
Abstract
Nuclear factor-kappa B (NF-κB) has been reported to play a pivotal role in many physiological processes including inflammation, apoptosis, and angiogenesis. We discovered a potent natural NF-κB inhibitor, dihydromyricetin, from the traditional herb Ampelopsis grossedentata, which has a long history of use in food and medicine. In this study, we demonstrated the effect of dihydromyricetin on NF-κB activation in TNF-α-induced HeLa cells. Dihydromyricetin was found to markedly inhibit the phosphorylation and degradation of the inhibitor of NF-κB alpha (IκBα), and subsequent nuclear translocation of p65. Dihydromyricetin also has an impact on upstream signaling of IKK through the inhibition of expression of adaptor proteins, TNF receptor-associated factor 2 (TRAF2), and receptor-interacting protein 1 (RIP1). Furthermore, the current results reveal that dihydromyricetin led to the downregulation of target genes involved in inflammation, proliferation, as well as potentiation of TNF-α-induced apoptosis through suppressing the activation of NF-κB. In conclusion, our data indicate that dihydromyricetin may be a potentially useful therapeutic agent for inflammatory diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The transcription factor NF-κB was discovered in 1986 as a nuclear factor that binds to the enhancer element of the immunoglobulin kappa light chain of activated B cells (thereby coining the abbreviation NF-κB) [1]. The NF-κB family of transcription factors consists of five members, p50, p52, p65 (RelA), c-Rel, and RelB, encoded by NF-κB1, NF-κB2, RelA, Rel, and RelB, respectively, which share an N-terminal Rel homology domain responsible for DNA binding and homo-and heterodimerization [2]. In most quiescent cells, these dimers are bound to inhibitory molecules of the IκB family of proteins (inhibitors of NF-κB) [3]. NF-κB is activated by a sequence involving the phosphorylation, ubiquitination, and degradation of IκBα, and the phosphorylation of p65, which in turn lead to the translocation of NF-κB to the nucleus, where it binds to specific response elements in the DNA [4]. In response to various stimuli, such as cytokines, DNA-damaging agents, bacterial wall or viral proteins, IκB dissociates via phosphorylation by the IκB kinase (IKK), and the activated transcription factor can translocate into the nucleus where is able to induce a large number of target genes involved in cell growth, apoptosis, cell adhesion and inflammation [5, 6].
TNF-α is a pleiotropic ligand of tumor necrosis factor receptor 1 and 2 (TNFR1 and TNFR2) that can signal both cell survival and cell death [7]. In the present study, we focused on the TNFR1 signaling pathway. Upon binding to its ligand, TNFR1 recruits the adaptor protein TNFR1-associated death domain protein (TRADD) directly to its cytoplasmic death domain, which later serves as an assembly platform for the recruitment of other molecules, such as TNFR-associated factor 2 (TRAF2) and receptor-interacting protein (RIP1) for the activation of NF-κB, or Fas-associated protein with death domain (FADD) for the activation of caspase-8 and induction of apoptosis [8–10].
Ampelopsis grossedentata, known as vine tea, is a plant in South China. Its main active ingredients are flavonoids, which have many bioactive properties such as scavenging free, radicals, antioxidant, anti-thrombus, anti-tumor, anti-inflammatory. Dihydromyricetin, a 2,3-dihydroflavonol compound, is a natural product extracted from A. grossedentata. Recently, it has been shown in some cancer cells that dihydromyricetin possesses anti-tumor effects, such as anti-proliferation, cell-cycle arrest, induction of apoptosis, and increased sensitivity to chemotherapeutic drugs [11, 12]. In this study, we researched the effects of dihydromyricetin on the NF-κB activation pathway and on the expression of TNF-α-induced NF-κB target genes. We found that dihydromyricetin inhibited the TNF-α-induced phosphorylation and degradation of IκBα, translocation of p65. This compound downregulates the expression of target genes involved in inflammation (iNOS), cell survival (c-IAP2, Bcl-2, TRAF1), proliferation (cyclin D1, COX-2), invasion (ICAM-1 and MMP-9), angiogenesis (VEGF), and potentiates TNF-α-induced apoptosis significantly via activation of caspase-8. Our finding may expand the application of dihydromyricetin to a valuable candidate for the intervention of NF-κB-dependent pathological conditions such as inflammation.
Materials and methods
Cell culture and reagents
HeLa, MDA-MB-231, HBL-100 and 293 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). HeLa, MDA-MB-231 and 293 cells were cultured in DMEM supplemented with 10 % heat-inactivated fetal bovine serum (Hyclone, Logan, UT, USA) and penicillin–streptomycin (100 U/ml) (Invitrogen, Carlsbad, CA, USA) at 37 °C in a humidified atmosphere with 5 % CO2. HBL-100 cells were maintained in RPMI medium supplemented as mentioned above. TNF-α was obtained from R&D Systems (Minneapolis, MN, USA). Dihydromyricetin was purchased from Beijing Yihua SGS Technology Company Ltd. (Beijing, China) and its structure is shown in Fig. 1a. The purity of dihydromyricetin was more than 98 % in high-performance liquid chromatography analysis.

Effect of dihydromyricetin (DMY) on the TNF-α-induced NF-κB-dependent reporter gene expression. a Structure of dihydromyricetin (DMY). b HeLa cells were transiently transfected with a NF-κB-dependent reporter gene for 48 h and then pretreated for 12 h with the indicated concentrations of dihydromyricetin (DMY) followed by stimulation for 12 h with 10 ng/ml TNF-α, and the luciferase activity was determined as described in “Materials and methods”. Data represented as mean ± standard deviation of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, significantly different when compared with TNF-α-stimulated normal cells. c HeLa cells were treated with the indicated concentrations of dihydromyricetin (DMY). After 24 h incubation, cell viability was determined by MTT assays. Data represented as mean ± S.D of three independent experiments
Transfections and luciferase reporter assay
A pNF-κB-Luc plasmid for NF-κB luciferase reporter assay was obtained from Stratagene (LaJolla, CA, USA). Transfections were performed as described previously [13]. NF-κB-dependent luciferase activity was measured using the Dual Luciferase Reporter Assay system. Briefly, HeLa cells (1 × 105 cells/well) were seeded in a well of 96-well plates for 24 h. The cells were then transfected with plasmids for each well and then incubated for a transfection period of 24 h. After that, the cell culture medium was removed and replaced with fresh medium containing various concentrations of dihydromyricetin for 12 h, followed by treatment with 10 ng/ml of TNF-α for 12 h. Luciferase activity was determined in Luminoskan™ Ascent Microplate Luminometer (Thermo Scientific, Waltham, MA, USA) by injecting 100 μl of assay buffer containing luciferin and measuring light emission for 10 s. Co-transfection with pRL-CMV (Promega, Madison, WI, USA), which expresses Renilla luciferase, was performed to enable normalization of data for transfection efficiency.
Measurement of cell viability by MTT assay
HeLa cells were seeded in 96-well plates and were pretreated for 24 h with dihydromyricetin at different concentrations (50–200 μM). At the end of the experiment, the media was removed and cells were cultured with MTT solution (5 mg/ml) [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (Sigma, St. Louis, MO, USA) for 4 h. The viable cells converted MTT to formazan, which generated a blue purple color after dissolving in 100 μl of DMSO. The absorbance at 570 nm was measured by Multiskan GO.
Western blot analysis
Treated HeLa cells were scraped off from the dish and lysed with lysis buffer (50 mM Tris–HCl, pH 7.5, 1 % Nonidet P-40, 1 mM EDTA, and 1 mM phenylmethylsulfonyl fluoride) supplemented with the protease inhibitor cocktail (Sigma, St. Louis, MO, USA). In certain experiments, the nuclear extracts were prepared using NE-PER nuclear and cytoplasmic extraction reagent. The protein concentrations were determined by Bradford assay. 50 μg of whole-cell extracts or 30 μg of nuclear extract protein per lane was separated by SDS–polyacrylamide gels and followed by transferring to a polyvinylidene fluoride membrane (Millipore, Bedford, MA, USA). The membrane was blocked with 5 % skim milk, and then incubated with the corresponding antibody. Proteins were probed with antibodies IκBα, phosphor (Ser32)-specific IκBα, p65, phosphor (Ser176)-IKKα/β, PARP, caspase-8 (Cell Signaling Technology, Beverly, MA, USA), cIAP-2, TRAF1, TRAF2, Bcl-2, cyclin D1, COX-2, VEGF, ICAM-1, RIP1, MMP-9, Topo-I (Santa Cruz Biotechnology, CA, USA) and α-tubulin (Sigma, St. Louis, MO, USA) overnight at 4 °C and then followed by secondary antibody-conjugated horseradish peroxidase (HRP) and detected by ECL solution according to the manufacturer’s instructions (Amersham Pharmacia Biotec, Buckinghamshire, UK).
Immunofluorescence assay
Cells were seeded into 24-well plates at 1 × 104 cells/well and pretreated with dihydromyricetin (200 μM) for 12 h, followed by treatment with 10 ng/ml TNF-α for 30 min. Cells pretreated with DMSO and 10 ng/ml TNF-α alone were used as negative and positive controls, respectively. Subsequently, cells were grown on coverslips to 80 % confluence and washed with PBS, fixed in fresh 4 % paraformaldehyde for 30 min followed by permeabilization in 0.2 % Triton X-100 at room temperature. Cells were blocked with 5 % BSA for 30 min and incubated overnight with the primary NF-κB p65 antibody at 4 °C, followed by incubation with Alexa Flour® 488 goat anti-rabbit lgG (H + L) for 30 min at room temperature. The cell nuclei were labeled with DAPI. The p65 protein appeared green under fluorescence microscopy and the nuclei appeared blue. The green and blue images were merged using Image J software to produce cyan fluorescence in areas of co-localization.
Reverse transcription-polymerase chain reaction (RT-PCR)
HeLa cells were grown to confluence in 6-cm2 culture plates, treated with the indicated concentrations of dihydromyricetin for 12 h and then stimulated with 10 ng/ml TNF-α for 12 h. Total RNA was isolated from cells using RNeasy Mini kits according to the manufacturer’s instructions (Qiagen, CA, USA). Complementary DNA was synthesized from 1 μg of total RNA in a 20 μl reverse transcription reaction mixture according to the manufacturer’s protocol (TaKaRa Bio, Kyoto, Japan). The following primer pairs were used for RT-PCR amplification: human VEGF, 5′-GCTCTACCTCCACCATGCCAA.
-3′ and 5′-TGGAAGATGTCCACCAGGGTC-3′; human iNOS, 5′-TTCTGTGCTAATGCGGAAGGT-.
3′ and 5′-GCTTCCGACTTTCCTGTCTCA-3′; human cyclin D1, 5′-CTGGCCATGAACTACCTGGA.
-3′ and 5′-GTCACACTTGATCACTCTGG-3′; GAPDH, 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′. GAPDH was used as an endogenous control to normalize input amount of each sample. PCR products were separated on 3 % agarose gel and visualized by ethidium bromide staining. Stained bands were visualized under UV light and photographed.
EdU labeling and immunofluorescence
The cells were seeded in 96-well culture plates. After 24 h, cells were treated with dihydromyricetin (200 μM) and incubated for 12 h. All cells were incubated with 5-ethynyl-2′-deoxyuridine (EdU, RIBOBIO; R11053) for 1 h and stained with Apollo®567 according to the manufacturer’s instruction. The stained cells were observed with Olympus IX83 inverted fluorescence microscope.
Apoptosis assays
Analyses for apoptosis were conducted with an Annexin V-FITC apoptosis detection kit (BD Biosciences, CA, USA) following the manufacturer’s instructions. Briefly, after incubation, cells were collected by centrifugation and resuspended in binding buffer (10 mM Hepes, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). The supernatant cells were incubated with Annexin V-FITC and PI for 15 min at room temperature in the dark. The samples were analyzed by flow cytometry using a FACScan flow cytometer. The CellQuest software was used to analyze the data (Becton–Dickinson).
Statistical analysis
All values are expressed as mean ± S.D. A comparison of the results was performed with one-way ANOVA and Tukey’s multiple comparison tests (Graphpad Software, Inc, San Diego, CA, USA). Statistically significant differences between groups were defined as p-values less than 0.05.
Results
Dihydromyricetin inhibits TNF-α-induced NF-κB-dependent reporter gene expression
In an effort to identify NF-κB inhibitors from natural resources, we have identified dihydromyricetin from A. grossedentata (Fig. 1a). To investigate the effect of dihydromyricetin on the induced NF-κB activation by TNF-α, we performed NF-κB reporter assay. After the cells were transiently transfected with the NF-κB-regulated luciferase reporter vector, the cells were further incubated with TNF-α in the presence of various concentrations of dihydromyricetin. We found that TNF-α-induced NF-κB reporter activity was substantially suppressed by dihydromyricetin in a dose-dependent manner (Fig. 1b). To evaluate the effect of dihydromyricetin on cell viability of HeLa cells, MTT assay was performed. Dihydromyricetin did not display significant cellular toxicity on HeLa cells up to 200 μM (Fig. 1c).
Dihydromyricetin inhibits TNF-α-induced phosphorylation and degradation of IκBα, phosphorylation and nuclear translocation of p65
The translocation of NF-κB to the nucleus is preceded by the phosphorylation, ubiquitination, and proteolytic degradation of IκBα [14]. To determine whether inhibition of TNF-α-induced NF-κB activation by dihydromyricetin is caused by inhibition of IκBα degradation, we exposed the HeLa cells to dihydromyricetin for 12 h and then treated them with TNF-α for 30 min. We then prepared cytoplasmic extracts and analyzed phosphorylation and degradation of IκBα by Western blot. Dihydromyricetin potently inhibited the TNF-α-induced phosphorylation and degradation of IκBα in a dose-dependent manner (Fig. 2a). Since the degradation of IκBα is known to cause the nuclear translocation of the p65 subunit of NF-κB, we next examined whether dihydromyricetin modulates TNF-α-induced nuclear translocation of p65. HeLa cells were preincubated with indicated concentrations of dihydromyricetin for 12 h and then treated with TNF-α (10 ng/ml) for 30 min. Nuclear extracts were analyzed for the presence of p65 with Western blot analysis. The results showed that dihydromyricetin also blocked TNF-α-induced nuclear translocation of p65 in a dose-dependent manner (Fig. 2b). To confirm these results, we performed immunofluorescence to observe p65 translocation in HeLa cells. The results indicated that in untreated cells, as well as dihydromyricetin (200 μM) pretreated followed by TNF-α treated cells, p65 was localized in the cytoplasm, whereas in cells treated with TNF-α alone, p65 was translocated to the nucleus (Fig. 2c). All of the results consistently support the idea that dihydromyricetin inhibits TNF-α-induced activation of NF-κB through inhibition of phosphorylation and degradation of IκBα, and then inhibition of p65 translocation to the nucleus.

Effect of dihydromyricetin (DMY) on the TNF-α-induced phosphorylation and degradation of IκBα, phosphorylation, and nuclear translocation of p65. a HeLa cells were preincubated with indicated concentrations of dihydromyricetin (DMY) for 12 h and then treated with TNF-α (10 ng/ml) for 30 min. Cytoplasmic extracts were analyzed by Western blot using indicated antibodies for p-IκBα, IκBα, and tubulin. b HeLa cells were preincubated with indicated concentrations of dihydromyricetin (DMY) for 12 h and then treated with 10 ng/ml TNF-α for 30 min. Nuclear extracts were analyzed by Western blot using indicated antibodies for p65 and Topo-I. c HeLa cells were incubated with 200 μM dihydromyricetin (DMY) for 12 h and followed by 10 ng/ml TNF-α stimulation for 30 min. After fixation, cells were stained with specific anti-p65 antibody followed Alexa Flour® 488, and the nucleus was counterstained with DAPI and examined by Fluorescence microscopy. Scale bars: 20 μm. Images were acquired for each fluorescence channel, using suitable filters with 40 × objective. The green and blue images were merged using J software
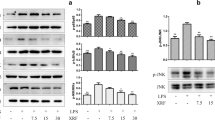
Dihydromyricetin inhibits TNF-α-induced phosphorylation of IKKα/β and the expression of adaptor protein cells of TNF receptor (TNFR) 1 signaling
Since IKK complex acts as a convergence point for a variety of activating signals for NF-κB and plays a critical role in degradation of IκB proteins, we investigated whether dihydromyricetin inhibits the TNF-α-induced phosphorylation of IKKα/β. As shown in Fig. 3a, dihydromyricetin suppressed TNF-α-induced phosphorylation of IKKα/β dose-dependently. In TNF-α-induced NF-κB activation, binding of TNF-α to TNFR1 induces receptor trimerization and recruitment of several downstream signaling proteins to their cytoplasmic domains [8]. Among them, TRAF2 and RIP1 play a critical role in the TNFR1 signaling leading to NF-κB activation. For this reason, we investigated whether dihydromyricetin affects the adaptor protein expression of TNFR1 signaling pathway. As shown in Fig. 3a, dihydromyricetin suppressed the expressions of TRAF2 and RIP1 dose-dependently.

Effect of dihydromyricetin (DMY) on TNF-α-induced phosphorylation of IKKα/β and expressions of adaptor proteins of TNF receptor (TNFR) 1 signaling. a HeLa cells were incubated with 200 μM dihydromyricetin (DMY) for 12 h and then incubated with 10 ng/ml TNF-α for 30 min. Cytoplasmic extracts were analyzed by Western blot using indicated antibodies for p-IKKα/β and tubulin. b HeLa cells were incubated with 200 μM DMY for 12 h and then incubated with 10 ng/ml TNF-α for 30 min. Cytoplasmic extracts were analyzed by Western blot using indicated antibodies for TRAF2, RIP1, and tubulin
Dihydromyricetin inhibits the expressions of TNF-α-induced NF-κB-regulated gene products
The expressions of VEGF, cyclin D1, and iNOS, which are involved in tumor cell proliferation, angiogenesis, metastasis and cycle, are known to be regulated by NF-κB. We examined whether dihydromyricetin can modulate the expression of these gene products induced by TNF-α. TNF-α significantly induced expression of VEGF, cyclin D1, and iNOS mRNA levels; however, the induced expression was blocked by dihydromyricetin in a dose-dependent manner (Fig. 4a). To investigate whether dihydromyricetin inhibits the induced expressions of NF-κB targets genes involved in early and late stages of aggressive cancers and inflammation, we analyzed TNF-α-induced expressions of MMP-9, cIAP-2, TRAF1, Bcl-2, cyclin D1, COX-2, ICAM-1, and VEGF. After HeLa cells were stimulated with 10 ng/ml TNF-α for 12 h in the presence or absence of dihydromyricetin, the induced expressions of anti-apoptotic proteins such as cIAP-2, TRAF1, and Bcl-2, protein involved in cell proliferation COX-2, and proteins involved in tumor angiogenesis, invasion, and metastasis ICAM-1 and VEGF, were analyzed by Western blot. Dihydromyricetin markedly suppressed TNF-α-induced expressions of all these proteins in a dose-dependent manner (Fig. 4b).

Effect of dihydromyricetin (DMY) on TNF-α-induced NF-κB-regulated gene products. a HeLa cells were preincubated with indicated concentrations of dihydromyricetin (DMY) for 12 h and then treated with 10 ng/ml TNF-α for an additional 12 h. RNA was isolated from cells, reverse-transcribed, and analyzed by RT-PCR for VEGF, cyclin D1, and iNOS. GAPDH was used to show equal loading of total RNA. b HeLa cells were incubated with 200 μM dihydromyricetin (DMY) for 12 h and then incubated with 10 ng/ml TNF-α for an additional 12 h. Whole-cell extracts were analyzed by Western blot using indicated antibodies for MMP-9, cIAP-2, TRAF1, Bcl-2, cyclin D1, COX-2, ICAM-1, VEGF, and tubulin
Dihydromyricetin inhibits the proliferation of cells
EdU is a thymidine analog that is incorporated into replicated chromosomal DNA during the S phase of the cell cycle. The EdU incorporation assay was performed to detect whether dihydromyricetin could affect the number of proliferating cells. Following 12-h treatment, dihydromyricetin (200 μM) exerted inhibition of the number of EdU-positive cells in HeLa and MDA-MB-231 cells. This indicated that dihydromyricetin inhibited the proliferation of HeLa and MDA-MB-231 cells in vitro (Fig. 5). Meanwhile, dihydromyricetin showed little inhibition effect on 293 and HBL-100 cells. This phenomenon indicated that dihydromyricetin may have less damage on normal cells.

Effect of dihydromyricetin (DMY) on the proliferation of cells. HeLa, MDA-MB-231, HBL-100, and 293 cells were incubated for 12 h in the presence or absence of dihydromyricetin (DMY, 200 μM). The EdU-labeled replicating cells were examined under a fluorescence microscope. Data represented as mean ± standard deviation of three independent experiments. **p < 0.01, significantly different when compared with control group
Dihydromyricetin potentiates TNF-a-induced apoptosis
Since TNF-α-induced expression of anti-apoptotic genes was downregulated by dihydromyricetin, we examined whether dihydromyricetin enhances apoptosis induced by TNF-α. Dihydromyricetin potentiated TNF-α-induced apoptosis, as assessed by Annexin V/PI double staining. As shown in Fig. 6a, treatment of HeLa cells with vehicle only, TNF-α alone, and dihydromyricetin alone induced apoptosis to 3.4, 10.0, and 18.1 %, respectively. However, combined treatment of the cells with TNF-α and dihydromyricetin resulted in a significant increase in the Annexin V-positive cell population (35.8 %). Since caspases are a group of cysteine proteases critical for apoptosis of eukaryotic cells [14], we investigated whether dihydromyricetin affects TNF-α-induced activation of caspase-8. TNF-α alone slightly and dihydromyricetin alone significantly affected the activation of caspase-8, whereas cotreatment with TNF-α and dihydromyricetin potentiated their activation, as indicated by the presence of cleaved caspase-8 (Fig. 6b, top panel). We also used the PARP cleavage assay to detect TNF-α-induced apoptosis. Again, dihydromyricetin potentiated the effect of TNF-α-induced PARP cleavage (Fig. 6b, middle panel). These results showed that dihydromyricetin enhances the apoptotic effects of TNF-α.

Effect of dihydromyricetin (DMY) on the TNF-α-induced apoptosis. a HeLa cells were pretreated with 200 μM dihydromyricetin (DMY) for 12 h and then incubated with 10 ng/ml TNF-α for 12 h, and subsequently stained with Annexin V-FITC and PI, followed by analysis using a flow cytometer. Representative plots of one set of triplicate experiments. Early apoptotic cell (Annexin V+ and PI−) were displayed in the lower right quadrant and late apoptotic cells (Annexin V+ and PI+) were shown in the upper right quadrant. b HeLa cells were pretreated with 200 μM dihydromyricetin (DMY) for 12 h and then incubated with 10 ng/ml TNF-α for 12 h. Whole-cell extracts were analyzed by Western blot using indicated antibodies for cleaved caspase-8, cleaved PARP, and tubulin
Discussion
Ampelopsis grossedentata was widely utilized not only as a healthy tea but also as a medicinal herb in traditional Chinese medicine to cure diseases including pharyngitis, sore throat, cold-related fever, and allergenic skin disease. Dihydromyricetin, derived from the Ampelopsis grossedentata, is a major active ingredient of flavonoid compounds. In this study, we identified dihydromyricetin as a potent inhibitor of TNF-α-induced NF-κB activation and further investigated how this compound suppressed NF-κB activation. Activation of NF-κB is induced by activation of IκB kinase (IKK) complex, which phosphorylates IκB, leading to its degradation and translocation of NF-κB to the nucleus, where it binds with DNA and activates the transcription of target genes [15–17]. Thus, it was suggested that degradation of IκB by the proteasome is an essential early step in the NF-κB activation pathway. In the present study, we demonstrated that dihydromyricetin suppressed not only the expression of RIP1 and TRAF2, the adaptor proteins of TNF receptor but also the TNF-α-induced phosphorylation of IKKα/β. This, in turn, inhibits the phosphorylation and degradation of IκBα which is required for NF-κB dimerization and maximal activation of transcription. The key regulatory step in the IκBα degradation involves the activation of IKK complex. Activation of IKK is mediated by phosphorylation through various upstream kinases including transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1), NF-κB inducing kinase, Akt or other kinases [18]. It is well known that the TNFR1 interacts with the signaling protein TNFR1-associated death domain protein (TRADD), which recruits the adaptor proteins, TRAF2, RIP1 and Fas-associated protein with death domain (FADD) [19–21]. Subsequently, TRAF2 and RIP1 are polyubiquitinated and induce the recruitment of upstream kinase such as TAK1 to activate IKK complex [22]. Therefore, our results suggest that the suppression of TRAF2 and RIP1 by dihydromyricetin could, at least in part, be responsible for the inhibition of upstream kinase in the activation of IKK complex, thereby repressing NF-κB signaling.
Caspases are a family of cysteine-aspartic proteases, which play an essential role in apoptosis. Poly (ADP-ribose) polymerase (PARP), a family of proteins involved in a variety of cellular processes including DNA repair and programmed cell death, is a downstream target of caspase-3 [23, 24]. Studies have shown that anti-apoptotic activity of NF-κB involves the inhibition of TNF-α-induced apoptosis through induction of a variety of anti-apoptotic proteins [3, 25]. We therefore tested the expressions of c-IAP2, TRAF1, and Bcl-2, all of which are important members involved in apoptosis, through Western blot after dihydromyricetin treatment. TNF-α binding to its receptor induces not only anti-apoptotic effects through the regulation of activation NF-κB-dependent genes for cell survival but also apoptotic effects via activation of caspases [9, 26]. The results confirmed that dihydromyricetin inhibits the TNF-α-induced expressions of these proteins. Furthermore, flow cytometric analysis with Annexin V staining indicated that dihydromyricetin potentiated TNF-α-induced apoptosis significantly via activation of caspase-8 and PARP.
NF-κB can regulate more than 150 genes, including those involved in cell proliferation, tumorigenesis, tumor cell invasion, angiogenesis, and cell cycle. COX-2 is also overexpressed in various types of cancer and involved in cellular proliferation, anti-apoptotic activity, angiogenesis, and an increase of metastasis [27]. ICAM-1 is known to be a major mediator of tumor cell invasion. Cyclin D1 exercises powerful control over the mechanisms that regulate the mitotic cell cycle. Our results showed that dihydromyricetin suppresses not only the expressions of COX-2, ICAM-1, and cyclin D1 but also the expressions of MMP-9 and VEGF, both of which have been shown to be expressed in response to NF-κB activation, and are known to be major mediators of tumor cell invasion and angiogenesis. iNOS is an enzyme that produces NO (nitric oxide), a molecule that acts as a signal for a variety of cellular functions throughout the body, including the triggering of inflammation and tumorigenesis [28]. The inhibition of the expression of iNOS by dihydromyricetin may be associated with inhibition of tumor cell proliferation as well.
In summary, our results suggest that dihydromyricetin could exhibit anti-proliferative, proapoptotic, anti-invasive, antiangiogenic, and anti-inflammatory effects through the suppression of NF-κB activation and NF-κB-regulated gene products. Our data indicate that dihydromyricetin may be useful for the development of alternative pharmacological strategies aimed at reducing the inflammatory process.
Abbreviations
- NF-κB:
-
Nuclear factor-κB
- IκBα:
-
Inhibitor of NF-κB alpha
- IKK:
-
IκB kinase
- TRAF2:
-
TNF receptor-associated factor 2
- RIP1:
-
Receptor-interacting protein 1
- Topo-I:
-
Topoisomerase-I
- TNF-α:
-
Tumor necrosis factor alpha
- MMP-9:
-
Matrix metalloproteinase-9
- iNOS:
-
Inducible nitric oxide synthase
- c-IAP2:
-
Cellular inhibitor of apoptosis-2
- Bcl-2:
-
B-cell lymphoma 2
- COX-2:
-
Cyclooxygenase-2
- ICAM-1:
-
Inter-cellular adhesion molecule-1
- VEGF:
-
Vascular endothelial growth factor
References
Sen R, Baltimore D (1986) Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 46:705–716
Hayden MS, Ghosh S (2008) Shared principles in NF-kappaB signaling. Cell 132:344–362. doi:10.1016/j.cell.2008.01.020
Hoesel B, Schmid JA (2013) The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer 12:86. doi:10.1186/1476-4598-12-86
Kumar A, Takada Y, Boriek AM, Aggarwal BB (2004) Nuclear factor-kappaB: its role in health and disease. J Mol Med (Berl) 82:434–448. doi:10.1007/s00109-004-0555-y
Karin M, Cao Y, Greten FR, Li ZW (2002) NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer 2:301–310. doi:10.1038/nrc780
Nakanishi C, Toi M (2005) Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer 5:297–309. doi:10.1038/nrc1588
Aggarwal BB (2003) Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol 3:745–756. doi:10.1038/nri1184
Hsu H, Xiong J, Goeddel DV (1995) The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 81:495–504
Hsu H, Shu HB, Pan MG, Goeddel DV (1996) TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 84:299–308
Schneider-Brachert W, Tchikov V, Neumeyer J, Jakob M, Winoto-Morbach S, Held-Feindt J, Heinrich M, Merkel O, Ehrenschwender M, Adam D, Mentlein R, Kabelitz D, Schutze S (2004) Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity 21:415–428. doi:10.1016/j.immuni.2004.08.017
Chen T, Zhu S, Lu Y, Cao H, Zhao Y, Jiang G, Zhu L, Lu T (2012) Probing the interaction of anti-cancer agent dihydromyricetin with human serum albumin: a typical method study. Anticancer Agents Med Chem 12:919–928
Wu S, Liu B, Zhang Q, Liu J, Zhou W, Wang C, Li M, Bao S, Zhu R (2013) Dihydromyricetin reduced Bcl-2 expression via p53 in human hepatoma HepG2 cells. PLoS One 8:e76886. doi:10.1371/journal.pone.0076886
Hwangbo C, Kim J, Lee JJ, Lee JH (2010) Activation of the integrin effector kinase focal adhesion kinase in cancer cells is regulated by crosstalk between protein kinase calpha and the PDZ adapter protein mda-9/Syntenin. Cancer Res 70:1645–1655. doi:10.1158/0008-5472.CAN-09-2447
Wang J, Lenardo MJ (2000) Roles of caspases in apoptosis, development, and cytokine maturation revealed by homozygous gene deficiencies. J Cell Sci 113(Pt 5):753–757
Baeuerle PA, Lenardo M, Pierce JW, Baltimore D (1988) Phorbol-ester-induced activation of the NF-kappa B transcription factor involves dissociation of an apparently cytoplasmic NF-kappa B/inhibitor complex. Cold Spring Harb Symp Quant Biol 53(Pt 2):789–798
Ghosh S, Baltimore D (1990) Activation in vitro of NF-kappa B by phosphorylation of its inhibitor I kappa B. Nature 344:678–682. doi:10.1038/344678a0
Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S (1995) Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes Dev 9:2723–2735
Viatour P, Merville MP, Bours V, Chariot A (2005) Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci 30:43–52. doi:10.1016/j.tibs.2004.11.009
Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV (1996) TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 4:387–396
Stanger BZ, Leder P, Lee TH, Kim E, Seed B (1995) RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell 81:513–523
Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM (1995) FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 81:505–512
Chen ZJ (2012) Ubiquitination in signaling to and activation of IKK. Immunol Rev 246:95–106. doi:10.1111/j.1600-065X.2012.01108.x
Burkle A, Brabeck C, Diefenbach J, Beneke S (2005) The emerging role of poly(ADP-ribose) polymerase-1 in longevity. Int J Biochem Cell Biol 37:1043–1053. doi:10.1016/j.biocel.2004.10.006
Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL (2006) Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci USA 103:18314–18319. doi:10.1073/pnas.0606528103
Karin M, Greten FR (2005) NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 5:749–759. doi:10.1038/nri1703
Chen G, Goeddel DV (2002) TNF-R1 signaling: a beautiful pathway. Science 296:1634–1635. doi:10.1126/science.1071924
Kipanyula MJ, Seke Etet PF, Vecchio L, Farahna M, Nukenine EN, Nwabo Kamdje AH (2013) Signaling pathways bridging microbial-triggered inflammation and cancer. Cell Signal 25:403–416. doi:10.1016/j.cellsig.2012.10.014
Chiang YM, Lo CP, Chen YP, Wang SY, Yang NS, Kuo YH, Shyur LF (2005) Ethyl caffeate suppresses NF-kappaB activation and its downstream inflammatory mediators, iNOS, COX-2, and PGE2 in vitro or in mouse skin. Br J Pharmacol 146:352–363. doi:10.1038/sj.bjp.0706343
Acknowledgments
This work was partially supported by the National Natural Science Foundation of China, No. 81360496. This study also received assistance from Jilin Province Science and Technology Development Plan item (20150101229JC) and Project of Education Department of Jilin Province 2016 (281).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts interest
The authors declare that there are no financial conflicts of interest in regard to this work.
Rights and permissions
About this article

Cite this article
Tang, N., Ma, J., Wang, K.S. et al. Dihydromyricetin suppresses TNF-α-induced NF-κB activation and target gene expression. Mol Cell Biochem 422, 11–20 (2016). https://doi.org/10.1007/s11010-016-2799-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-016-2799-6