
Abstract
Celecoxib is the only FDA selective cyclooxygenase-2 (COX-2) inhibitor approved nowadays. Studies showed that its therapeutic efficacy and toxicity may be related to inter-individual variability in pharmacodynamics and pharmacokinetics. This review aims to give an updated overview on celecoxib pharmacodynamics and pharmacokinetics in relation to genetics. For this purpose, a Medline search was performed to collect relevant literature between 2004 and 2014. Studies showed that single nucleotide polymorphism (SNP) in PTGS2-765G>C does not control COX-2 inhibitory effect of celecoxib. PGTS1 mutations may have an impact on the selectivity of celecoxib and as such on its gastrointestinal adverse events. Moreover, CYP2C9*2 and *3 allele were identified in bleeding patients taking celecoxib versus control patients. CYP2C9*2, CYP2C*3, two PTGS1 SNPs, and other variant genotypes have shown an association with acute coronary syndromes in patients taking celecoxib. As for the metabolism of celecoxib, in vitro and ex vivo studies showed a reduced clearance of celecoxib in individuals carrier of CYP2C9*3 allele and to a lower extent with CYP2C9*2 carriers. Studies also demonstrated that CYP2C8 does not have a major role in the metabolism of celecoxib. Non-conclusive data are found on the Uridine diphosphate glucuronosyltransferases (UGTs) which catalyze the second phase of the metabolism of celecoxib. As a conclusion, celecoxib should be used with caution in patients known or suspected to be poor CYP2C9 metabolizers. As for CYP2C8, UGTs and other genotypes further studies are still needed to confirm their role in the administration of celecoxib to the right person at the right dose and right time.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Several non-steroidal anti-inflammatory drugs (NSAIDs) are available in the market to treat inflammation, relieve pain, and reduce fever [30]. Those drugs exert their action mainly by inhibiting prostaglandins biosynthesis through preventing arachidonic acid (AA) to bind to the cyclooxygenase (COX) enzyme active site. Two main isoforms of COX enzymes exist: COX-1 and COX-2. COX-1 has a role in numerous physiological functions. It maintains normal renal function, protects the mucosa of the gastrointestinal tract, and produces proaggregatory thromboxane A2 in the platelets. COX-2 is present in negligible amounts at normal physiologic conditions. Genetically, it is regulated through several steps namely gene transcription and post transcription. Overexpression of COX-2 can lead to various pathologic conditions including inflammatory diseases and cancers [16]. Although traditional NSAIDs are effective in relieving pain and reducing inflammation mediated by COX-2, their usefulness is limited in many patients due to their gastrointestinal side effects. As a consequence, in order to produce potent anti-inflammatory and analgesic agents with fewer side effects compared to traditional NSAIDs, scientist in the 1990s intensified their research in order to design selective COX-2 inhibitors. From different drugs launched, only etoricoxib and celecoxib were allowed to remain in the market. In 2007, etoricoxib received a non-approvable letter from the Food and Drug Administration (FDA) due to safety concerns of an increased risk of cardiovascular events. Even though this is true, Merck marketed it outside the United States. On the other hand, celecoxib remained in the US market but with a black box warning indicating a risk of cardiovascular events [1, 3–5, 22]. Pharmacogenetics found that serious adverse events can be anticipated and thus avoided by knowing the presence or absence of certain alleles in patients to be treated [6, 14]. For example, celecoxib is known to be a substrate of CYP2C9, thus any polymorphism that affects this enzyme activity or expression may affect celecoxib clearance [2]. Other polymorphisms are also involved in the pharmacodynamics of celecoxib such as Prostaglandin-endoperoxide synthase (PTGS) 1 and 2, the genes that encode COX-1 and COX-2, respectively [9, 24].
Aim of the Work
Knowing that the therapeutic efficacy and toxicity of celecoxib are related to inter-individual variability [2, 15], the aim of this review is to have a complete genetic overview on celecoxib pharmacodynamics and pharmacokinetics as to implement the right drug, right dose, and right time for the right person.
Method
A Medline search was performed to collect all relevant literature between 2004 and 2014. Key words used were NSAIDs, celecoxib, pharmacodynamics, pharmacokinetics, and pharmacogenetics.
Celecoxib Pharmacodynamics
Following stimulation of the cell membrane by inflammatory signal, AA, the precursor of Prostaglandin (PG), is released through the action of phospholipases. The free AA is converted to eicosanoids catalyzed by COX, lipoxygenase, and cytochrome P450 enzyme. In the COX pathway, the initial reaction converts AA to prostaglandin G2 (PGG2). The latter is reduced to prostaglandin H2 which under the action of various cell-specific isomerases and synthases is converted to prostaglandin D2, prostaglandin E2 (PGE2), prostaglandin F2α, prostacyclin, and thromboxane A2 (TXA2). Celecoxib, a non-steroidal anti-inflammatory drug, which FDA indicated for osteoarthritis, rheumatoid arthritis, ankylosing spondylitis, acute pain, and primary dysmenorrhea, exerts its action by inhibiting the COX-2 pathway [7, 12, 15, 22]. Thus, the genetic variation of COX-2 that decreases the net activity of this enzyme may decrease the therapeutic success of celecoxib by shifting the inflammatory process to the other pathways. According to Cipollone et al. [9], single nucleotide polymorphism (SNP)-765G>C in the PTGS2 gene, which codes for COX-2, is accompanied with more than two times decrease in COX-2 expression. On the other hand, Skarte et al. [28] showed by in vivo whole blood assay that SNP in PTGS2-765G>C did not modulate COX-2 inhibitory effect of celecoxib. In fact, there was no statistical significance in the inhibition of PGE2 by celecoxib in the homozygous carriers of the PTGS2 wild-type 765G allele and the homozygous carriers of the PTGS2 variant-type 765G allele. COX-2 expression was also similar in the two groups. Consequently, as shown by Shi et al. [26], the difference in effectiveness of celecoxib is only attributed to the dose and not to genetic factors.
The most common side effects encountered with celecoxib related to the gastrointestinal (GI) tract include nausea, dyspepsia, flatulence, diarrhea, and abdominal pain. Upper GI ulcers and bleeding are also been reported even though they occur fewer than with traditional NSAIDs. This was shown in the Successive Celecoxib Efficacy and Safety Study I (SUCCESS-I) as well as by Singh et al [27]. The odds ratio of the occurrence of ulcer due to traditional NSAIDs as compared to celecoxib is equal to 7.02 (95 % CI 1.46–33.80, p = 0.008) [27]. The GI side effect of celecoxib may be attributed to the fact that the latter retains some COX-1 inhibitory activity which differs from patient to patient [22]. Data demonstrate that PTGS1 mutations may have an impact on the selectivity of celecoxib. PTGS1 SNP (c50C>T (rs3842787)) has shown to reduce COX-1 inhibition in T variant carriers taking celecoxib and thus leads to less GI side effects in those patients [13]. GI bleeding is also attributed to the amount of celecoxib present at the site of action. Thus, any mutation that decreases the metabolism of celecoxib may increase this side effect. According to Pilotto et al [21], CYP2C9*1/*2 and *3 allele were identified in GI bleeding patients taking celecoxib versus control patients with an odds ratio equal to 3.8 (95 % CI 1.090–13.19, p = 0.036) and 12.9 (95 % CI 2.917–57.922, p < 0.001), respectively.
TXA2 is synthesized by COX-1 in platelet. It stimulates smooth muscle proliferation, vasoconstriction, and platelet aggregation. Conversely, prostacyclin (PGI2) is synthesized from conversion of AA by COX-2 in vessel walls. It promotes vasodilation and inhibition of platelet aggregation. By selectively inhibiting COX-2, celecoxib creates an imbalance between TXA2 and PGI2 and thus leads to an increase in the risk of thrombosis [11]. In fact, CLASS study showed no significant difference between celecoxib and traditional NSAIDs in regard to serious cardiovascular events, while an increase in the cardiovascular events in patient taking celecoxib was shown in the APC trial as mentioned by Shi and Klotz [25]. This may be attributed to genetic differences between patients. In fact, two PTGS1 SNPs (rs10303135 and rs12353214) were significantly associated with acute coronary syndrome in patients taking celecoxib as found by St Germaine et al. [29]. Kraus et al. [20] found two variants of the prostaglandin E2 synthase gene (rs2241271 and rs2302821), one in the C-reactive protein gene (rs1800947) and three in the epidermal growth factor receptor signaling gene (rs6017996, rs6018256, and rs6018257), to be associated with celecoxib cardiovascular side effect. Moreover, CYP2C9*2 and CYP2C9*3 genotypes carriers have higher risk of cardiovascular adverse events with 400 mg celecoxib twice daily as compared to the wild carriers (RR 2.76; 95 % CI 1.08–7.06) as stated by Chan et al. [8]. Nevertheless, this correlation was not shown with patients randomized to celecoxib 200 mg twice daily [8]. Thus, dose administered may be another factor. As mentioned in a pooled analysis of six randomized trials comparing celecoxib with placebo, cardiovascular risk is twice greater in patients taking celecoxib twice per day as compared to once per day [15].
Celecoxib Pharmacokinetics
After oral administration, celecoxib reaches peak plasma level at approximately 3 h. Peak plasma level and area under the curve are dose dependent up to 200 mg twice daily under fasting conditions. High-fat meal may delay peak plasma level to about 1–2 h. A reduction in plasma celecoxib concentration is seen in patients co-administering aluminum- and magnesium-containing antacid [12, 15].
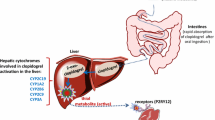
Celecoxib is extensively metabolized to inactive form in the liver and only less than 3 % is excreted unchanged through urine and feces. Seventy to ninety percent of the administered drug is metabolized via CYP2C9 to hydroxycelecoxib. The remaining part is catalyzed by CYP3A4. Carboxycelecoxib is formed by oxidation of hydroxycelecoxib under the influence of cytosolic alcohol dehydrogenases ADH1 and ADH2. By Uridine diphosphate glucuronosyltransferases (UGTs), carboxycelecoxib is conjugated to glucuronic acid to form 1-O-glucuronide. Approximately 27 % of the drug is excreted in the urine and 57 % excreted in the feces [12, 15, 23–25]. Elimination half-life ranges from 11 to 16 h. In patients with liver disease, the plasma level is increased by twofold [25].
Since celecoxib metabolism is primarily mediated by CYP2C9, the FDA has warned that patients known to be poor CYP2C9 metabolizers should be administered celecoxib with caution [15]. In fact, in several studies, homozygous CYP2C9*3/*3 mutants showed a higher area under the curve than wild-type CYP2C9*1/*1 subjects. Thus, carriers of CYP2C9*3 allele have an impaired hepatic clearance of celecoxib [1, 18, 23–25]. CYP2C9*3 allele has been reported in 5.8 % of the Caucasian, 5.3 % of the British, and 10 % of the Turkish individuals [17, 23]. CYP2C9*2 has also been linked to significant reduction in COX-2 activity. In vitro study showed that, in the presence of CYP2C9*2 allele, methyl hydroxylation of celecoxib is decreased by 34 %. Nevertheless, the effect of CYP2C9*2 is much weaker than that of CYP2C9*3 [15, 23].
CYP2C8 can have a role in the metabolism of celecoxib since its sequence is highly homologous to CYP2C9. Frequent CYP2C8 polymorphisms encountered are CYP2C8*2, CYP2C8*3, and CYP2C8*4. CYP2C8*2 allele is found in 18 % of African American, CYP2C8*3 in 13.7 % of Caucasians, and CYP2C8*4 in 6.6 % of Caucasians. Several allele variants of CYP2C8 have been associated with decreased metabolism of celecoxib [10, 23]. Nevertheless, according to Prieto-Perez et al. [23], an association between the pharmacokinetic of celecoxib and polymorphisms of CYP2C8 exists but with no statistical significance. This result suggests that this gene does not play a major role in the metabolism of celecoxib.
Uridine diphosphate glucuronosyltransferases catalyzes the second phase of the metabolism of celecoxib. Even though it is clear that celecoxib is metabolized by UGTs, the specific UGT involved is still unknown [24].
According to Karjalainen et al. [19], celecoxib inhibits CYP2D6 and thus increases its substrates such as metoprolol and dextromethorphan. In fact, in vivo, celecoxib has increased the area under the curves of metoprolol by 64 % and dextromethorphan by 13.6 %.
Conclusion
PTGS1 mutations may have an impact on the selectivity of celecoxib and as such on the gastrointestinal adverse events of celecoxib. CYP2C9*2 and *3 allele were identified in bleeding patients taking celecoxib versus control patients. CYP2C9*2, CYP2C*3, two PTGS1 SNPs, and others variant genotypes have shown an association with acute coronary syndrome in patients taking celecoxib. As for the metabolism of celecoxib, in vitro and ex vivo studies showed a reduced clearance of celecoxib in individuals carrier of CYP2C9*3 allele and to a lower extent with CYP2C9*2 carriers. These data are conclusive with the recommendation of the FDA requiring patients suspected to be poor CYP2C9 metabolizers based on genotype or previous history to be administered celecoxib with caution since the latter may lead to undesirable side effects such as gastrointestinal bleeding, myocardial infarction, and stroke. As for CYP2C8 and UGTs, data are still inconclusive and further studies are needed.
References
ADAPT Research Group (2006) Cardiovascular and cerebrovascular events in the randomized controlled Alzheimer’s disease anti-inflammatory prevention trial (ADAPT). PLoS Clin Trials 1:e33. doi:10.1371/journal.pctr.0010033
Ali ZK, Kim RJ, Ysla FM (2009) CYP2C9 polymorphisms: considerations in NSAID therapy. Curr Opin Drug Discov Dev 12(1):108–114
Arber N, Eagle CJ, Spicak J, Racz I, Dite P, Hajer J et al (2006) Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med 355:885–895. doi:10.1056/NEJMoa061652
Barkin RL (2012) Topical nonsteroidal anti-inflammatory drugs: the importance of drug, delivery, and therapeutic outcome. Am J Ther. doi:10.1097/MJT.0b013e3182459abd
Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K et al (2006) Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med 355:873–884. doi:10.1056/NEJMoa061355
Bielinski SJ, Olson JE, Pathak J, Weinshilboum RM, Wang L et al (2014) Preemptive genotyping for personalized medicine: design of the right drug, right dose, right time-using genomic data to individualize treatment protocol. Mayo Clin Proc 89(1):25–33. doi:10.1016/j.mayocp.2013.10.021
Brunton LL, Chabner BA, Knollmann BC (eds) (2011). Anti-inflammatory, antipyretic & analgesic agents; pharmacotherapy of gout. In: Goodman & Gilman’s the pharmacological basis of therapeutics, 12th edn, Chapter 34, McGraw-Hill, New York
Chan AT, Zauber AN, Hsu M, Breazna A, Hunter DJ, Rosenstein RB, Eagle CJ, Hawk ET, Bertagnolli MM (2009) Cytochrome P450 2C9 variants influence response to celecoxib for prevention of colorectal adenoma. Gatroenterology 136:2127–2136. doi:10.1053/j.gastro.2009.02.045
Cipollone F, Toniato E, Martinotti S, Fazia M, Iezzi A, Cuccurullo C et al (2004) A polymorphism in the cyclooxygenase 2 gene as an inherited protective factor against myocardial infarction and stroke. J Am Med Assoc 291:2221–2228. doi:10.1001/jama.291.18.2221
Daily EB, Aquilante CL (2009) Cytochrome P450 2C8 pharmacogenetics: a review of clinical studies. Pharmacogenomics 10(9):1489–1510. doi:10.2217/pgs.09.82
Dogne JM, De Leval X, Hanson J, Frederich M, Lambermont B, Ghuysen A et al (2004) New developments on thromboxane and prostacyclin modulators. Part I: thromboxane modulators. Curr Med Chem 11:1223–1241. doi:10.2174/0929867043365260
FDA (2011). Celebrex® (Celecoxib) capsules. http://www.fda.gov
Fries S, Grosser T, Price TS, Lawson JA, Kapoor S, DeMarco S, Pletcher MT, Wiltshire T, FitzGerald GA (2006) Marked interindividual variability in the response to selective inhibitors of cyclooxygenase-2. Gastroenterology 130(1):55–64. doi:10.1053/j.gastro.2005.10.002
Gharani N, Keller MA, Stack CB, Hodges LM, Schmidlen TJ et al (2013) The Coriell personalized medicine collaborative pharmacogenetics appraisal, evidence scoring and interpretation system. Genome Med 5:93. doi:10.1186/gm499
Gong L, Thorn CF, Bertagnolli MM, Grosser T, Altman RN, Klein TE (2012) Celecoxib pathways: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics 22(4):310–318. doi:10.1097/FPC.0b013e32834f94cb
Harper K, Tyson-Capper AJ (2008) Complexity of COX-2 gene regulation. Biochem Soc Trans 36(Pt 3):543–545. doi:10.1042/BST0360543
Ishizaki T (2002) CYP2C pharmacogenetics and tailor-made therapeutic implications. Int Congr Ser 1244:1–9
Kapur BM, Lala PK, Shaw JLV (2014) Pharmacogenetics of chronic pain management. Clin Biochem 47:1169–1187. doi:10.1016/j.clinbiochem.2014.05.065
Karjalainen MJ, Neuvonen PJ, Backman JT (2008) In vitro inhibition of CYP1A2 by model inhibitors, anti-inflammatory analgesics and female sex steroids: predictability of in vivo interactions. Basic Clin Pharmacol Toxicol 103:157–165. doi:10.1111/j.1742-7843.2008.00252.x
Kraus S, Hummler S, Toriola AT, Poole EM, Scherer D, Kotzmann J et al (2013) Impact of genetic polymorphisms on adenoma recurrence and toxicity in a COX2 inhibitor (celecoxib) trial: results from a pilot study. Pharmacogenet Genomics 23(8):428–437. doi:10.1097/FPC.0b013e3283631784
Pilotto A, Seripa D, Franceschi M, Scarcelli C, Colaizzo D, Grandone E, Nitro V, Andriulli A, Leandro G, Di Mario F, Dallapiccola B (2007) Genetic susceptibility to nonsteroidal anti-inflammatory drug-related gastroduodenal bleeding: role of cytochrome P450 2C9 polymorphisms. Gatroenterology 133(2):465–471. doi:10.1053/j.gastro.2007.05.025
Praveen Rao PN, Knaus EE (2008) Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): cyclooxygenase (COX) inhibition and beyond. J Pharm Pharm Sci 11(2):81s–110s
Prieto-Perez R, Ochoa D, Cabaleiro T, Roman M, Sanchez-Rojas SD et al (2013) Evaluation of the relationship between polymophisms in CYP2C8 and CYP2C9 and the pharmacokinetics of celecoxib. J Clin Pharmacol 53(12):1261–1267. doi:10.1002/jcph.169
Rollason V, Samer CF, Daali Y, Desmeules JA (2014) Prediction by pharmacogenetics of safety and efficacy of non-steroidal anti-inflammatory drugs: a review. Curr Drug Metab 15:326–343. doi:10.2174/1389200215666140202214454
Shi S, Klotz U (2008) Clinical use and pharmacological properties of selective COX-2 inhibitors. Eur J Clin Pharmacol 64:233–252. doi:10.1007/s00228-007-0400-7
Shi W, Wang YM, Li D, Chen NN, Chen BY (2004) Safety and efficacy of oral non-steroidal anti-inflammatory drugs in patients with rheumatoid arthritis: a six-month randomized study. Clin Drug Investig 24:89–101
Singh G, Fort JG, Goldstein JL, Levy RA, Hanrahan PS et al (2006) Celecoxib versus naproxen and diclofenac in osteoarthritis patients: SUCCESS-I study. Am J Med 119:255–266. doi:10.1016/j.amjmed.2005.09.054
Skarte C, Reus M, Schmidt R, Grundei I, Schuss P et al (2006) The cyclooxygenase 2 genetic variant-765G>C does not modulate the effects of celecoxib on prostaglandin E2 production. Clin Pharmacol Ther 80(6):621–632. doi:10.1016/j.clpt.2006.05.021
St. Germaine CG, Bogaty P, Boyer L, Hanley J, Engert JC, Brophy JM (2010) Genetic polymorphisms and the cardiovascular risk of non-steroidal anti-inflammatory drugs. Am J Cardiol 105:1740–1745. doi:10.1016/j.amjcard.2010.01.352
Wyatt JE, Pettit WL, Harirforoosh S (2012) Pharmacogenetics of nonsteroidal anti-inflammatory drugs. Pharmacogenomics J 12:462–467. doi:10.1038/tpj.2012.40
Author information
Authors and Affiliations
Corresponding author
Additional information
Endorsed by Asser Ghoneim.
Rights and permissions
About this article

Cite this article
Domiati, S., Ghoneim, A. Celecoxib for the Right Person at the Right Dose and Right Time: An Updated Overview. Springer Science Reviews 3, 137–140 (2015). https://doi.org/10.1007/s40362-015-0034-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40362-015-0034-6