
Abstract
Hepatocellular carcinoma (HCC) is still a leading cancer killer in the community. Molecular targeted therapy with celecoxib (CXB) has shown promising antitumor effects; however, its use may be limited due to serious side effects. Curcumin (CUR) has also shown beneficial effects against HCC. Then, it was aimed to investigate the effects of adding CUR to CXB on HCC HepG2 cells. HepG2 cells were treated with CXB and/or CUR at increasing concentrations to investigate synergistic drug interactions, as calculated combination index (CI). Combination treatment effects on cell viability and caspase-3 activation were assessed. The levels of Akt, nuclear factor-kappa B (NF-κB), prostaglandin E2 (PGE2), malondialdehyde (MDA), cyclin D1 (CD1), and vascular endothelial growth factor (VEGF) were also evaluated. CXB (3.13–100 μM) and/or CUR (1.25–40 μM) reduced HepG2 cell viability dose-dependently. Nevertheless, lower combined concentrations showed higher synergism (CI < 1) and higher CXB dose reduction index (DRI > 1). Also, the addition of CUR to CXB resulted in increased cytotoxicity and caspase-3 activation, as compared to CXB alone. In addition, the selected combination significantly reduced the levels of Akt, NF-κB, PGE2, MDA, CD1, and VEGF, as compared to either agent alone. In conclusion, CUR augmented the CXB-mediated antitumor effects in HepG2 cells through, at least in part, antiproliferative, antioxidant, and pro-apoptotic mechanisms. This may allow the further use of CXB at lower concentrations, combined with CUR, as a promising safer targeted strategy for HCC management.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC), as the most common form of liver cancer, represents the third main cause of worldwide cancer deaths annually. In Egypt, there is a high incidence of HCC where it also represents the principal cause of death from all cancers. HCC prognosis is still extremely poor and advanced HCC is highly aggressive with a low response to traditional therapies (El–Serag and Rudolph 2007; Shaker et al. 2013). In this context, the significance relies on the massive worldwide burden that HCC represents, and particularly to the Egyptian community. The recent success of the Egyptian model experience in the treatment of HCV in collaboration with the WHO’s Global Strategy has highlighted the importance of managing remaining complications including HCC (Elgharably et al. 2017; Waked et al. 2016). Therefore, new strategies are urgently needed, including chemotherapy that remains a principal treatment method, especially the targeted one.
Targeted strategies include selective COX-2 inhibitors (COXIBs) that have been evaluated as potential anticancer agents for various malignancies. Specifically, celecoxib (CXB) has demonstrated anticancer effects in tumors, particularly, colon carcinoma and HCC both in vitro and in vivo (Cui et al. 2005; Harris 2009). The antiproliferative and pro-apoptotic effects of COXIBs on human HCC cell lines are likely to be multifactorial as they may use both cyclooxygenase (COX)-2 and non-COX-2 biochemical targets to mediate their antitumor activities (Cui et al. 2005; Grösch et al. 2006). It is reported that CXB may inhibit Akt phosphorylation through the COX-2-PGE2-PI3K pathway (Kulp et al. 2004). Also, the nuclear transcription factor-kappa B (NF-κB) and reactive oxygen species (ROS), among others, are targets for COXIBs (Cervello et al. 2013; Maeda and Omata 2008; Wyrebska et al. 2014). In addition, CXB induces cytochrome c release, activates caspase-9 and caspase-3, and eventually stimulates apoptosis in HCC (Grösch et al. 2006). Moreover, most COX-independent effects of CXB in vitro were only observed at supra-therapeutic concentrations that were 10–100 times higher than plasma concentrations measured clinically (Grösch et al. 2006). Nevertheless, the use of COXIBs and their dose have been associated with an increased incidence of cardiovascular toxicity (Bertagnolli et al. 2009). Several pharmacogenetics factors that may contribute to the toxicity profile of CXB were reviewed (Domiati and Ghoneim 2015).
A novel concept in anticancer pharmacology is the use of combination therapy. A combination consisting of CXB with agents that specifically affect relevant molecular targets may take advantage of increased antitumor effects and could reduce the toxicity associated with CXB (Cervello et al. 2013; Du et al. 2013; El-Awady et al. 2011; Lev-Ari et al. 2005a; Lev-Ari et al. 2005b; Morisaki et al. 2013; Narayanan et al. 2005). Then, aiming to keep the anticancer effect of CXB with minimal toxicity profile, we combined CXB at low concentration with curcumin (CUR), keeping in mind that CUR has a good safety profile (Cheng et al. 2001).
The pharmacological terms combination index (CI) and coefficient of drug interaction are used to analyze effects of drug combinations to indicate whether the combined drugs are synergistic (CI < 1), additive (CI = 1), or antagonistic (CI > 1) (Cervello et al. 2013; Chou 2006). Synergistic drug combination is able to reduce the dose of the drug used, thereby reducing toxicity while maintaining efficacy. The concept of the dose-reduction index (DRI) is a measure of how many folds the dose of a drug in combination may be reduced at a given effect compared with the dose of drug alone. Thus, DRI is important in clinical situations, in which dose reduction may reduce toxicity in patient while retaining therapeutic efficacy. The greater DRI > 1 value indicates a greater dose reduction for a given therapeutic effect (Chou 2006). In order to calculate CI, computerized algorithms were based on general theories for biological applications of mass-action law and receptor occupancy. These have established a dose-response theory of basic equations in biomedical sciences guided by Henderson, Michaelis, Hill, and Scatchard (Chou 2006; Chou and Martin 2007).
The HepG2 liver cancer cell line was selected as a HCC model due to several reasons. Firstly, it is of wild apoptotic p53 gene (Müller et al. 1997), and highly expresses the COX-2 enzyme needed to investigate both COX-2-dependent and COX-2-independent antiproliferative pathways in human liver cancer cells (Bae et al. 2001). Secondly, HepG2 represents the most widely used human liver cancer cell line in pharmacological research aiming to develop new potential drugs and combinations. It is phenotypically more hepatocytic than others and expresses many differentiated essential hepatic functions including drug-metabolizing enzymes (Donato et al. 2015; Duan et al. 2014; Fan et al. 2014; Feo et al. 2007; Khalil et al. 2015; Qiu et al. 2015).
CUR is the most important of three main curcuminoids present in turmeric and has a potential role in liver cancer (S Darvesh et al. 2012). CUR is a potent antioxidant having neuroprotective, hepatoprotective, anti-inflammatory, and antitumor activities in normal and cancer cells, as previously demonstrated by our teams and by others (Ghoneim 2012; Ghoneim 2009; Ghoneim et al. 2002; Ghoneim and Eldahshan 2012; Teiten et al. 2010). CUR also targets negatively NF-κB and positively caspase-3 (Ghoneim 2009; Shishodia et al. 2005). Furthermore, CUR has been shown to inhibit several signaling proteins such as COX-2, cyclin D1, vascular endothelial growth factor receptor (VEGF), and phosphoinositol-3 (PI)3/Akt/mammalian target of rapamycin (mTOR) pathway (Aggarwal et al. 2003; Beevers et al. 2009).
Both CUR and CXB inhibit COX-2 via diverse mechanisms. CUR downregulates COX-2 mRNA and decreases its protein levels (Goel et al. 2001), whereas, CXB inhibits COX-2 directly by occupying its binding location (Hood et al. 2003). Furthermore, the combination of CUR and CXB was found to demonstrate a synergistic anticancer effect in colon cancer (Lev-Ari et al. 2005a) and in pancreatic cancer (Lev-Ari et al. 2005b). However, the combined effect of CUR and CXB against HCC cells remains unknown. Hence, the present study aimed to evaluate the anticancer effect of CUR combined with CXB in HCC HepG2 cells. The ultimate goal was to test the validity of the hypothesis stipulating the attainment of a better anticancer efficacy of this combination using a lower and safer dose of CXB. To better understand the molecular mechanisms underlying the interactive cytotoxic effects of this combination, several biomarkers were investigated along the proliferative signaling pathway of the Akt/NF-κB/PGE2/ROS axis, such as, proliferation (CD1), angiogenesis (VEGF), and apoptosis (caspase-3).
Materials and methods
Chemicals
Celecoxib (CXB) was obtained as a gift from Amriya Pharmaceutical Industries, PHARCO Co. Alexandria, Egypt). Curcumin (CUR), 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT), fetal bovine serum (FBS), and dimethyl sulfoxide (DMSO) were procured from Sigma-Aldrich (St. Louis, USA). Trypsin, Dulbecco’s modified eagle medium (DMEM), phosphate buffer saline, and penicillin/streptomycin antibiotic mixtures were purchased from Maadi medical supplies (Cairo, Egypt). Ethanol was bought from El-Nasr Pharmaceutical Chemicals Co. (Cairo, Egypt).
Cell lines and cell culture
HepG2 cell line was procured from the American Type Culture Collection (ATCC; USA). The cells (5 passages after thawing) were cultured in 75-cm2 flasks in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin antibiotic mixture and kept in a humidified incubator with 95% air and 5% CO2 at 37 °C (Thermo Electron Co., Waltham, Massachusetts, USA). The media were continuously changed after 3–4 days, and the cells passaged after reaching 80–90% confluence.
Cell viability assay
The effects of CUR and CXB and their combination on cell proliferation of Hep G2 were assessed by the MTT assay (van Meerloo et al. 2011). Cells were seeded at 5000 cell/well in 96-well plates containing 100 μL of DMEM and allowed to adhere to the plate overnight. DMEM was replaced by fresh medium comprising serially diluted concentrations of CUR (1.25–40 μM) and/or CXB (3.13–100 μM) dissolved in vehicle DMSO at a final concentration of 0.1%. Cells were protected from light for the duration of CUR treatment. The plates were incubated for 72 h at 37 °C and the medium was then aspirated; the cells were rinsed with PBS, and 200 μL of the MTT (0.5 mg/ml in DMEM) was incubated with cells for further 4 h in the dark. After removal of supernatant, 100 μL of DMSO was added to dissolve the crystals formed by maintaining agitation for 15 min. Absorbance was detected at 570 nm using a microplate reader (Bio-Rad, USA). Values were means of two separate experiments, each done in triplicate. Cell viability was expressed as percentage of the untreated control wells. The median inhibitory concentrations (IC50) and the 30% inhibitory concentration (IC30) values were calculated using the CompuSyn software (ComboSyn, Inc.) (Chou 2006).
Synergism experiments
Drug-drug interactions between CXB and CUR were evaluated in HepG2 cells by MTT assay. Cells were incubated with each drug independently and in combination for 72 h before assessment of cytotoxicity. The combination index (CI) was produced by Compusyn software (version 1.0.1), where CI < 1 indicate synergism, CI = 1 designate additive effect, and CI > 1 means antagonism as developed by Chou and Talalay (Chou 2006; Chou and Martin 2007).
Cells were then cultured in 75-cm2 flasks to adhere for 24 h under routine conditions. After this period, each three of 75-cm2 flasks containing cells were incubated with CXB (IC30), CUR (IC50), or a combination of both (Lev-Ari et al. 2005a; Lev-Ari et al. 2005b; Morisaki et al. 2013) for 72 h. Then, the cells were harvested, resuspended in 500 μl PBS, and kept frozen till measuring the following biomarkers.
Morphological changes in HepG2 cells
HepG2 cells normal appearance was observed under light microscope, and after being treated with CXB, CUR, or a combination of CXB and CUR. Cells were investigated for typical polygonal and intact cellular appearance. Otherwise, observation took place for any morphological changes characteristic of cell death, either by apoptosis and/or necrosis, particularly rounding, detachment from the surface of the plates and/or shrinkage and condensation.
Assay of caspase-3 activity
HepG2 cell pellets were lysed with lysis buffer for 30 min at 4 °C and the supernatants were collected and used to detect caspase-3 levels. A colorimetric test kit (Sigma-Aldrich, St. Louis, USA) was used. The hydrolysis of a peptide substrate (Ac-DEVD-pNA) by the enzyme released a p-nitroaniline part. Concentration of the cleaved moiety was calculated from a calibration curve with absorbance at 405 nm using a microtiter plate reader as previously described (Ghoneim 2009; Nicholson et al. 1995). Data were expressed as means ± SEM of three separate experiments, each done in triplicate.
Measurement of the levels of Akt, NF-κB, CD1, and VEGF
The Sandwich ELISA technique was used as a reliable quantitative method having high sensitivity and specificity (Gan and Patel 2013). Several ELISA kits were used according to the manufacturers’ instructions for the detection of the following biomarkers in HepG2 cells. Cell pellet lysates were used for analysis of phospho-Akt (Akt [pS473] kit, DRG International, Inc., Massachusetts, USA), phospho-NF-κB (p-NF-κB p65 (S536) kit, Ray Biotech, Georgia, USA), CD1 (MBS724349 kit, MyBioSource, CA, USA), and VEGF (CSB-E11718h kit, CUSABIO, Maryland, USA). The Sandwich ELISA kit used monoclonal antibody specific for each of the following biomarkers coated onto the wells provided. Firstly, the marker antigen binds to the monoclonal antibody. After washing, an antigen-specific antibody acted as a detector by binding to the captured marker. Finally, a horseradish peroxidase (HRP) antirabbit IgG was added, then a substrate solution was added to produce color after being acted upon by the peroxidase enzyme (Morisaki et al. 2013; Sasaki et al. 2013).
Assessment of prostaglandin-E2 (PGE2) and malondialdehyde (MDA) levels
Both PGE2 and MDA production levels were assessed based on the competitive binding of enzyme immunoassay using two specific ELISA kits from MyBioSource, CA, USA. A color change was detected at 450 nm. The concentration of target antigen was then determined using a standard curve (Bassiouny et al. 2010; Lev-Ari et al. 2005a; Morisaki et al. 2013).
Statistical analysis
Data were analyzed by one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test for multiple comparisons using GraphPad Prism 3.0 (Graph Pad Software Inc., CA, USA) as appropriate. Statistical difference significance was set at p < 0.05. Compusyn software (version 1.0.1) (Chou and Martin 2007) was used to estimate the synergistic effects of different drug combinations by generating the combination index (CI), where CI < 1, CI = 1, and CI > 1 designated synergism, additive effect, and antagonism, respectively (Chou 2006; Chou and Martin 2007).
Results
Effects of CXB and/or CUR on HepG2 cell growth
To evaluate the effect of CXB and/or CUR on the growth of the human HCC cell lines, HepG2 cell growth inhibition assay was done by MTT. Cells were treated with increasing concentrations of CXB (3.25–100 μmol/l) or CUR (1.25–40 μmol/l) (Fig. 1). The IC50 value, at which growth was half of that in the vehicle-treated control, was calculated using Compusyn software (version 1.0.1). CXB reduced cell viability dose-dependently with an IC50 of 89.9 μM and an IC30 of 42.5 μM in HepG2 cells. CUR also inhibited cell proliferation dose-dependently (IC50 13 μM). It was clear that increasing concentrations of CXB began to significantly inhibit growth of cells only from 50 μmol/l, whereas, increasing concentrations of CUR began to significantly inhibit growth of cells from 10 μmol/l (Fig. 1). Interestingly, the IC30 of CXB (42.5 μM) is between its minimum effective concentration (50 μM) and its no-observed effect level concentration (25 μM), as illustrated by the line chart in Fig. 1.

Effects of CXB or CUR on viability of HepG2 cells. Cells were cultured in the presence of increasing concentrations of celecoxib (CXB) (3.125–100 μmol/l) or curcumin (CUR) (1.25–40 μmol/l) for 72 h in DMEM containing 10% FBS. Cell viability was assessed by MTT assay. Data points represent the means ± SEM of two separate experiments, each done in triplicate. *p < 0.05 indicated significant difference for CXB or CUR versus corresponding control group. Statistical analysis was determined by one-way ANOVA followed by Tukey post hoc test
Also, the addition of increasing concentrations of CUR (1.25–40 μmol/l) to increasing concentrations of CXB (3.125–100 μmol/l) consistently increased growth inhibition in HepG2 cells compared to the corresponding CXB-alone group (Fig. 2). Moreover, the combination treatment was found to be synergistic as calculated using Compusyn software (version 1.0.1) (Chou and Martin 2007).

Effects of adding CUR to CXB in combination on the viability of HepG2 cells. Cells were incubated for 72 h in the presence of the indicated increasing concentrations of celecoxib (CXB) (3.125–100 μmol/l) and curcumin (CUR) (1.25–40 μmol/l) alone or in combination. Data are expressed as the percentage of control cells and are the means ± SEM of three separate experiments, each of which was performed in triplicate.*p < 0.05 indicated significant difference for the combination versus corresponding CXB-alone group. Statistical analysis was determined by one-way ANOVA followed by the Tukey post hoc test
Synergistic antiproliferative effects of celecoxib and curcumin in HepG2 cells
To examine the combined effects of CXB and CUR on HepG2 cells, these were treated with CXB, CUR, or both agents in an arbitrary ratio of 1 CUR: 2.5 CXB. Concerning synergy experiments, HepG2 cells were treated with the combination of CXB and CUR at doses indicated.
CompuSyn software was used to determine the type of drug interaction between the agents. Table 1 presents the combination indices (CIs) detected after treatment of HepG2 cells with different combinations of the two agents and indicated their interaction pattern. The CI values were estimated according to the method of Chou using CompuSyn software, where CI < 1, CI = 1, and CI > 1 designated synergism, additive effect, and antagonism, respectively (Chou 2006; Chou and Martin 2007; Cusimano et al. 2007; Morisaki et al. 2013). The results in Table 1 showed the addition of 100 μM CXB to 40 μM CUR significantly inhibited cell growth by 79% in HepG2 cells with CI = 1.15 which showed slight antagonistic effect with dose reduction index (DRI) of CXB = 2.8, while 50 μM CXB plus 20 μM CUR significantly inhibited cell growth by 70% in HepG2 cells with CI = 0.9; which began to show slight synergism that increased with lower concentrations, and with higher DRI of CXB = 3.8. The CI value of 25 μM CXB plus 10 μM CUR was 0.7 which meant better synergism, with much higher DRI of CXB = 5.1 (Table 1). Based on these experiments and observations, the CUR IC50 (13 μm) added to the CXB IC30 (42.5 μM) was selected as a novel combination to be investigated mechanistically for its underlying pharmacologic effects on the subsequent molecular biomarkers of HepG2 cytotoxicity. This combination is complying more with our approach to reduce CXB dose to decrease its side effects as much as possible.
Effects of CXB and/or CUR on apoptosis of HepG2 cells
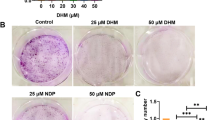
Firstly, the morphological effects of CXB and/or CUR in the HepG2 cells were determined by preliminary morphological assay. The HepG2 cells were treated with CXB (IC30: 42.5 μM) and/or CUR (IC50: 13 μM) for 72 h, and observed under a light microscope. The control normal HepG2 cells showed a typical polygonal and intact appearance. Treatment of cells with CXB and/or CUR induced morphological changes characteristic of cell death, particularly when exposed to combination of CXB and CUR, where the cells appeared spares, and detached from the surface of the plates with progressive cytoplasmic shrinkage and condensation (the typical morphologic signs of apoptosis under light microscope) (Fig. 3).

Effects of CXB and/or CUR on the morphological changes in HepG2 cells. a Represents the control normal HepG2 cells which showed a typical polygonal and intact appearance under light microscope. Treatment of cells with CXB IC30 (b), CUR IC50 (c) or a combination of CXB and CUR (d) induced morphological changes characteristic of cell death, particularly in (d) where the cells appeared spares, and detached from the surface of the plates. The cells lost their usual morphology and looked smaller in size, rounded, and shrunken (characteristic of apoptosis)
Secondly, to confirm apoptosis biochemically, the treatment of cells with combination of CXB and CUR led to a significant increase in caspase-3 activity as shown in Fig. 5c. Caspase-3 is a key biochemical apoptosis marker. The results showed that the combination of IC30 of CXB with IC50 of CUR led to increased caspase-3 activation in HepG2 cells by 42% compared to CXB-alone group.
Effects of CXB and/or CUR on the levels of Akt, NF-κB, PGE2, and MDA
The effects of combined drugs on the tumor proliferation signaling pathway of the Akt/NF-κB/PGE2/ROS axis were investigated. The levels of phospho-Akt, phosphorylated NF-κB, PGE2, and MDA were all reduced by CUR or CXB compared to the control group. It was found that treatment with the combination of IC30 of CXB and IC50 of CUR resulted in a greater reduction in Akt, NF-κB, PGE2, and MDA than what was observed by either agent alone (Fig. 4a–d). The addition of CUR to CXB decreased the levels of p-Akt, NF-κBp65, PGE2, and MDA by about 40, 29, 37, and 47%, respectively, compared to CXB-alone group, in HepG2 cells.

Effects of CXB and/or CUR on Akt/NF-κB/PGE2/ROS axis in HepG2 cells. Cells were treated with CXB IC30 and/or CUR IC50 for 72 h. The levels of p-Akt (a), NF-κBp65 (b), PGE2 (c), and MDA (d) were measured using different ELISA techniques as described in “Materials and Methods” section. Columns represent the means ± standard error of mean (SEM) of three samples each performed in triplicate. Statistically significant differences between groups are designated as *p < 0.05 vs. control, #p < 0.05 vs. curcumin (CUR) and $p < 0.05 vs. celecoxib (CXB) group. Statistical analysis was performed using ANOVA followed by the Tukey post hoc test. HepG2 human liver-derived hepatoma G2, NF-κB nuclear factor kappa B, PGE2 prostaglandin E2, ROS reactive oxygen species measured as malondialdehyde (MDA)
Effects of CXB and/or CUR on CD1, VEGF levels, and caspase-3 activity
The effects of combined drugs on the levels of effector tumor markers of proliferation (CD1), angiogenesis (VEGF), or apoptosis (active caspase-3) were assessed. As shown in Fig. 5, the results showed a significant decrease in the levels of CD1 and VEGF by CUR or CXB compared to the control group. Also, the addition of CUR to CXB decreased CD1 and VEGF levels by 38 and 43%, respectively, in HepG2 cells, as compared to CXB-alone group. Moreover, the results showed that the combination of CXB with of CUR led to increased caspase-3 activation in HepG2 cells by 42% compared to CXB-alone group.

Effects of CXB and/or CUR on markers of proliferation, angiogenesis, or apoptosis in HepG2 cells. The levels of effector tumor markers of proliferation (cyclin D1; CD1) (a), angiogenesis (vascular endothelial growth factor; VEGF) (b), or apoptosis (active caspase-3) (c) were measured using ELISA technique or colorimetrically as described in “Materials and Methods” section. Columns represent the means ± standard error of mean (SEM) of three samples each performed in triplicate. Statistically significant differences between groups are designated as *p < 0.05 vs. control, #p < 0.05 vs. curcumin (CUR) and $p < 0.05 vs. celecoxib (CXB) group. Statistical analysis was performed using ANOVA followed by the Tukey post hoc test
Discussion
Combination therapy may take advantage of synergistic inhibitory effects against cancer, as well as, reduced toxicity associated with CXB. In this context, several combinations of CXB with several chemotherapeutic compounds have been investigated (Cervello et al. 2013; Cusimano et al. 2007; Narayanan et al. 2003). Similarly, CUR has been demonstrated to possess antiproliferative, pro-apoptotic, and anti-oxidative properties against a diversity of organ cancers including the liver (Cao et al. 2007). For that reason, the development of a treatment that comprises a safe phytochemical in combination with a safer low dose of CXB seems highly promising. Mechanistically, both CUR and CXB inhibit COX-2 and PGE2 (Lev-Ari et al. 2005a; Yoysungnoen et al. 2006). Interestingly, numerous non-COX-2 pathways are also targeted by both drugs, such as Akt (Kulp et al. 2004), NF-κB (Shishodia et al. 2003), oxidative stress (Lev-Ari et al. 2005a), CD1 (Liu et al. 2011), VEGF (Liu et al. 2011; Morisaki et al. 2013; Zhang et al. 2018), and apoptosis (Maeda and Omata 2008; Park and Hong 2016). Consequently, the present study aimed to investigate the anticancer effects and underlying mechanisms of the promising combination of CUR and CXB on HepG2 cells. Aiming to keep the anticancer effect of CXB with minimal toxicity profile, a minimal effective concentration (IC30) was combined with CUR at its half maximal inhibitory concentration (IC50), keeping in mind that CUR showed a good safety profile (Cheng et al. 2001; Cheng et al. 2010; Wang 2012).
The present results showed that CUR inhibited HepG2 cells proliferation dose-dependently (IC50 13 μM), in parallel with previous reports (Fan et al. 2014). CXB also reduced HepG2 cells viability in a dose-dependent manner (IC50 89.9 μM; IC30 42.5 μM), in line with previous reports (Cervello et al. 2013; Cui et al. 2005; Cusimano et al. 2007; Naruse et al. 2002). In addition, an increase of growth inhibition was observed for the combination of CUR and CXB, as previously documented for other combinations in different cancer cell lines (Banerjee et al. 2007; Gong et al. 2004; Narayanan et al. 2003; Somers-Edgar et al. 2008). When the IC30 of CXB was combined with the IC50 of CUR, the results showed enhanced cytotoxicity and apoptosis in HepG2 cells (Figs. 1, 2, 3, and 5C). Also, the molecular signaling pathways in HepG2 cells were significantly affected by the addition of CUR to CXB. The results showed greater reduction in p-Akt, NF-κB, PGE2, MDA, CD1, and VEGF, than with either agent alone (Figs. 4 and 5).
The present research was actually based on the previous studies of the combination of CXB and CUR in colorectal and pancreatic cancer cells, so as to perform more detailed pharmacologic, drug interaction, and mechanistic insights into HepG2 HCC cell lines. Previous studies focused only on COX-2-dependent pathway (Lev-Ari et al. 2005a; Lev-Ari et al. 2005b). They proved that the combination of CXB (5–25 μM) and CUR (10–15 μM) resulted in several-fold reduction in cell number. When they studied the colorectal HT-29 and pancreatic P-34 cell lines, the celecoxib IC30 was 25 and 10 μM in cancer cells of the pancreas and colon, respectively. When combined with CUR, it resulted in about 80% growth inhibitory effect in both cancer cells (Lev-Ari et al. 2005a; Lev-Ari et al. 2005b). In the present study, the IC30 of CXB was found to be 42.5 μM. These differences in the results may be due to the difference in the cell lines used. We studied HCC cell lines (HepG2) while Levi-Ari et al. studied the pancreatic P-34 cell lines with high COX-2 expression (Lev-Ari et al. 2005b) and the colorectal cancer cells (HT29) which are more sensitive for CXB effect as they were showing higher COX-2 expression than HepG2 cells when previously compared together (Bae et al. 2001). This reason is clear when Levi-Ari et al. themselves studied the effect of different concentrations of CXB and CUR combinations on different cell lines showing different sensitivities towards concentrations of either single or combined drugs (Lev-Ari et al. 2005a; Lev-Ari et al. 2005b). Moreover, the present research was based on the recommendations of Levi-Ari et al. for additional experiments needed for the identification of the precise synergistic (CI) and underlying non-COX-2 mechanisms that may be targeted by CUR and CXB, such as, Akt, NF-κB, CD1, VEGF, caspase-3, and ROS.
In the present results, the CI values were < 1 for most of the concentrations tested (Table 1), indicating a synergistic effect between CXB and CUR in HepG2 cells, particularly at low concentrations < IC50, while in high concentrations, CI values showed additive to slight antagonistic effect between CUR and CXB in HepG2 cells. Synergistic drug combination is able to reduce the dose of the drug used, thereby maintaining safety with efficacy. In our synergy experiments, the DRI values showed the great benefits of adding CUR to CXB, particularly at low doses which allow us to avoid the toxicity of CXB to great extent (Table 1). Based on these results, the CXB IC30 (42.5 μM) and CUR IC50 (13 μM) were selected to study the subsequent biomarkers as it was found a higher synergism using the (IC30 42.5 μM) of CXB than combining with its higher (IC50 89.9 μM). In addition, there is a great advantage of reducing the CXB dose to prevent the serious side effects of large doses of CXB in contrast to CUR which is known to be safe even at large doses (IC50 13 μM). These concentrations showed higher synergism with better CXB DRI lying between 3.8 and 5.1 (Table 1), and this is complying more with our approach to reduce CXB dose to decrease its side effects as much as possible. These findings agreed with the previous studies proving that CUR synergistically potentiated the inhibitory effect of CXB on cell growth in P-34 cells (Lev-Ari et al. 2005b). On the other hand, CXB showed antagonistic antiproliferative effects when combined with cisplatin in ovarian and colon cancer cell lines (Bijman et al. 2008). This difference may be due to the difference in the cell lines studied. El-Awady et al. reported an interaction of CXB with different anticancer agents as antagonistic in breast cell lines but not in cells of other cancers (El-Awady et al. 2011). The effect of the used cells on the studied drugs was proved by Lim et al. who showed that CXB attenuated the cytotoxic effect of 5-FU in human colon cancer cells proving that CXB may act through blocking of cell cycle advancement, and stopping apoptosis induced by the drug (Lim et al. 2007). Conversely, and while studying the effect of the same drugs by Bassiouny et al., a synergism of CXB on drug-induced apoptosis was observed in HCC patients (Bassiouny et al. 2010).
Non-COX hypotheses are supported by the fact that antitumor effectiveness has almost been attributed to concentrations of COXIBs usually exceeding clinical margin (5 μM) (Davies et al. 2000; Gong et al. 2012). Moreover, previous reports demonstrate that COX-2-deficient cells underwent apoptosis when exposed to CXB at concentrations of 50–100 μM (Hawk et al. 2002). Based on that, the selected CXB dose was decreased to its IC30 (42.5 μM) to be combined with the IC50 of CUR (Figs. 1 and 2) to investigate the combination effects on HepG2 cell death signaling pathways. HepG2 cells treated with this combination showed an increase in apoptosis, in which the cells lost their usual morphology, and looked smaller in size, rounded and shrunken, compared to use of either agent alone (Fig. 3), as was observed in similar situations (Cheng et al. 2010; Du et al. 2013; Ghoneim and Eldahshan 2012; Khalil et al. 2015). These progressive cytoplasmic shrinkage and nuclear condensation as typical morphologic signs of apoptosis were previously established by Kerr et al. (Kerr et al. 1972). Biochemically, caspase-3 is a key apoptosis marker (Cui et al. 2005; Du et al. 2013; Ghoneim 2009; Khalil et al. 2015; Sato et al. 2011). It was found a rise in caspase-3 activity in HepG2 cells treated with CUR and CXB combination rather than either CUR or CXB alone (Fig. 5c). This indicated that CUR potentiated the apoptotic effect of CXB leading to an increase in apoptosis in line with studies in colorectal cancer cell lines (Lev-Ari et al. 2005a).
Concerning the signaling pathway of the Akt/NF-κB/PGE2/ROS axis, it is known that Akt activation is documented to be involved in COX-2-mediated HCC growth (Leng et al. 2003). The present results (Fig. 4a) agreed with studies of CXB effects on Akt phosphorylation in HCC cells alone and in combination with sorafenib (Morisaki et al. 2013; Sui et al. 2014). CXB, in turn, showed anticancer activity in different cancer cells by impeding the NF-κB pathway (Huang et al. 2010; Vaish et al. 2010), as well as, CUR (De Porras et al. 2016; Sato et al. 2011). In the present study, the levels of NF-κB p65 and MDA revealed (Fig. 4b, d) a greater reduction by CUR and CXB combination. CUR, in turn, scavenges oxygen radicals that represent a key player in carcinogenesis (FUJISAWA et al. 2004; Ghoneim 2009; Patial et al. 2015). Studies have also proven the significant inhibition of lipid peroxidation (LPO) produced by CXB (Ekor et al. 2013). These findings suggested that CUR potentiated the antioxidant effect of CXB. Nevertheless, this seemed to be different from a previous study on CXB and CUR (Lev-Ari et al. 2005a), which showed that synergism may be due to that MDA is a by-product of prostaglandin breakdown (VanderVeen et al. 2003). Regarding COX-dependent HepG2 cell death signaling pathways, the PGE2 levels showed (Fig. 4c) a significant decrease by combined drugs. That is comparable to the CXB and CUR combination effect in colorectal cancer cells (Lev-Ari et al. 2005a) and in pancreatic adenocarcinoma cells (Lev-Ari et al. 2005b).
Moreover, the present study investigated the effects of CXB and CUR on HepG2 cell markers of proliferation (CD1) and angiogenesis (VEGF). The levels of CD1 (Fig. 5a) indicated that CUR increased the antiproliferative effect of CXB in HepG2 cells. This was thought to be due to the enhanced effect of the combination on PI3K/Akt pathway, as Akt promotes G1-S phase progression by preventing the phosphorylation and degradation of cyclin D1 (Alao 2007). Furthermore, COX inhibition has been demonstrated to be a potential anti-angiogenic approach in various HCC tumors by inhibiting p-Akt and VEGF (Sui et al. 2014). Evaluating the levels of VEGF (Fig. 5b) gave evidence that CUR increased the anti-angiogenic effect of CXB. In connection with these results, both CUR and CXB have previously been shown to inhibit VEGF in HCC cells including the HepG2 ones (Liu et al. 2011; Morisaki et al. 2013; Zhang et al. 2018).
Previous signaling studies have generally established that Akt pathway is actively involved in the regulation of NFκB essential for oncogenic transformation (Kane et al. 1999). Indeed, inhibitors of Akt interfere with overexpression of NFκB, as previously proven (Bai et al. 2009). Crosstalk and direct relation between p-Akt and NF-κB is already proven in several cancer cells (Oeckinghaus et al. 2011), including HepG2 cells (Hu et al. 2017; Shan et al. 2014). Actually, it is already proven that CXB significantly inhibits the phosphorylation of Akt using Akt inhibitors in HepG2 cells (Leng et al. 2003). Many references built on that and further assessed the direct effects of CXB on p-Akt (Cui et al. 2014; Morisaki et al. 2013; Sui et al. 2014). The same complies also with CUR which is already proven to inhibit the Akt/NF-κB pathway, especially in liver cancer and HepG2 cells (S Darvesh et al. 2012; Shen and Tergaonkar 2009). That is why, many studies also assessed the direct effects of CXB or CUR on Akt/NF-κB pathway in HepG2 cells, particularly (Cusimano et al. 2007; Morisaki et al. 2013; Zhang et al. 2018).
In turn, NF-κB is one of the most important molecular targets in cancer therapy (Shen and Tergaonkar 2009) that is especially overexpressed in HCC (Tai et al. 2000). Additionally, NF-κB activation directly affects molecular hallmarks of cancer through modulating cell proliferation (CD1), angiogenesis (VEGF), inflammation (COX-2/PGE2), apoptosis (caspase-3), and oxidative stress (MDA) (Maeda and Omata 2008; Park and Hong 2016; Xia et al. 2014). Many studies assessed the anticancer effects of different drugs acting on the NF-κB signaling pathways (Oeckinghaus et al. 2011), particularly, in HepG2 cells (Ozaki et al. 2007; Xia et al. 2013; Zhang et al. 2018). In fact, it is already proven that both CXB and CUR have already been proven to be NF-κB inhibitors, especially in liver cancer and HepG2 cells (Cervello et al. 2011; Reuter et al. 2008; S Darvesh et al. 2012; Shen and Tergaonkar 2009; Wyrebska et al. 2014). Based on the above-mentioned facts, and similarly to the present research, many other studies directly assessed the effects of CXB or CUR on the NF-κB, CD1, VEGF, PGE2, and ROS in HepG2 cells, particularly (Liu et al. 2011; Morisaki et al. 2013; Ramyaa and Padma 2014; Zhang et al. 2018).
To our knowledge, evidence was provided for the first time that the combination of CXB (a COXIB) and CUR (a phytochemical) led to synergistic cytotoxic effect in the HepG2 cell line as demonstrated by increased apoptosis and inhibition of proliferation. It appeared also in this context that this combination acted via the signaling pathway of the Akt/NF-κB/PGE2/ROS axis that affects proliferation (CD1) and angiogenesis (VEGF) by induction, and apoptosis (caspase-3) by inhibition. Both CUR and CXB acted at different points in this signaling pathway that affected proliferation, angiogenesis, and apoptosis. This may, at least in part, underlie the synergistic cytotoxic effects of these interacting drugs on the viability of HepG2 cells (Fig. 6; graphical abstract).

Proposed underlying mechanisms of the synergistic antiproliferative effects of curcumin and celecoxib on HepG2 cells. Both curcumin and celecoxib act at different points in the signaling pathway of the Akt/NF-κB/PGE2/ROS axis that affects proliferation (CD1) and angiogenesis (VEGF) by induction and apoptosis (caspase-3) by inhibition. This may underlie the synergistic effect of the combined drugs on the viability of HepG2 cells when exposed to this combination. CD1 cyclin D1, HepG2 human liver-derived hepatoma G2, NF-κB nuclear factor kappa B, PGE2 prostaglandin E2, ROS reactive oxygen species measured as malondialdehyde (MDA), VEGF vascular endothelial growth factor
Conclusions
CUR showed synergistic antiproliferative interactions with CXB in HepG2 cells. This might further allow for the use of lower and safer doses of CXB than those currently used though linked to cardiovascular risk. Therefore, this novel liver cancer combination may represent promising adjuvant targeted chemotherapy in treating HCC patients, a finding that needs further clinical investigations. This would better follow similar preclinical research on liver cancer cell lines other than the presently used HepG2 cells.
Abbreviations
- CD1:
-
Cyclin D1
- CI:
-
Combination index
- CUR:
-
Curcumin
- CXB:
-
Celecoxib
- DRI:
-
Dose reduction index
- HCC:
-
Hepatocellular carcinoma
- HepG2:
-
Human liver-derived hepatoma G2
- DMEM:
-
Dulbecco’s modified eagle medium
- FBS:
-
Fetal bovine serum
- IC50 :
-
Median inhibitory concentration
- MDA:
-
Malondialdehyde
- MTT:
-
3-[4, 5-Dimethylthiazol-2-yl]2,5-diphenyltetrazolium bromide
- NF-κB:
-
Nuclear factor kappa B
- p-Akt:
-
Phospho-Akt
- PGE2 :
-
Prostaglandin E2
- SEM:
-
Standard error of the mean
- VEGF:
-
Vascular endothelial growth factor
References
Aggarwal BB, Kumar A, Bharti AC (2003) Anticancer potential of curcumin: preclinical and clinical studies. Anticancer Res 23:363–398
Alao JP (2007) The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention Molecular cancer 6:24
Bae SH, Jung ES, Park YM, Kim BS, Kim BK, Kim DG, Ryu WS (2001) Expression of cyclooxygenase-2 (COX-2) in hepatocellular carcinoma and growth inhibition of hepatoma cell lines by a COX-2 inhibitor, NS-398 Clinical Cancer Research 7:1410–1418
Bai D, Ueno L, Vogt PK (2009) Akt-mediated regulation of NFκB and the essentialness of NFκB for the oncogenicity of PI3K and Akt. Int J Cancer 125:2863–2870
Banerjee S et al (2007) In vitro and in vivo molecular evidence for better therapeutic efficacy of ABT-627 and taxotere combination in prostate cancer. Cancer Res 67:3818–3826
Bassiouny AR, Zaky A, Neenaa HM (2010) Synergistic effect of celecoxib on 5-fluorouracil-induced apoptosis in hepatocellular carcinoma patients. Ann Hepatol 9:410–418
Beevers CS, Chen L, Liu L, Luo Y, Webster NJ, Huang S (2009) Curcumin disrupts the mammalian target of rapamycin-raptor complex. Cancer Res 69:1000–1008
Bertagnolli MM et al. (2009) Five-year efficacy and safety analysis of the Adenoma Prevention with Celecoxib Trial Cancer prevention research:1940–6207. CAPR-1908-0206
Bijman MN, Hermelink CA, van Berkel MP, Laan AC, Janmaat ML, Peters GJ, Boven E (2008) Interaction between celecoxib and docetaxel or cisplatin in human cell lines of ovarian cancer and colon cancer is independent of COX-2 expression levels Biochemical pharmacology 75:427–437
Cao J et al. (2007) Curcumin induces apoptosis through mitochondrial hyperpolarization and mtDNA damage in human hepatoma G2 cells Free Radical Biology and Medicine 43:968–975
Cervello M et al (2011) COX-2-dependent and COX-2-independent mode of action of celecoxib in human liver cancer cells Omics. A J Integrative Biology 15:383–392
Cervello M, Bachvarov D, Lampiasi N, Cusimano A, Azzolina A, McCubrey JA, Montalto G (2013) Novel combination of sorafenib and celecoxib provides synergistic anti-proliferative and pro-apoptotic effects in human liver cancer cells. PloS One 8:e65569
Cheng A-L et al (2001) Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res 21:2895–2900
Cheng C-Y, Lin Y-H, Su C-C (2010) Curcumin inhibits the proliferation of human hepatocellular carcinoma J5 cells by inducing endoplasmic reticulum stress and mitochondrial dysfunction. Int J Molecular Med 26:673–678
Chou T-C (2006) Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacological Rev 58:621–681
Chou T, Martin N (2007) CompuSyn software for drug combinations and for general dose-effect analysis, and user’s guide Paramus: ComboSyn Inc
Cui W, Yu C-H, Hu K-Q (2005) In vitro and in vivo effects and mechanisms of celecoxib-induced growth inhibition of human hepatocellular carcinoma cells. Clinical Cancer Res 11:8213–8221
Cui J et al (2014) Cyclooxygenase-2 inhibitor is a robust enhancer of anticancer agents against hepatocellular carcinoma multicellular spheroids. OncoTargets Therapy 7:–353
Cusimano A, Foderà D, D’Alessandro N, Lampiasi N, Azzolina A, Montalto G, Cervello M (2007) Potentiation of the antitumor effects of both selective cyclooxygenase-1 and cyclooxygenase-2 inhibitors in human hepatic cancer cells by inhibition of the MEK/ERK pathway Cancer biology & therapy 6:1457–1464
Darvesh AS, Aggarwal B, Bishayee A (2012) Curcumin and liver cancer: a review. Current Pharmaceutical Biotechnology 13:218–228
Davies NM, McLachlan AJ, Day RO, Williams KM (2000) Clinical pharmacokinetics and pharmacodynamics of celecoxib. Clinical Pharmacokinetics 38:225–242
De Porras VR et al. (2016) Curcumin mediates oxaliplatin-acquired resistance reversion in colorectal cancer cell lines through modulation of CXC-chemokine/NF-κB signalling pathway Scientific reports 6:24675
Domiati S, Ghoneim A (2015) Celecoxib for the right person at the right dose and right time: an updated overview. Springer Sci Rev 3:137–140
Donato MT, Tolosa L, Gómez-Lechón MJ (2015) Culture and functional characterization of human hepatoma HepG2 cells Protocols in In Vitro Hepatocyte Research:77–93
Du Q, Hu B, An H-M, Shen K-P, Xu L, Deng S, Wei M-M (2013) Synergistic anticancer effects of curcumin and resveratrol in Hepa1-6 hepatocellular carcinoma cells. Oncology Reports 29:1851–1858
Duan W et al (2014) Curcumin inhibits hypoxia inducible factor-1α-induced epithelial-mesenchymal transition in HepG2 hepatocellular carcinoma cells. Molecular Med Reports 10:2505–2510
Ekor M, Odewabi AO, Kale OE, Adesanoye OA, Bamidele TO (2013) Celecoxib, a selective cyclooxygenase-2 inhibitor, lowers plasma cholesterol and attenuates hepatic lipid peroxidation during carbon-tetrachloride–associated hepatotoxicity in rats Drug and chemical toxicology 36:1–8
El-Serag HB, Rudolph KL (2007) Hepatocellular carcinoma: epidemiology and molecular carcinogenesis Gastroenterology 132:2557–2576
El-Awady RA, Saleh EM, Ezz M, Elsayed AM (2011) Interaction of celecoxib with different anti-cancer drugs is antagonistic in breast but not in other cancer cells. Toxicology Appl Pharmacology 255:271–286
Elgharably A, Gomaa AI, Crossey MME, Norsworthy PJ, Waked I, Taylor-Robinson SD (2017) Hepatitis C in Egypt—past, present, and future. Int J General Med 10:1–6. https://doi.org/10.2147/ijgm.s119301
Fan H, Tian W, Ma X (2014) Curcumin induces apoptosis of HepG2 cells via inhibiting fatty acid synthase. Targeted Oncology 9:279–286
Feo F, Pascale RM, Calvisi DF (2007) Models for liver cancer. Cancer Handbook 2:1118–1133
Fujisawa S, Atsumi T, Ishihara M, Kadoma Y (2004) Cytotoxicity, ROS-generation activity and radical-scavenging activity of curcumin and related compounds. Anticancer Res 24:563–570
Gan SD, Patel KR (2013) Enzyme immunoassay and enzyme-linked immunosorbent assay. J Invest Dermatol 133:e12
Ghoneim AI (2009) Effects of curcumin on ethanol-induced hepatocyte necrosis and apoptosis: implication of lipid peroxidation and cytochrome c Naunyn-Schmiedeberg's archives of pharmacology 379:47
Ghoneim A (2012) Phytochemicals and amino acids: inducers or inhibitors of cell death? In: Natural compounds as inducers of cell death. Springer, pp 3–32
Ghoneim AI, Eldahshan OA (2012) Anti-apoptotic effects of tamarind leaves against ethanol-induced rat liver injury. J Pharmacy Pharmacology 64:430–438
Ghoneim AI, Abdel-Naim AB, Khalifa AE, El-Denshary ES (2002) Protective effects of curcumin against ischaemia/reperfusion insult in rat forebrain. Pharmacological Res 46:273–279
Goel A, Boland CR, Chauhan DP (2001) Specific inhibition of cyclooxygenase-2 (COX-2) expression by dietary curcumin in HT-29 human colon cancer cells. Cancer Letters 172:111–118
Gong SJ, Jin CJ, Rha SY, Chung HC (2004) Growth inhibitory effects of trastuzumab and chemotherapeutic drugs in gastric cancer cell lines. Cancer Letters 214:215–224
Gong L, Thorn CF, Bertagnolli MM, Grosser T, Altman RB, Klein TE (2012) Celecoxib pathways: pharmacokinetics and pharmacodynamics. Pharmacogenetics Genomics 22:310
Grösch S, Maier TJ, Schiffmann S, Geisslinger G (2006) Cyclooxygenase-2 (COX-2)–independent anticarcinogenic effects of selective COX-2 inhibitors. J National Cancer Institute 98:736–747
Harris R (2009) Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology 17:55–67
Hawk ET, Viner JL, Dannenberg A, DuBois RN (2002) COX-2 in cancer—a player that's defining the rules. Oxford University Press,
Hood WF, Gierse JK, Isakson PC, Kiefer JR, Kurumbail RG, Seibert K, Monahan JB (2003) Characterization of celecoxib and valdecoxib binding to cyclooxygenase. Molecular Pharmacology 63:870–877
Hu Z, Jiang K, Chang Q, Zhang Y, Zhou B, Zhang Z, Tao R (2017) Effect of talin1 on apoptosis in hepatoma carcinoma cells via the PI3K/Akt/NF-κB signaling pathway. RSC Advances 7:40179–40188
Huang C-H, Guh J-H, Chen GS, Lu P-H, Chern J-W (2010) Anticancer activity of a cyclooxygenase inhibitor, CX9051, in human prostate cancer cells: the roles of NF-κB and crosstalk between the extrinsic and intrinsic apoptotic pathways. Naunyn-Schmiedeberg’s Archives Pharmacology 382:159–169
Kane LP, Shapiro VS, Stokoe D, Weiss A (1999) Induction of NF-κB by the Akt/PKB kinase. Current Biology 9:601–S601
Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wideranging implications in tissue kinetics. British J Cancer 26:239
Khalil MI, Ibrahim MM, El-Gaaly GA, Sultan AS (2015) Trigonella foenum (Fenugreek) induced apoptosis in hepatocellular carcinoma cell line, HepG2, mediated by upregulation of p53 and proliferating cell nuclear antigen BioMed research international 2015
Kulp SK et al (2004) 3-phosphoinositide-dependent protein kinase-1/Akt signaling represents a major cyclooxygenase-2-independent target for celecoxib in prostate cancer cells. Cancer Res 64:1444–1451
Leng J, Han C, Demetris AJ, Michalopoulos GK, Wu T (2003) Cyclooxygenase-2 promotes hepatocellular carcinoma cell growth through Akt activation: evidence for Akt inhibition in celecoxib-induced apoptosis. Hepatology 38:756–768
Lev-Ari S et al (2005a) Celecoxib and curcumin synergistically inhibit the growth of colorectal cancer cells. Clinical Cancer Research 11:6738–6744
Lev-Ari S et al (2005b) Curcumin synergistically potentiates the growth inhibitory and pro-apoptotic effects of celecoxib in pancreatic adenocarcinoma cells. Biomedicine Pharmacotherapy 59:S276–S280
Lim YJ, Rhee JC, Bae YM, Chun WJ (2007) Celecoxib attenuates 5-fluorouracil-induced apoptosis in HCT-15 and HT-29 human colon cancer cells. World J Gastroenterology: WJG 13:1947
Liu Y, Liu A, Li H, Li C, Lin J (2011) Celecoxib inhibits interleukin-6/interleukin-6 receptor-induced JAK2/STAT3 phosphorylation in human hepatocellular carcinoma cells Cancer Prevention Research:canprevres. 0317.2010
Maeda S, Omata M (2008) Inflammation and cancer: role of nuclear factor-kappaB activation. Cancer Sci 99:836–842
Morisaki T, Umebayashi M, Kiyota A, Koya N, Tanaka H, Onishi H, Katano M (2013) Combining celecoxib with sorafenib synergistically inhibits hepatocellular carcinoma cells in vitro. Anticancer Res 33:1387–1395
Müller M et al (1997) Drug-induced apoptosis in hepatoma cells is mediated by the CD95 (APO-1/Fas) receptor/ligand system and involves activation of wild-type p53. J Clinical Investigation 99:403–413
Narayanan BA, Condon MS, Bosland MC, Narayanan NK, Reddy BS (2003) Suppression of n-methyl-n-nitrosourea/testosterone-induced rat prostate cancer growth by celecoxib. Clinical Cancer res 9:3503–3513
Narayanan NK, Narayanan BA, Reddy BS (2005) A combination of docosahexaenoic acid and celecoxib prevents prostate cancer cell growth in vitro and is associated with modulation of nuclear factor-κB, and steroid hormone receptors. Int J Oncology 26:785–792
Naruse I, Fukumoto H, Saijo N, Nishio K (2002) Enhanced anti-tumor effect of trastuzumab in combination with cisplatin. Cancer Sci 93:574–581
Nicholson DW et al (1995) Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 376:37–43
Oeckinghaus A, Hayden MS, Ghosh S (2011) Crosstalk in NF-κB signaling pathways. Nature Immunology 12:695
Ozaki I et al (2007) Menatetrenone, a vitamin K2 analogue, inhibits hepatocellular carcinoma cell growth by suppressing cyclin D1 expression through inhibition of nuclear factor κB activation. Clinical Cancer Research 13:2236–2245
Park MH, Hong JT (2016) Roles of NF-κB in cancer and inflammatory diseases and their therapeutic approaches. Cells 5:15
Patial V, Mahesh S, Sharma S, Pratap K, Singh D, Padwad YS (2015) Synergistic effect of curcumin and piperine in suppression of DENA-induced hepatocellular carcinoma in rats. Environ Toxicology Pharmacology 40:445–452
Qiu G-H, Xie X, Xu F, Shi X, Wang Y, Deng L (2015) Distinctive pharmacological differences between liver cancer cell lines HepG2 and Hep3B. Cytotechnology 67:1–12
Ramyaa P, Padma VV (2014) Quercetin modulates OTA-induced oxidative stress and redox signalling in HepG2 cells—up regulation of Nrf2 expression and down regulation of NF-κB and COX-2 Biochimica et Biophysica Acta (BBA)-General Subjects 1840:681–692
Reuter S, Eifes S, Dicato M, Aggarwal BB, Diederich M (2008) Modulation of anti-apoptotic and survival pathways by curcumin as a strategy to induce apoptosis in cancer cells. Biochemical Pharmacology 76:1340–1351
Sasaki T et al (2013) Increased phosphorylation of AKT in high-risk gastric mucosa. Anticancer Res 33:3295–3300
Sato A et al (2011) Curcumin analog GO-Y030 is a novel inhibitor of IKKβ that suppresses NF-κB signaling and induces apoptosis. Cancer Science 102:1045–1051
Shaker MK, Abdella HM, Khalifa MO, Dorry AKE (2013) Epidemiological characteristics of hepatocellular carcinoma in Egypt: a retrospective analysis of 1313 cases. Liver Int 33:1601–1606
Shan RF, Zhou YF, Peng AF, Jie ZG (2014) Inhibition of Aurora-B suppresses HepG2 cell invasion and migration via the PI3K/Akt/NF-κB signaling pathway in vitro. Experimental Therapeutic Medicine 8:1005–1009
Shen H-M, Tergaonkar V (2009) NFκB signaling in carcinogenesis and as a potential molecular target for cancer therapy. Apoptosis 14:348–363
Shishodia S, Potdar P, Gairola CG, Aggarwal BB (2003) Curcumin (diferuloylmethane) down-regulates cigarette smoke-induced NF-κB activation through inhibition of IκBα kinase in human lung epithelial cells: correlation with suppression of COX-2, MMP-9 and cyclin D1. Carcinogenesis 24:1269–1279
Shishodia S, Amin HM, Lai R, Aggarwal BB (2005) Curcumin (diferuloylmethane) inhibits constitutive NF-κB activation, induces G1/S arrest, suppresses proliferation, and induces apoptosis in mantle cell lymphoma. Biochemical Pharmacology 70:700–713
Somers-Edgar TJ, Scandlyn MJ, Stuart EC, Le Nedelec MJ, Valentine SP, Rosengren RJ (2008) The combination of epigallocatechin gallate and curcumin suppresses ERα-breast cancer cell growth in vitro and in vivo. Int J Cancer 122:1966–1971
Sui W, Zhang Y, Wang Z, Wang Z, Jia Q, Wu L, Zhang W (2014) Antitumor effect of a selective COX-2 inhibitor, celecoxib, may be attributed to angiogenesis inhibition through modulating the PTEN/PI3K/Akt/HIF-1 pathway in an H22 murine hepatocarcinoma model. Oncology Reports 31:2252–2260
Tai DI, Tsai SL, Chang YH, Huang SN, Chen TC, Chang KS, Liaw YF (2000) Constitutive activation of nuclear factor κB in hepatocellular carcinoma Cancer 89:2274–2281
Teiten M-H, Eifes S, Dicato M, Diederich M (2010) Curcumin―the paradigm of a multi-target natural compound with applications in cancer prevention and treatment. Toxins 2:128–162
van Meerloo J, Kaspers GJ, Cloos J (2011) Cell sensitivity assays: the MTT assay Cancer cell culture: methods and protocols:237–245
Vaish V, Tanwar L, Sanyal SN (2010) The role of NF-κB and PPARγ in experimentally induced colorectal cancer and chemoprevention by cyclooxygenase-2 inhibitors. Tumor Biology 31:427–436
VanderVeen LA, Hashim MF, Shyr Y, Marnett LJ (2003) Induction of frameshift and base pair substitution mutations by the major DNA adduct of the endogenous carcinogen malondialdehyde. Proceedings National Academy Sci 100:14247–14252
Waked I et al (2016) Ombitasvir, paritaprevir, and ritonavir plus ribavirin for chronic hepatitis C virus genotype 4 infection in Egyptian patients with or without compensated cirrhosis (AGATE-II): a multicentre, phase 3, partly randomised open-label trial. Lancet Gastroenterology Hepatology 1:36–44. https://doi.org/10.1016/S2468-1253(16)30002-4
Wang W (2012) Enhanced antitumor effect via combination of triptolide with 5-fluorouracil in pancreatic cancer cell lines. J Clinical Oncology 30:352–352
Wyrebska A, Gach K, Janecka A (2014) Combined effect of parthenolide and various anti-cancer drugs or anticancer candidate substances on malignant cells in vitro and in vivo. Mini Rev Medicinal Chemistry 14:222–228
Xia J, Gao J, Inagaki Y, Kokudo N, Nakata M, Tang W (2013) Flavonoids as potential anti-hepatocellular carcinoma agents: recent approaches using HepG2 cell line Drug discoveries & therapeutics 7:1–8
Xia Y, Shen S, Verma IM (2014) NF-κB, an active player in human cancers. Cancer Immunology Res 2:823–830
Yoysungnoen P, Wirachwong P, Bhattarakosol P, Niimi H, Patumraj S (2006) Effects of curcumin on tumor angiogenesis and biomarkers, COX-2 and VEGF, in hepatocellular carcinoma cell-implanted nude mice. Clinical Hemorheology Microcirculation 34:109–115
Zhang HH, Zhang Y, Cheng YN, Gong FL, Cao ZQ, Yu LG, Guo XL (2018) Metformin incombination with curcumin inhibits the growth, metastasis, and angiogenesis of hepatocellular carcinoma in vitro and in vivo. Molecular Carcinogenesis 57:44–56
Acknowledgments
The authors would like to thank the researchers at the Medical Technology Center of the Medical Research Institute (Alexandria, Egypt), as well as, Prof. Abdel-Hamid Zaki at the National Research Center (Cairo, Egypt) for technical support.
Author contribution statement
AG, MH, MK, and FA conceived and designed research. FA and MH conducted experiments. FA, MH, MK, and AG contributed materials, technical and analytical tools. FA, MH, and AG analyzed data. AG and FA wrote the manuscript. All authors read and approved the manuscript.
Funding
The current research did not receive any grants from funding agencies in the public or commercial sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The current research has followed accepted principles of ethical and professional conduct according to approval reference number (1116PO1) by the Research Ethics Committee of the Faculty of Pharmacy, Damanhour University, regarding originality, risk control, and community service.
Conflict of interest
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Abdallah, F.M., Helmy, M.W., Katary, M.A. et al. Synergistic antiproliferative effects of curcumin and celecoxib in hepatocellular carcinoma HepG2 cells. Naunyn-Schmiedeberg's Arch Pharmacol 391, 1399–1410 (2018). https://doi.org/10.1007/s00210-018-1557-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-018-1557-6