
Abstract
Aim
The purpose of this study was to determine if acute nicotine attenuated ketamine-induced regional cerebral blood flow (rCBF).
Method
Following 2–4 h of nicotine abstinence, healthy chronic smokers participated in four sets of rCBF studies, H 152 O positron emission tomography, during a simple sensory motor control task. The four drug conditions studied were placebo, ketamine alone, nicotine alone, and ketamine + nicotine.
Results
Intravenous ketamine increased rCBF in frontal, orbital–frontal, and anterior cingulate areas. Nicotine alone induced marked rCBF elevations in the lateral occipital cortex and rCBF suppressions in the basal ganglia and anterior cingulate cortex. Nicotine added to ketamine attenuated the ketamine-induced elevated rCBF in the anterior cingulate cortex but caused a marked rCBF increase in the orbital frontal region.
Conclusion
This study illustrates the interactive effects of ketamine, an NMDA receptor antagonist, and nicotine in multiple brain regions. Nicotine substantially ameliorated the effects of ketamine on anterior cingulate rCBF and, when given alone, markedly suppressed anterior cingulate rCBF. The enhanced, synergistic orbitofrontal effects observed with ketamine and nicotine together suggest a marked increase in excitatory neurotransmission in a brain region often linked to psychosis, reward, and addictive behaviors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Accumulating evidence indicates that hypofunction of the glutamate N-methyl-d-aspartate receptor (NMDAR) is involved in the pathophysiology of schizophrenia (Coyle et al. 2002; Krystal et al. 2003;Olney et al. 1999; Tamminga and Holcomb 2005). NMDAR antagonists, such as phencyclidine (PCP) and ketamine, induce positive, negative, and cognitive symptoms in healthy adult humans similar to that observed in schizophrenia (Krystal et al. 1994; Newcomer et al. 1999; Rowland et al. 2005a,b; Rowland 2005). Ketamine exacerbates existing or dormant symptoms in patients with schizophrenia (Lahti et al. 1995a,b; Malhotra et al. 1997). In nonhuman animal research, NMDAR antagonists induce behavioral alterations (Jentsch et al. 1997) and neurochemical changes (Adams et al. 2002; Gao et al. 1993) that translate well to features of schizophrenia. Hence, NMDAR antagonism appears to be a good model of schizophrenia and provides a framework to test novel drug agents for the treatment of schizophrenia.
Glutamatergic and cholinergic systems, specifically the NMDA and nicotinic receptors, interact in a complex manner (Deutsch et al. 2003; McGehee et al. 1995; Toth et al. 1992). Nicotine has been shown to increase frontal extracellular glutamate concentrations through the stimulation of α4β2 nicotinic receptors (Gioanni et al. 1999). Animal studies show that nicotine ameliorates some behavioral and electrophysiological (Cromwell and Woodward 2007) effects of NMDAR antagonism (Levin et al. 1996; Tizabi et al. 1998). As such, drugs that target the nicotinic system may be good candidates to test with the NMDAR hypofunction model (Buccafusco and Terry 2009). This is supported by the implication that the nicotinic system is involved in the pathophysiology of schizophrenia. Patients exhibit a higher smoking rate, heavier smoking patterns, and extract more nicotine when smoking than the general population (Dalack et al. 1998; Strassnig et al. 2006). Postmortem studies have revealed decreased nicotinic receptor number in brains of patients with schizophrenia (Ripoll et al. 2004). Acute nicotine administration has been shown to improve negative symptoms (Smith et al. 2002) and various cognitive impairments (Depatie et al. 2002; Harris et al. 2004; Murphy and Glanzman 1999; Smith et al. 2002) in patients with schizophrenia. In another study of non-psychiatric subjects, chronic smokers and nonsmokers exhibited markedly different electrophysiological responses to intravenous ketamine (Knott et al. 2006). These studies point toward important relationships between nicotine and glutamate interactions, especially as they relate to negative symptoms and cognition.
Nicotine’s action on local cerebral blood flow in human studies has demonstrated consistent results (Domino et al. 2000; London 1990; Stapleton et al. 2003; Zubieta et al. 2005). Its administration to chronic smokers reliably enhances blood flow in visual cortex and suppresses flow to the anterior cingulate cortex. In marked contrast, intravenous ketamine, which elicits robust glutamate release, promotes blood flow elevations in anterior cingulate, orbital frontal, and dorsal frontal regions (Breier et al. 1997; Holcomb et al. 2005; Rowland 2005; Vollenweider et al. 1997).
The purpose of this study was to determine if nicotine attenuated the ketamine-induced regional cerebral blood flow (rCBF) alterations in healthy humans (Holcomb et al. 2001). We hypothesized that acute nicotine would normalize the frontal rCBF elevations and diminish psychomimetic behaviors associated with NMDAR antagonism. Nicotine alone was expected to induce those patterns described above. But because nicotine’s actions are diverse, we also expected to observe nicotine’s indirect actions through its stimulation of dopamine release in the basal ganglia. Whereas ketamine is expected to precipitate glutamate release by blocking NMDA receptors on inhibitory GABA interneurons, dopamine released secondary to nicotine action is expected to inhibit glutamate release. This dynamic interaction, one drug-promoting glutamate release and the other suppressing it, was the focus of this study.
This study is unique in providing brain activity information arising from the interaction between glutamatergic and cholinergic systems, which are especially important to schizophrenia and drug abuse research.
Methods
The University of Maryland School of Medicine Human Institutional Review Board and the Johns Hopkins Joint Committee on Clinical Investigation approved this study.
Subjects
Nine healthy subjects (five female and four male; mean age, 30.8; right-handed; chronic smokers for 10 years or longer, five cigarettes per day or more) participated in this study. Inclusion/exclusion criteria were as follows: (1) no past or present psychiatric disorder as determined with the Structured Clinical Interview for Diagnostic and Statistical Manual of Mental Disorders, fourth edition, Non-Patient Version (Spitzer et al. 1992); (2) no first-degree relatives with a diagnosis of a psychotic disorder; (3) no current medical illnesses as determined with a physical exam and laboratory tests; and (4) no previous exposure to ketamine or PCP. All subjects gave written informed consent prior to the study and were paid for their participation.
Task description
A simple visual task, with low cognitive demand, was employed (Fig. 1). Pilot testing showed that ketamine did not disrupt accuracy or response time on this task. We expected the low demand task to provide a common behavioral state among subjects. The potential confound of performance-related rCBF activity differing across the drug conditions was minimized. For this task, subjects indicated by right- or left-hand button press the displacement side of a rectangular block on the computer screen when the “test image” was presented following a cueing image. The hand used to press the button was congruent with the direction of the displaced object’s location. The number of right and left trials were balanced within a given scan. Accuracy and reaction time were recorded for each trial.

Subjects were required to press a button in the right or left hand to indicate the displacement side of the rectangle
Ketamine screening
Prior to scanning, all subjects underwent a ketamine test infusion to ensure drug tolerability. Subjects performed variations of the visual task during placebo and ketamine conditions. A test dose of nicotine spray (0.5 mg in each nostril) was administered 20 min after the ketamine infusion was stopped. This helped to demonstrate the subject’s tolerance of the intranasal nicotine. Blood pressure and heart rate were monitored throughout the session.
PET scanning
Positron emission tomography (PET) procedures were conducted at the Johns Hopkins University Cyclotron Facility. We studied participants on two separate days and used radiolabeled water (H 152 O) to measure rCBF (Herscovitch et al. 1983; Raichle et al. 1983). Subjects fasted overnight and refrained from smoking for at least 2 h (2–4 h) before the study. Tobacco withdrawal symptoms were not assessed prior to the PET scans. Subjects also abstained from alcohol, caffeine, and medications overnight prior to the study. PET procedures were performed on a GE Advance Tomograph (Waukesha, WI, USA) in 3D acquisition mode. Catheters were placed in the antecubital veins of both arms: one for isotope injection and drug level blood sampling and one for i.v. ketamine/saline infusions. Transmission scans were obtained prior to tracer infusions to provide attenuation correction information; 12 mCi of H 152 O were injected per scan. Task performance began 20 s before isotope infusion and continued throughout each scan, which lasted 90 s. Scans were separated by 7 to 8 min except when nicotine was given. On those occasions, the interscan interval was approximately 12 min.
Placebo (saline), nicotine, ketamine, and ketamine + nicotine conditions were assessed (Fig. 2). Four PET blood flow scans were obtained in association with each drug condition. In the analyses discussed here, only two scans from each drug condition are considered. This is due to the fact that only two of the four drug condition scans were associated with a control behavior. The other two scans were associated with a match-to-sample task that required working memory and are not presented.

The number of subjects first given PET session 1 was balanced with those given session 2 initially. The entire PET session lasted about 80 min
During one session subjects received placebo infusion followed by intranasal nicotine administration. During the other session, subjects received ketamine infusion followed by intranasal nicotine spray. Session order, placebo-first, or ketamine-first session was pseudo-randomized and counterbalanced across subjects. Ketamine was administered in a double-blinded fashion. Ketamine was administered with a computerized infusion pump starting with a loading dose of 0.2 mg/kg over 10 min followed by a maintenance dose of 0.4 mg kg−1 h−1. After four ketamine scans were obtained, subjects were removed from the scanner and given nicotine intranasal spray (0.5 mg/nostril = 1.0 mg total); scanning resumed immediately afterward. Psychiatric assessment was completed at baseline, after the ketamine loading dose, during ketamine steady-state infusion, post nicotine, and during recovery. The rating scale consisted of the Brief Psychiatric Rating Scale (Overall and Gorham 1962). Blood samples for ketamine level determination were collected at the same time as the ratings. A second transmission PET scan was acquired at the end of each day’s session to provide additional attenuation information for image reconstruction.
Behavioral assessment
To determine if participants experienced schizophrenia-like features associated with ketamine administration, a 2 (drug group: ketamine and placebo) × 5 (time: baseline, loading, steady-state, post-nicotine, and recovery) repeated measures analysis of variance (ANOVA) was performed for the BPRS total rating scores and positive and negative subscale scores. When appropriate, significant findings were investigated with follow-up tests (Table 1).
PET data analysis
All scans were realigned and spatially normalized into the stereotactic space of Talairach and Tournoux (1988). Images were smoothed with a full width at half maximum of 10 × 10 × 10 mm in the x, y, and z planes. Pixel rCBF values were scaled using the ratio adjustment method. The image data were analyzed using Statistical Parametric Mapping (SPM99; Welcome Department of Cognitive Neurology, London, England), where voxel by voxel comparisons determined significant changes in rCBF between the drug conditions (p ≤ 0.01, corrected at the cluster level). The significance of each cluster of activated voxels was based on the magnitude of activation (z = 2.36) and spatial extent (corrected, p ≤ 0.05, or uncorrected at 0.005; Friston et al. 1994, 1996; Poline et al. 1997).
Results
Behavioral and psychiatric rating scales
Three 2 (drug group: ketamine, placebo) × 5(time: baseline, loading, steady state, post-nicotine, and recovery) repeated measures ANOVAs were performed for the BPRS total rating scores and positive and negative symptom subscale scores. Results revealed significant interactions for the total score [F (1, 8) = 7.0, p < 0.05] and negative subscale [F (1, 8) = 11.7, p < 0.05] and a trend interaction for the positive subscale [F(1, 8) = 3.8, p < 0.06]. These scores increased during the ketamine loading dose and steady-state time points and decreased following nicotine administration, but these scores did not change in the placebo condition. Follow-up tests revealed a significant difference between ketamine and placebo conditions during the loading dose time point [total, t (8) = 3.1, p < 0.05; negative symptoms, t (8) = 3.6, p < 0.05]. Measurements taken at the steady-state time points approached trend significance [total, t(8) = 1.8, p = 0.1; negative, t(8) = 2.3, p = 0.051]. There were no significant differences on psychiatric ratings between the ketamine + nicotine and placebo + nicotine conditions. This suggests that nicotine may attenuate the psychotomimetic reaction associated with ketamine administration. Means and standard deviations (SD) are presented in Table 2. No subject reported adverse effects or recreational ketamine-like substance use during follow-up phone contact.
The visual task elicited similar accuracy and response times across all four conditions. The accuracy ranged from 97.5% to 100%, and the reaction times ranged from 731 to 748 ms.
Ketamine and nicotine blood levels
Ketamine plasma levels were not detectable at baseline but rose to 65 ± 24 ng/ml (mean ± SD) following the loading dose. At the mid-point of the study, immediately prior to nicotine administration, the level averaged 88 ± 22 ng/ml, and at the completion of the nicotine + ketamine phase, the blood level averaged 110 ± 29 ng/ml. Nicotine blood levels did not change significantly across conditions. In the four conditions, the blood levels ranged from 5.2 to 6.5, ±0.5 ng/ml.
PET data
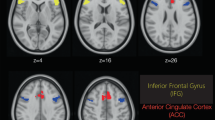
Regional activations are described in terms of Brodmann areas from the Talairach atlas (Talairach and Tournoux 1988). Detailed activation results are shown in Table 2 and Fig. 3. Increased activity associated with ketamine, relative to placebo, was observed primarily in ventral frontal brain regions. Ketamine caused bilateral rCBF activation in the superior (BA 8/10/11/21), middle (BA 11), and inferior frontal (BA 47) regions as well as the insula. In the right hemisphere, orbitofrontal (BA 11) and anterior cingulate (BA 32) regions were increased. In the left hemisphere, middle temporal (BA 21) regions were increased. Decreased activity with ketamine, relative to placebo, was observed in bilateral cerebellum and hippocampal/parahippocampal systems.

Regional cerebral blood flow maps were generated by various pharmacological contrasts. Whereas nicotine’s actions are predominantly suppressive and ketamine’s predominantly enhancing, the two drugs together caused a marked elevation in OFC rCBF
Nicotine, relative to placebo, evoked increased rCBF activity bilaterally in the inferior occipital region (BA 18). Decreased rCBF activity was observed in the right medial frontal (BA 8/32), right anterior cingulate (BA 24), left inferior frontal (BA 47), ventral striatum, left caudate/putamen, and left cerebellum.
Ketamine + nicotine, relative to placebo, resulted in increased activity in the bilateral orbitofrontal (BA 11), right superior frontal (BA 9/10), bilateral superior temporal (BA 22/38), and right inferior temporal (BA 20). Decreased activity was observed in bilateral cerebellum, amygdala, precentral gyrus, and anterior cingulate.
Discussion
The purpose of this study was to determine whether acute nicotine administration diminishes the neural and behavioral effects of intravenous ketamine. Because ketamine induces psychomimetic, “schizophrenia” symptoms, by eliciting asynchronous and elevated glutamatergic output, (Krystal et al. 2003; Moghaddam 2003), and because subjects with schizophrenia are disproportionately addicted to cigarettes, we asked whether nicotine might ameliorate some of ketamine’s actions. The results show that nicotine affects ketamine’s neural actions in two disparate ways. First, it reverses and normalizes the neural hyperactivity of the rostral and dorsal anterior cingulate cortex. Second, it enhances excitatory activity in the orbital frontal cortex. The study does not, however, permit a clear assessment of nicotine’s effect on ketamine-induced behavioral changes. Although BPRS total and subscale scores diminished after nicotine was added during the ketamine infusion, this may have occurred coincidentally due to the time it was given. Psychomimetic symptoms tend to resolve with time even when ketamine elicited blood flow changes are still present and ketamine blood levels are still high (Newcomer et al. 1999). Nonetheless, this study is noteworthy in showing robust nicotine–ketamine interactions in the orbital frontal cortex (OFC) and the anterior cingulate cortex, two regions that are extensively implicated in addiction and psychiatric illness.
When given alone, intranasal nicotine administration induced marked rCBF suppression throughout the rostral and dorsal anterior cingulate cortex, the ventral striatum and the globus pallidus. The only area that showed enhanced activity with nicotine was the lateral occipital cortex. In marked contrast with the suppressant actions of nicotine, ketamine elicited widespread rCBF elevations, especially in the orbital frontal and anterior cingulate cortices. Although noteworthy, these individual changes have been described by investigators previously (Breier et al. 1997; Holcomb et al. 2001; Vollenweider et al. 1997). The interactions between nicotine and ketamine, however, have only been studied in experimental animal models (Fu et al. 2000; Rodvelt et al. 2008), not in human subjects
Ketamine + nicotine induces greater rCBF elevations in the OFC than ketamine alone. The following discussion briefly considers how nicotine, ketamine and the two drugs combined might alter glutamate release, the crucial determinant of local blood flow in the brain (Patel et al. 2003; Rothman et al. 1999; Shulman et al. 2002). The OFC, anterior cingulate, and striatum are emphasized due to their well-documented roles in addiction, reward, psychiatric illness, and planning behaviors.
Replicated by multiple investigators, nicotine’s suppressant action on local brain blood flow has been documented over the last 10 years (Domino et al. 2000; Ghatan et al. 1998; Rose et al. 2003; Stapleton et al. 2003; Zubieta et al. 2005). Nicotine lowered rCBF in the anterior cingulate, orbital frontal cortex, and ventral striatum/nucleus accumbens. Although an extensive body of work has demonstrated nicotine’s pharmacological actions on dopamine and glutamate release (Fu et al. 2000; Jones and Wonnacott 2004; Lambe et al. 2003; Mansvelder and McGehee 2000; Reid et al. 2000), neuroimaging studies with nicotine have not offered mechanistic models to suggest how and why nicotine would suppress rCBF in OFC, cingulate, and striatal regions. There is now overwhelming evidence for glutamate’s primary role in local vascular regulation and neuroimaging signal generation (Patel et al. 2003; Rothman et al. 1999; Shulman et al. 2002). This consensus prompts us to hypothesize how nicotine and ketamine interact to modify glutamate release.
Nicotinic stimulation of presynaptic cholinergic receptors located on glutamatergic afferents in the ventral tegmental area elicits robust dopamine release in the ventral striatum and frontal cortex (Jones and Wonnacott 2004; Mansvelder and McGehee 2000). Dopaminergic modulation of glutamatergic networks in forebrain regions may inhibit (Seamans and Yang 2004) excitatory transmission. This may occur by direct inhibitory dopaminergic action on pyramidal neurons (Bandyopadhyay and Hablitz 2007; Mair and Kauer 2007) or by dopamine’s activation of GABA interneurons, which subsequently inhibit pyramidal neurons (Tseng and O’Donnell 2007).
Ketamine, an NMDAR antagonist, promotes elevated blood flow and glucose metabolism secondary to enhanced glutamate release throughout the human frontal cortex (Breier et al. 1997; Holcomb et al. 2005; Vollenweider et al. 1997). For unknown reasons, ketamine-induced glutamate release is particularly robust in the OFC and ventral cingulate cortices. NMDAR antagonism has a potent effect on GABA inhibitory control of glutamatergic pyramidal neurons. By antagonizing the NMDARs on inhibitory neurons ketamine indirectly promotes excitatory neurotransmission (Farber 2003; Moghaddam 2003). The resulting elevated, asynchronous glutamatergic activity is apparently associated with diminished cognitive abilities and a worsening of psychotic symptoms in subjects with schizophrenia and induced psychotic and cognitive symptoms in healthy volunteers (Krystal et al. 1994; Lahti et al. 1995a,b; Newcomer et al. 1999).
Our study measured changes in local cerebral blood flow in subjects treated with ketamine who were given nicotine 40 min after starting the ketamine infusion. Nicotine, therefore, acted on the central nervous systems of subjects with altered glutamatergic activity. Nicotine administration may have further enhanced dopaminergic and glutamatergic release in the OFC. In this “activated” state, dopamine may have contributed to greater glutamatergic transmission. It may have amplified the state initially created by ketamine. These mixed neurotransmitter actions may have contributed to an intensely active glutamatergic state in the OFC. A previous, unpublished ketamine continuous infusion study of three healthy volunteers, by our group, followed rCBF responses out to 55 min. That pilot study did not find elevations in the OFC beyond the first 30 min of the infusion. It is, therefore, extremely unlikely that the OFC’s response to ketamine–nicotine together is a “late” response to ketamine alone.
The rostral and dorsal anterior cingulate response to nicotine, during ketamine infusion, did not reflect greater glutamate transmission. Instead, the anterior cingulate had a reduction in rCBF. This suggests that nicotine may be especially potent in its inhibitory action in the anterior cingulate cortex. It also suggests that nicotine may be particularly helpful in reversing some of the attentional and memory deficits induced by NMDAR antagonism (Buccafusco and Terry 2009).
Nicotine’s suppression of glutamate release in the anterior cingulate occurs in conjunction with robust glutamate suppression in the basal ganglia. This dual suppression pattern may be an important indicator of what the person addicted to nicotine is “seeking” from her smoking habit. To the extent that cortico-striate glutamate neurotransmission is controlled by nicotine, it may provide the smoker with a way to modulate her mood and anxiety state, and this may be an important reason why those with schizophrenia are especially heavy nicotine users. Excessive glutamate release may sustain schizophrenic symptoms, and nicotine acquired from cigarettes may help modulate those symptoms by inhibiting glutamate transmission in limbic cortex. In kind, the effectiveness of antipsychotic medication may depend, in part, on suppression of glutamate release in the anterior cingulate cortex (Holcomb et al. 1996; Lahti et al. 2009).
NMDAR antagonism may promote greater dopamine release in some brain regions than in others (Kosowski and Liljequist 2004; Lorrain et al. 2003). If ketamine enhances dopamine and glutamate release in the OFC more than in the anterior cingulate, then nicotine’s addition may substantially facilitate dopamine’s excitatory actions in the OFC. Clarity on this question will only come through animal and neuroimaging studies specifically designed to assess metabolite changes in this region under multiple drug treatment combinations created across a range of temporal epochs.
The shortcomings of this study are important. First, the results are preliminary. Data must be acquired on more subjects to substantiate these findings. Second, because subjects were given the ketamine–nicotine only when in the PET scanning apparatus, it was difficult to evaluate their subjective drug response. Furthermore, nicotine was given after ketamine’s most robust behavioral actions (evident during the first 20 min) had subsided. This makes it impossible to know to what extent nicotine might directly antagonize ketamine’s psychomimetic actions. The randomization was incomplete. Ketamine was not given following a placebo scan, but nicotine was. This may make the comparison between placebo and nicotine more accurate than the comparison between ketamine and placebo.
The study does, however, draw attention to a fundamentally important interaction between dopamine, glutamate, and nicotine in the OFC and cingulate cortices. In light of the extensive body of work associated with these structures in the behavioral/psychiatric literature, it is reasonable to pursue additional studies that control for time of administration and the environment of the intervention.
References
Adams BW, Bradberry CW, Moghaddam B (2002) NMDA antagonist effects on striatal dopamine release: microdialysis studies in awake monkeys. Synapse 43:12–18
Bandyopadhyay S, Hablitz JJ (2007) Dopaminergic modulation of local network activity in rat prefrontal cortex. J Neurophysiol 97:4120–4128
Breier A, Malhotra AK, Pinals DA, Weisenfeld NI, Pickar D (1997) Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am J Psychiatry 154:805–811
Buccafusco JJ, Terry AV Jr (2009) A reversible model of the cognitive impairment associated with schizophrenia in monkeys: potential therapeutic effects of two nicotinic acetylcholine receptor agonists. Biochem Pharmacol 78:852–862
Coyle JT, Tsai G, Goff DC (2002) Ionotropic glutamate receptors as therapeutic targets in schizophrenia. Curr Drug Target CNS Neurol Disord 1:183–189
Cromwell HC, Woodward DJ (2007) Inhibitory gating of single unit activity in amygdala: effects of ketamine, haloperidol, or nicotine. Biol Psychiatry 61:880–889
Dalack GW, Healy DJ, Meador-Woodruff JH (1998) Nicotine dependence in schizophrenia: clinical phenomena and laboratory findings. Am J Psychiatry 155:1490–1501
Depatie L, O’Driscoll GA, Holahan AL, Atkinson V, Thavundayil JX, Kin NN, Lal S (2002) Nicotine and behavioral markers of risk for schizophrenia: a double-blind, placebo-controlled, cross-over study. Neuropsychopharmacology 27:1056–1070
Deutsch SI, Rosse RB, Billingslea EN, Bellack AS, Mastropaolo J (2003) Modulation of MK-801-elicited mouse popping behavior by galantamine is complex and dose-dependent. Life Sci 73:2355–2361
Domino EF, Minoshima S, Guthrie S, Ohl L, Ni L, Koeppe RA, Zubieta JK (2000) Nicotine effects on regional cerebral blood flow in awake, resting tobacco smokers. Synapse 38:313–321
Farber NB (2003) The NMDA receptor hypofunction model of psychosis. In: Moghaddam B, Wolf ME (eds) Glutamate and disorders of cognition and motivation. New York Academy of Sciences, New York, pp 119–130
Friston KJ, Worsley KJ, Frackowiak RSJ, Mazziotta JC, Evans AC (1994) Assessing the significance of focal activations using their spatial extent. Hum Brain Mapp 1:210–220
Friston KJ, Holmes A, Poline JB, Price CJ, Frith CD (1996) Detecting activations in PET and fMRI: levels of inference and power. Neuroimage 4:223–235
Fu Y, Matta SG, Gao W, Brower VG, Sharp BM (2000) Systemic nicotine stimulates dopamine release in nucleus accumbens: re-evaluation of the role of N-methyl-D-aspartate receptors in the ventral tegmental area. J Pharmacol Exp Ther 294:458–465
Gao XM, Shirakawa O, Du F, Tamminga CA (1993) Delayed regional metabolic actions of phencyclidine. Eur J Pharmacol 241:7–15
Ghatan PH, Ingvar M, Eriksson L, Stone-Elander S, Serrander M, Ekberg K, Wahren J (1998) Cerebral effects of nicotine during cognition in smokers and non-smokers. Psychopharmacology (Berl) 136:179–189
Gioanni Y, Rougeot C, Clarke PB, Lepouse C, Thierry AM, Vidal C (1999) Nicotinic receptors in the rat prefrontal cortex: increase in glutamate release and facilitation of mediodorsal thalamo-cortical transmission. Eur. J Neurosci 11:18–30
Harris JG, Kongs S, Allensworth D, Martin L, Tregellas J, Sullivan B, Zerbe G, Freedman R (2004) Effects of nicotine on cognitive deficits in schizophrenia. Neuropsychopharmacology 29:1378–1385
Herscovitch P, Markham J, Raichle ME (1983) Brain blood flow measured with intravenous H2(15)O. I. Theory and error analysis. J Nucl Med 24:782–789
Holcomb HH, Cascella NG, Thaker GK, Medoff DR, Dannals RF, Tamminga CA (1996) Functional sites of neuroleptic drug action in the human brain: PET/FDG studies with and without haloperidol. Am J Psychiatry 153:41–49
Holcomb HH, Lahti AC, Medoff DR, Weiler M, Tamminga CA (2001) Sequential regional cerebral blood flow brain scans using PET with H2(15)O demonstrate ketamine actions in CNS dynamically. Neuropsychopharmacology 25:165–172
Holcomb HH, Lahti AC, Medoff DR, Cullen T, Tamminga CA (2005) Effects of noncompetitive NMDA receptor blockade on anterior cingulate cerebral blood flow in volunteers with schizophrenia. Neuropsychopharmacology 30:2275–2282
Jentsch JD, Redmond DE Jr, Elsworth JD, Taylor JR, Youngren KD, Roth RH (1997) Enduring cognitive deficits and cortical dopamine dysfunction in monkeys after long-term administration of phencyclidine. Science 277:953–955
Jones IW, Wonnacott S (2004) Precise localization of alpha7 nicotinic acetylcholine receptors on glutamatergic axon terminals in the rat ventral tegmental area. J Neurosci 24:11244–11252
Knott V, McIntosh J, Millar A, Fisher D, Villeneuve C, Ilivitsky V, Horn E (2006) Nicotine and smoker status moderate brain electric and mood activation induced by ketamine, an N-methyl-D-aspartate (NMDA) receptor antagonist. Pharmacol Biochem Behav 85:228–242
Kosowski AR, Liljequist S (2004) The NR2B-selective N-methyl-D-aspartate receptor antagonist Ro 25–6981 [(+/−)-(R*, S*)-alpha-(4-hydroxyphenyl)-beta-methyl-4-(phenylmethyl)-1-pipe ridine propanol] potentiates the effect of nicotine on locomotor activity and dopamine release in the nucleus accumbens. J Pharmacol Exp Ther 311:560–567
Krystal JH, D’Souza DC, Mathalon D, Perry E, Belger A, Hoffman R (2003) NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology (Berl) 169:215–233
Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB Jr, Charney DS (1994) Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51:199–214
Lahti AC, Holcomb HH, Medoff DR, Tamminga CA (1995a) Ketamine activates psychosis and alters limbic blood flow in schizophrenia. Neuroreport 6:869–872
Lahti AC, Koffel B, LaPorte D, Tamminga CA (1995b) Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology 13:9–19
Lahti AC, Weiler MA, Holcomb HH, Tamminga CA, Cropsey KL (2009) Modulation of limbic circuitry predicts treatment response to antipsychotic medication: a functional imaging study in schizophrenia. Neuropsychopharmacology 34:2675–2690
Lambe EK, Picciotto MR, Aghajanian GK (2003) Nicotine induces glutamate release from thalamocortical terminals in prefrontal cortex. Neuropsychopharmacology 28:216–225
Levin ED, Wilson W, Rose JE, McEvoy J (1996) Nicotine-haloperidol interactions and cognitive performance in schizophrenics. Neuropsychopharmacology 15:429–436
London ED (1990) Effects of nicotine on cerebral metabolism. Ciba Found Symp 152:131–140
Lorrain DS, Baccei CS, Bristow LJ, Anderson JJ, Varney MA (2003) Effects of ketamine and N-methyl-D-aspartate on glutamate and dopamine release in the rat prefrontal cortex: modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience 117:697–706
Mair RD, Kauer JA (2007) Amphetamine depresses excitatory synaptic transmission at prefrontal cortical layer V synapses. Neuropharmacology 52:193–199
Malhotra AK, Pinals DA, Adler CM, Elman I, Clifton A, Pickar D, Breier A (1997) Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology 17:141–150
Mansvelder HD, McGehee DS (2000) Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron 27:349–357
McGehee DS, Heath MJ, Gelber S, Devay P, Role LW (1995) Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science 269:1692–1696
Moghaddam B (2003) Bringing order to the glutamate chaos in schizophrenia. Neuron 40:881–884
Murphy GG, Glanzman DL (1999) Cellular analog of differential classical conditioning in aplysia: disruption by the NMDA receptor antagonist DL-2-amino-5- phosphonovalerate. J Neurosci 19:10595–10602
Newcomer JW, Farber NB, Jevtovic-Todorovic V, Selke G, Melson AK, Hershey T, Craft S, Olney JW (1999) Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology 20:106–118
Olney JW, Newcomer JW, Farber NB (1999) NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res 33:523–533
Overall JE, Gorham DR (1962) The brief psychiatric rating scale. Psychol Rep 10:799–812
Ref Type: Generic
Patel AB, De Graaf RA, Mason GF, Rothman DL, Shulman RG, Behar KL (2003) Coupling of glutamatergic neurotransmission and neuronal glucose oxidation over the entire range of cerebral cortex activity. Ann NY Acad Sci 1003:452–453
Poline JB, Worsley KJ, Evans AC, Friston KJ (1997) Combining spatial extent and peak intensity to test for activations in functional imaging. Neuroimage 5:83–96
Raichle ME, Martin WR, Herscovitch P, Mintun MA, Markham J (1983) Brain blood flow measured with intravenous H2(15)O. II. Implementation and validation. J Nucl Med 24:790–798
Reid MS, Fox L, Ho LB, Berger SP (2000) Nicotine stimulation of extracellular glutamate levels in the nucleus accumbens: neuropharmacological characterization. Synapse 35:129–136
Ripoll N, Bronnec M, Bourin M (2004) Nicotinic receptors and schizophrenia. Curr Med Res Opin 20:1057–1074
Rodvelt KR, Kracke GR, Schachtman TR, Miller DK (2008) Ketamine induces hyperactivity in rats and hypersensitivity to nicotine in rat striatal slices. Pharmacol Biochem Behav 91:71–76
Rose JE, Behm FM, Westman EC, Mathew RJ, London ED, Hawk TC, Turkington TG, Coleman RE (2003) PET studies of the influences of nicotine on neural systems in cigarette smokers. Am J Psychiatry 160:323–333
Rothman DL, Sibson NR, Hyder F, Shen J, Behar KL, Shulman RG (1999) In vivo nuclear magnetic resonance spectroscopy studies of the relationship between the glutamate-glutamine neurotransmitter cycle and functional neuroenergetics. Philos Trans R Soc Lond B Biol Sci 354:1165–1177
Rowland LM (2005) Subanesthetic ketamine: how it alters physiology and behavior in humans. Aviat Space Environ Med 76:C52–C58
Rowland LM, Astur RS, Jung RE, Bustillo JR, Lauriello J, Yeo RA (2005a) Selective cognitive impairments associated with NMDA receptor blockade in humans. Neuropsychopharmacology 30:633–639
Rowland LM, Bustillo JR, Mullins PG, Jung RE, Lenroot R, Landgraf E, Barrow R, Yeo R, Lauriello J, Brooks WM (2005b) Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: a 4-T Proton MRS study. Am J Psychiatry 162:394–396
Seamans JK, Yang CR (2004) The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol 74:1–58
Shulman RG, Hyder F, Rothman DL (2002) Biophysical basis of brain activity: implications for neuroimaging. Q Rev Biophys 35:287–325
Smith RC, Singh A, Infante M, Khandat A, Kloos A (2002) Effects of cigarette smoking and nicotine nasal spray on psychiatric symptoms and cognition in schizophrenia. Neuropsychopharmacology 27:479–497
Spitzer RL, Williams JB, Gibbon M, First MB (1992) The Structured Clinical Interview for DSM-III-R (SCID). I: history, rationale, and description. Arch Gen Psychiatry 49:624–629
Stapleton JM, Gilson SF, Wong DF, Villemagne VL, Dannals RF, Grayson RF, Henningfield JE, London ED (2003) Intravenous nicotine reduces cerebral glucose metabolism: a preliminary study. Neuropsychopharmacology 28:765–772
Strassnig M, Brar JS, Ganguli R (2006) Increased caffeine and nicotine consumption in community-dwelling patients with schizophrenia. Schizophr Res 86:269–275
Talairach J, Tournoux P (1988) Co-planar stereotaxic atlas of the human brain. Thieme Medical, New York
Tamminga CA, Holcomb HH (2005) Phenotype of schizophrenia: a review and formulation. Mol Psychiatry 10:27–39
Tizabi Y, Mastropaolo J, Park CH, Riggs RL, Powell D, Rosse RB, Deutsch SI (1998) Both nicotine and mecamylamine block dizocilpine-induced explosive jumping behavior in mice: psychiatric implications. Psychopharmacology (Berl) 140:202–205
Toth E, Sershen H, Hashim A, Vizi ES, Lajtha A (1992) Effect of nicotine on extracellular levels of neurotransmitters assessed by microdialysis in various brain regions: role of glutamic acid. Neurochem Res 17:265–271
Tseng KY, O’Donnell P (2007) D2 dopamine receptors recruit a GABA component for their attenuation of excitatory synaptic transmission in the adult rat prefrontal cortex. Synapse 61:843–850
Vollenweider FX, Leenders KL, Scharfetter C, Antonini A, Maguire P, Missimer J, Angst J (1997) Metabolic hyperfrontality and psychopathology in the ketamine model of psychosis using positron emission tomography (PET) and [18F]fluorodeoxyglucose (FDG). Eur Neuropsychopharmacol 7:9–24
Zubieta JK, Heitzeg MM, Xu Y, Koeppe RA, Ni L, Guthrie S, Domino EF (2005) Regional cerebral blood flow responses to smoking in tobacco smokers after overnight abstinence. Am J Psychiatry 162:567–577
Acknowledgments
We thank Robert F. Dannals, David Clough, Hayden Ravert, Robert Smoot, and Karen Edmonds of the Johns Hopkins Cyclotron Facility, and Nancy Kakoyannis, Ning Ma, and Adrienne Lahti of the University of Maryland School of Medicine, Department of Psychiatry for their valuable help. This study was funded by a research grant from Novartis Pharmaceuticals, Basel, Switzerland, to William T. Carpenter and the University of Maryland School of Medicine, and an NIMH grant (K01MH077230) to Laura M. Rowland. We also thank the volunteers for participating in the study. Lori Beason-Held is supported by the Intramural Research Program, National Institute on Aging (Baltimore, MD), National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rowland, L.M., Beason-Held, L., Tamminga, C.A. et al. The interactive effects of ketamine and nicotine on human cerebral blood flow. Psychopharmacology 208, 575–584 (2010). https://doi.org/10.1007/s00213-009-1758-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-009-1758-2