral mixtures containing goethite, As recovery from the crystalline HFO fraction is overestimated (Table 4, Fig. 2). Part of the As which was not sufficiently mobilised in the first three extraction steps is released by the strong mineral acids used for dissolution of the residual fractions (Table 4). In sample F, with the exception of the dissolution of arsenopyrite, the recoveries for the individual fractions were overestimated (Table 4, Fig. 2). Arsenopyrite was probably slightly oxidised in previous extraction steps. Again, the As recovery from the amorphous HFO fraction probably corresponds to the dissolution of calcium arsenate.
With the exception of sample F, the As recoveries from the AEC fraction are underestimated with respect to the expected values (Table 4, Fig. 2). On the other hand, the recovery of As from subsequent fractions of the crystalline HFO is generally twice that of the expected value (about 200%, Table 4, Fig. 2); this phenomenon probably corresponds to the inefficient reaction during the first extraction step. The high As recovery in the amorphous HFO fraction observed in samples E and F corresponds either (i) to residual dissolution of calcium arsenate which was not completely dissolved during the first extraction step (samples E and F) or (ii) to the dissolution of newly formed amorphous HFO, which may form during the rapid alteration of arsenopyrite grains (sample F). Another common phenomenon is a strong As leaching in the residual silicate fraction, which was observed in all the samples studied. This fact is likely to be related to the inefficient extraction in previous steps (Table 4, Fig. 2). However, the totals of extracted As during the Hall procedure correspond well to the expected values (Table 4).
The origin of material (soils, stream sediments), mineral composition, grain size, Eh and pH of the interacting solution must be considered before the application of the sequential extraction procedure. The synthetic mixtures used for the extraction experiments corresponded to soils formed in oxidising and slightly alkaline conditions above sulphidic ores with carbonates. However, the amount of As extracted from real soils may be significantly lower, because the soil fraction <2 mm typically used for sequential extraction corresponds to a lower reactive surface area. Compared to our synthetic samples, the real soils may contain larger mineral grains, which require longer extraction time for their complete dissolution (carbonate and sulphide grains). This would result in an underestimation of the leached amount of As in the carbonate and sulphide extraction step. On the other hand, a comparison of our experimental data with real soils may be satisfactory for the HFO fraction. In soils, HFO are often present in very thin coatings on mineral grains and, having a large reactive surface, they can be more easily dissolved. To compare synthetic and real natural samples, the same fraction (i.e. <60 μm) should be used.
Conclusions
Synthesis of artificial phases with admixtures of As was found to be a successful way for testing the extraction method. The use of extraction procedures for trace elements occurring as anionic complexes under normal conditions encounters considerable difficulties. On simple mineral mixtures containing calcium arsenate, As-bearing goethite and arsenopyrite, it was found that the extraction methods of Tessier et al. [1], Borovec et al. [2] and Zhang and Moore [3] are not suitable for the determination of As speciation. Quantitative oxidation of arsenopyrite did not occur using the oxidation agents recommended by these procedures. On the contrary, a good fit between observed and expected extractions was found for the method of Hall et al. [4] performed on simple mineral mixtures. Subsequent detailed study of the Hall method was carried out on more complex synthetic mineral mixtures with varying amounts of As-containing kaolinite, carbonate, calcium arsenate, As-bearing goethite and arsenopyrite. Although such a method cannot be employed for an accurate differentiation amongst adsorbed As and As bound to arsenates and carbonates in the first extraction step, the results show that it is relatively suitable for the route identification of As distribution in similar heavily polluted soils or sediments found in real environments. Using the Hall method, a current differentiation between "labile", "medium-labile" and "residual" forms of As can be performed and subsequently used for further environmental evaluation.
References
Tessier A, Campbell PGC, Bisson M (1979) Anal Chem 51:844–851
Borovec Z, Tolar V, Mráz L (1993) Ambio 22:200–205
Zhang Y, Moore JN (1997) Appl Geochem 12:685–691
Hall GEM, Vaive JE, Beer R, Hoashi M (1996) J Geochem Explor 56:59–78
Bowell RJ (1994) Appl Geochem 9:279–286
Pichler T, Hendry MJ, Hall GEM (2001) Environ Geol 40:495–506
Dutré V, Vandecasteele C (1998) Environ Sci Technol 32:2782–2787
Pierrot J (1964) Bull Soc Fr Minéral Cristallogr 87:169–211
Filippi M (2001) Mineralia Slovaca 33:13
Juillot F, Ildefonse P, Morin G, Calas G, de Kersabiec AM, Benedetti M (1999) Appl Geochem 14:1031–1048
Wenzel WW, Kirchbaumer N, Prohaska T, Stingeder G, Lombi E, Adriano DC (2001) Anal Chim Acta 436:309–323
Raven KP, Jain A, Loeppert RH (1998) Environ Sci Technol 32:344–349
Davis JA, Leckie JO (1978) Environ Sci Technol 12:1309–1315
Wang XH, Ahlberg E, Forssberg KSE (1992) J Appl Electrochem 22:1095–1103
Sverjensky DA (1994) Geochim Cosmochim Acta 58:3123–3129
Acknowledgements
This study was financially supported by Rio Tinto Technology, Granting Agency of the Charles University (No. 273/97/B GEO) and Ministry of Education of the Czech Republic (No. 113100005). The authors thank Dr. Madeleine Štulíková for translation of this manuscript, Marie Fayadová for assistance in analytical work and Marek Chvátal for XRD analyses. The reviews of two anonymous referees helped to improve the first version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mihaljevič, M., Poňavič, M., Ettler, V. et al. A comparison of sequential extraction techniques for determining arsenic fractionation in synthetic mineral mixtures. Anal Bioanal Chem 377, 723–729 (2003). https://doi.org/10.1007/s00216-003-2115-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-003-2115-7