
Abstract
Summary
We identified a novel large fragment deletion from intron 9 to 3’UTR in PLS3 (E10-E16del) in one Chinese boy with X-linked early-onset osteoporosis and vertebral fractures, which expanded the pathogenic spectrum of X-linked early-onset osteoporosis. Treatment with zoledronic acid was beneficial for increasing BMD and reshaping the vertebral bodies of this patient.
Introduction
X-linked early-onset osteoporosis is a rare disease, which is characterized by low bone mineral density (BMD), vertebral compression fractures (VCFs), and/or long bone fractures. We aimed to detect the phenotype and the underlying pathogenic mutation of X-linked early-onset osteoporosis in a boy from a nonconsanguineous Chinese family.
Methods
We investigated the pathogenic mutation of the patient with X-linked early-onset osteoporosis by targeted next-generation sequencing and confirmed it by Sanger sequencing. We also observed the effects of zoledronic acid on fracture frequency and BMD of the patient.
Results
Low BMD and multiple VCFs were the main phenotypes of X-linked early-onset osteoporosis. We identified a total of 12,229 bp deletion in PLS3, involving intron 9 to the 3’UTR (E10-E16 del). This large fragment deletion might be mediated by Alu repeats and microhomology of 26 bp at each breakpoint junction. Zoledronic acid treatment could significantly increase the Z-score of BMD and reshape the compressed vertebral bodies.
Conclusion
We identified a large fragment deletion mutation in PLS3 for the first time and elucidated the possible mechanism of the deletion, which led to X-linked early-onset osteoporosis and multiple vertebral fractures. Our findings would enrich the etiology spectrum of this rare disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
X-linked early-onset osteoporosis is a rare disorder, which is predominantly characterized by low bone mineral density (BMD), vertebral compression fractures (VCFs), and long bone fractures under minor trauma [1,2,3,4,5]. However, typical extraskeletal manifestations of osteogenesis imperfecta (OI) are usually absent in this disease, such as blue sclerae, dentinogenesis imperfecta, joint hyperlaxity, hearing loss, and so on.
In 2013, PLS3 (MIM 300131) was detected for the first time as a pathogenic gene of X-linked early-onset osteoporosis in five families [1]. PLS3 has 16 exons and is located on chromosome Xq23 (chrX: 115,561,182-115,650,861 bp, UCSC GRCh38/hg38). PLS3 encodes plastin 3, also known as T-plastin, a 630-amino-acid protein. It is almost ubiquitously expressed in solid tissues, which is involved in the binding and bundling of actin filaments in the cytoskeleton [6]. Plastin 3 has two N-terminal EF hand motifs and two C-terminal actin-binding domains (ABDs) that are composed of two calponin-homology (CH) domains [7]. In the full-length structure, a nuclear export signal (NES) overlaps an N-terminal EF hand motif. EF hands can regulate protein function through binding calcium. Upon binding of ABD1 and ABD2 to each actin filament, plastin 3 can cross-link the filaments to form actin bundles, which is important for osteocyte dendrites [8].
Previous research showed that PLS3 knockdown zebrafish manifested a malformation of craniofacial bone structure [1]. A PLS3 polymorphism (rs140121121) was associated with a risk of fracture among elderly heterozygous women that was two times as high as that among noncarriers [1]. It has been hypothesized that deficiency or decreased expression of plastin 3 caused by pathogenic mutations in PLS3 may disturb osteocyte mechanosensing [1, 9]. As far as we know, only nine families have been reported to suffer from X-linked early-onset osteoporosis induced by PLS3 mutations [1,2,3,4,5], with the majority of the affected individuals being male. As of yet, no information about PLS3 mutations is available in Chinese patients.
Therefore, we set out to investigate the phenotype of a boy with early-onset osteoporosis from a nonconsanguineous family of Chinese Han origin. We aimed to detect pathogenic mutation in PLS3, and to prospectively evaluate the effects of zoledronic acid on fracture frequency and bone mineral density (BMD) of the patient.
Material and methods
Subjects
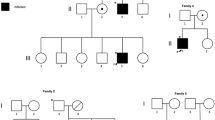
The proband, a 13-year-old boy, of Chinese Han origin, suffered from back pain for 8.5 years and gradually developed kyphosis. The proband, his parents, and his sister were included in this study. The pedigree of the family was shown in Fig. 1a. The proband suffered from VCFs and was diagnosed as osteoporosis by department of pediatrics and department of endocrinology, Peking Union Medical College Hospital (PUMCH) in 2012.

Confirmation of the large fragment deletion mutation in PLS3 and prediction of the possible mechanism of the rearrangement. a Pedigree of the early-onset osteoporosis family in this study. The proband was designated with an arrow. b Schematic partial representation of the region (intron 9 to 3’URT) around which the gDNA-walking strategy was designed and performed. The number and series of forward and reverse primers had been depicted with arrows. The deletion breakpoints were depicted by slanted lines, which were located in introns 9 and 3’UTR. Blue arrows: forward primers, purple arrows: reverse primers. c Sanger sequencing results by three primer sets (P-1F/P-7R, P-1F/P-1R, and P-1F/P-1R). The sequence amplified using the primer set P-1F/P-7R came from the proband. The sequence amplified using the primer set P-1F/P-1R and P-1F/P-1R both came from his sister. In the proband, genomic DNA could be amplified using the special primer set (P-1F/P-7R). In his father and sister, DNA could be amplified by PCR using P-1F/P-1R and P-7F/P-7R, but not by P-1F/P-7R. In his mother, DNA could be amplified by P-1F/P-7R, P-1F/P-1R, and P-7F/P-7R. d Mechanistic model of microhomology-mediated end joining (MMEJ) of the large fragment deletion in PLS3. The microhomology sequences surrounding the deletions (chrX: 115,641,420-115,641,445 for the proximal element, and chrX: 115,653,649-115,653,674 for the distal one) and the AluSx1 elements at the breakpoint junctions were shown in rectangular frames
Clinical evaluation
Clinical data were collected in detail, including bone fracture history, growth speed, and development. The bone, teeth, joint, sclerae, and neuromuscular functions were examined carefully. Fractures were suspected by medical history and were confirmed by X-ray films of bone. The height of the patient was measured with Harpenden stadiometer (Seritex Inc., East Rutherford, NJ). Age- and sex-adjusted height was calculated as Z-score based on a growth curve of normal Chinese children [10].
Biochemical measurements
Serum concentrations of calcium (Ca), phosphate (P), total alkaline phosphatase (ALP), alanine aminotransferase (ALT), and creatinine (Cr) were measured using an automatic biochemical analyzer in central laboratory of PUMCH. Serum levels of β-cross linked C-telopeptide of type I collagen (β-CTX), 25-hydroxyvitamin D (25OHD), and intact parathyroid hormone (PTH) were measured using an automated electrochemiluminescence system (Roche Diagnostics, Switzerland). Serum concentration of pigment epithelium-derived factor (PEDF) was determined by DuoSet ELISA Kit (R&D Systems, Minneapolis, MN, USA).
Radiographic assessments
Bone mineral density at lumbar spine and proximal hip was measured by dual-energy X-ray absorptiometry (DXA, Lunar Prodigy, USA) with appropriate pediatric software. BMD data were transformed to age- and gender-specific Z-score based on normal reference data of BMD in Asian children [11]. X-ray films of the skull, lower extremities, pelvis, and thoracolumbar spine were examined. Bone age was assessed according to X-ray film of hand with method of Greulich and Pyle (GP) [12]. A vertebral fracture was defined as a 20% or greater reduction in vertebral height [13].
Treatment and follow-up
Infusion of zoledronic acid (Aclasta, Novartis Pharma Stein AG) was given to the boy at a dose of 0.1–0.125 mg per kilogram body weight annually from the age of 10.5 years. Supplementation with 500 mg calcium plus 200 IU vitamin D3 (Calcichew D, GE pharmaceutical co., LTD), and 0.25 μg calcitriol (Roche Pharma Ltd.) were prescribed daily. Changes in bone turnover biomarkers, BMD, and X-ray films of thoracolumbar spine were evaluated every 6 months to investigate the effects of zoledronic acid. All adverse events were recorded in detail during the follow-up.
Detection of pathogenic mutation by targeted next-generation sequencing
Peripheral venous blood samples were obtained from the proband, his parents, and sister. In order to confirm that the gene mutation was not merely polymorphisms, blood samples were also drawn from 100 unaffected, unrelated, and healthy subjects. Genomic DNA (gDNA) was extracted from white blood cells using a DNA extraction kit (QIAamp DNA; Qiagen, Germany).
In order to identify the pathogenic genetic defect, the gDNA of the proband was sequenced using a targeted next-generation sequencing (NGS) panel (Illumina HiSeq2000 platform, Illumina, Inc., San Diego, CA, USA). The known pathogenic genes of OI (COL1A1, COL1A2, PPIB, IFITM5, SERPINF1, CRTAP, LEPRE1, SERPINH1, FKBP10, SP7, BMP1, TMEM38B, and WNT1) and early-onset osteoporosis (LRP5 and PLS3) were included in this panel [1,2,3,4,5, 14,15,16]. Bioinformatic analysis of the NGS data was carried out by previously described method [17]. Only those mutations expected to produce damaged protein were further analyzed, which included frame shift variants, nonsense variants, missense variants, and acceptor and donor splice sites variants.
Investigation of recombination breakpoint junctions
Exon 10 to exon 16 of PLS3, as well as their exon/intron boundaries were amplified from the gDNA of the proband using polymerase chain reaction (PCR). To identify the deletion breakpoint junctions, we adopted a primer-walking approach to test the gDNA. A series of primers, mapping around the suspicious deletion region, were used in a PCR assay in order to amplify the junction fragment. Finally, a specific primer set (P-1F/P-7R) was identified (Fig. 1b). Another two primer sets (P-1F/P-1R and P-7F/P-7R) were designed to detect the normal sequence at the breakpoint junctions (Fig. 1b). Primers for standard PCR and sequencing were designed from the genomic sequence (NG_012518.2), using the web-based Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/). The primer sequences used to amplify exon 9 to 16, and the specific primer sets were listed in Table S. PCR was conducted with TaqMix DNA polymerase (Biomed, China) and its standard buffer, under the following conditions: initial denaturation at 95 °C for 3 min, followed by 35 cycles at 95 °C for 30 s, 58–61 °C for 30 s, and 72 °C for 40–100 s. PCR products were purified and sequenced using an ABI 377 DNA automated sequencer with dye terminator cycle sequencing kits (Applied Biosystems).
In normal individuals, gDNA could not be PCR-amplified using the special primer set (P-1F/P-7R) due to the large amplicon size. However, DNA could be amplified in hemizygous man and heterozygous woman because the large deletion would lead to an amplicon size suitable for PCR [18]. Upon identification of the breakpoint junctions, we subjected our findings to BLAT (UCSC Genome Browser, GRCh38/hg38 assembly) to identify the length and location of the large fragment deletion.
We analyzed the 300 bp sequences at the proximal and distal side of each breakpoint junction to elucidate the potential mechanism of the large fragment deletion. Each sequence was analyzed for repetitive DNA with RepeatMasker (http://www.repeatmasker.org/), and for tandem repeats with Tandem Repeats Finder (http://tandem.bu.edu/trf/trf.html) [19,20,21]. The sequences were aligned with each other in order to detect identical regions (microhomologies) that could result in a large fragment deletion [22, 23].
Targeted NGS was only performed in the proband. Biochemical index were measured and X-ray films were taken in four members of the family. PCR of PLS3 was performed in 4 members of the family and in 100 healthy controls, who were selected from the volunteers of the study of Chinese bone turnover markers study (CHBTMS) [24]. The study protocol was approved by the Scientific Research Ethics Committee of PUMCH, and signed informed consents were obtained from the patient, his sister, his parents, and all healthy controls before they participated in this study.
Results
Clinical phenotype of the patient
The proband, a 13-year-old boy, was the second child of a healthy nonconsanguineous family. He had no skeletal deformities at birth. Cognitive and motor development was normal. From 2 years old, he suffered from back pain and gradually manifested kyphosis. The symptoms aggravated at 9 years old. He had no fractures of the extremities. He came to our clinic at 10.5 years old. He had no blue sclerae, dentinogenesis imperfecta, joint hyperlaxity, deafness, and neuromuscular abnormalities. His height was 142 cm (50th percentile) and weight was 40 kg (50th to 75th percentile).
Serum levels of calcium, phosphate, ALP, and PEDF were within age-matched normal range. As the normal reference range of β-CTX was not available in Chinese children, we could not judge if the level of β-CTX of our patient was normal. Serum 25OHD level of the patient was 11.3 ng/ml, indicating vitamin D deficiency. Serum PTH level of the patient was normal. No secondary hyperparathyroidism was found because the patient had received treatment of calcitrol and calcium. As the patient had compressed fracture at the first lumbar vertebra, we only measured the BMD of lumbar spine 2–4 (L2–4). The areal BMD at L2–4 and femoral neck were 0.409 and 0.454 g/cm2, respectively. BMD Z-score were obviously low as −3.0 at L2–4 and −3.4 at the femoral neck, which indicated that the patient had a lower BMD than age- and sex-matched healthy controls. X-ray films revealed osteoporosis of the spine with multiple thoracic and lumbar VCFs. No wormian bones were observed. The cortical width and shape of the long bones were roughly normal. The bone age was appropriate for the chronological age (Fig. 2).

Phenotype of the proband and effects of zoledronic acid treatment. a For the proband: A no wormian bones at the occipital region; B the bone age was appropriate to chronological age; C–D no abnormities of lower extremities and pelvis; E lateral X-ray of the spine showed several vertebral compressions in the proband, indicated by arrows; F–G lateral X-ray of spine showed reshape of several compressed vertebral bodies after 12 and 24 months of zoledronic acid treatment. b Changes in BMD Z-score at lumbar spine and femoral neck after treatment with zoledronic acid. ZOL zoledronic acid
Moreover, the patient’s father was 48 years old and his height was 171 cm. The BMD Z-score of the father was 0.4 at lumbar spine and −0.4 at the femoral neck. His mother was 47 years old, 3 years since menopause. Her BMD T-score was −1.4 at lumbar spine and −0.8 at the femoral neck. Secondary hyperparathyroidism was found in his mother because of vitamin D deficiency and no supplementation with vitamin D. No vertebral fracture was found in the parents of the patient. The boy’s sister, 23 years old, had normal BMD and height of 163 cm. The clinical, biochemical characteristics, and bone mineral density of the patient, his parents, and sister were presented in Table 1 and Fig. S.
Mutations of PLS3
A large fragment deletion from exon 10 to exon 16 in PLS3 gene was identified by NGS in the proband. Regular PCR primers designed to amplify exon 10 to exon 16 did not yield any products, which suggested the presence of a large fragment deletion. In the proband, the primer-walking approach could not amplify gDNA using P-1, P-2, P-3, P-4, P-5, P-6, and P-7, while gDNA could be amplified with P-E9, P-8, and P-9 primers (Fig. 1b). Therefore, we suspected that the breakpoint junctions were located in sequences that could be amplified by P-1F/P-1R and P-7F/P-7R in a normal person. We used the specific primer set, P-1F/P-7R to amplify the proband gDNA and could identify the sequence around the breakpoint junctions. DNA from the proband’s father and sister could be amplified by PCR using the primers P-1F/P-1R and P-7F/P-7R, but not with the primer P-1F/P-7R. DNA from the proband’s mother could be amplified using the primers P-1F/P-7R, P-1F/P-1R, and P-7F/P-7R. The sequences amplified by P-1F/P-7R, P-1F/P-1R, and P-7F/P-7R were shown in Fig. 1c. Because PLS3 was located on the X-chromosome, we suspected the proband’s mother was heterozygous, and the patient was hemizygous carrier of mutant PLS3.
Using the UCSC blast search, a total of 12,229 bp deletion was identified at Xq23. The proximal breakpoint of this large deletion was located in intron 9, and the distal one mapped downstream of the 3’UTR. We mapped a 26-bp identical sequence (chrX: 115,641,420-115,641,445 for the proximal element, and chrX: 115, 653, 649-115, 653, 674 for the distal one) at each breakpoint junction. The exact breakpoint could not be determined at a nucleotide level due to the presence of microhomology in both segments (Fig. 1c, d). Bioinformatic analysis demonstrated that the proximal and distal breakpoint junctions were both within an AluSx1 element. No tandem repeats were identified at either segment.
The PLS3 mutation identified in this boy was absent in the 100 healthy controls, which did not match polymorphisms in any of the public databases. By NGS, we did not detect mutation in the other pathogenetic genes of autosomal dominant or recessive inherited OI in this patient.
Treatment and follow-up
After 24 months of treatment with zoledronic acid, the BMD Z-score significantly increased from −3.0 to 2.1 at lumbar spine and from −3.4 to −0.4 at femoral neck (Fig. 2b). Back pain and kyphosis were significantly relieved. During treatment, the height of the proband arrived at 166 cm (90th–97th percentile) when he was 12.5 years old. X-ray films indicated significant improvement of osteoporosis in the spine. Compressed vertebrae were reshaped and no bone fracture occurred during a 2-year treatment of zoledronate acid.
The patient had an acute-phase reaction within the second day after the first infusion of zoledronic acid, with the highest body temperature of 39 °C. No other adverse effect was observed.
Discussion
We identified a large fragment deletion mutation in PLS3 for the first time in a 13-year-old Chinese boy with X-linked early-onset osteoporosis. The proband presented with early-onset multiple vertebral compressed fractures and low BMD. The longitudinal growth was normal. Deformities of long bone or typical extraskeletal manifestations of OI were not observed. We identified a novel large fragment deletion mutation in PLS3 spanning intron 9 to the 3’UTR, which could be mediated by Alu elements and microhomology. His mother was heterozygous carrier of this mutation. Treatment with zoledronic acid was beneficial in increasing BMD and reshaping the compressed vertebrae.
The exact mechanism of PLS3 mutations leading early-onset primary osteoporosis had not been completely elucidated. It was thought that plastin 3 was involved in cytoskeleton modification [7], which was important for osteocytes sensing local mechanical strains, releasing and receiving signals to control bone matrix production, mineralization, and resorption [12, 25]. Actin filaments could also influence nuclear shape, allowing direct transmission of physical stimuli to the nuclear region [12]. In addition, expression of plastin 3 increased when osteoblastic MC3T3-E1cells started to mineralize, which indicated that plastin 3 may play a role in modulating bone mineralization [2]. However, the mechanism of PLS3 mutations inducing early-onset osteoporosis remained to be investigated.
Including the novel mutation in our study, only ten mutations in PLS3 had been reported in X-linked osteoporotic patients [1,2,3,4,5]. They were distributed in exon 3, 8, 10, 13, 14, and 15, intron 2 and 7 in PLS3, including 2 missense mutations (family 8 and 10), 1 nonsense mutation (family 3), 3 deletion mutations (family 2, 6, and 7), 1 insertion mutation (family 5), 2 splice-site mutations (family 4 and 9), and 1 large fragment deletion mutation (family 1 in our study) (Table 2 and Fig. 3). This was the first case of X-linked early onset osteoporosis induced by a novel large fragment deletion in PLS3. The identified mutations in PLS3 were shown in Fig. 3. The protein produced by the mutated PLS3 in our study was likely to be severely functionally compromised. A truncated protein would cause the deletion of CH2, CH3, and CH4, abolishing part of the first and the entire second actin-binding domain [7].

Schematic presentation of the coding exons of PLS3 cDNA (NM_005032.6) and of PLS3 protein (NP_005023.2). The locations of the exons were aligned relatively to the encoding regions of the PLS3 protein. The positions of the mutations reported in the previous studies were indicated by black words. The mutation in our patient was indicated by red words. NES nuclear export signal; EF EF hand motifs, CH calponin-homology domains, ABD actin-binding domains
As we know, microhomologies were a series of nucleotides (<70 bp) that were identical at the breakpoint junctions and could contribute to genomic rearrangement. They had been identified in many diseases such as X-linked nephrogenic diabetes insipidus [18], recurrent hydatidiform moles [26], and intellectual disability [20]. Of the short interspersed elements (300 bp) found in the human genome, Alu elements were the most abundant and contributed to genomic diversity and instability [26]. Using the primer-walking approach, we identified the precise breakpoint junctions of the mutation in PLS3. The breakpoints were within Alu elements and were flanked on both sides by 26-bp microhomology, one of which was completely removed by the deletion, forming a chimeric Alu hybrid. The presence of Alu elements and microhomology suggested that this large fragment deletion might be caused by microhomology-mediated end joining (MMEJ) [21, 26] (Fig. 1d).
We reviewed the phenotypes of patients with PLS3 mutations [1,2,3,4,5] (Table 2). All of the previously reported patients with PLS3 mutations did not have extraskeletal manifestations, except for two patients from family 10 with blue sclerae and hearing loss [4]. Patients with PLS3 mutations could present with waddling gait (family 2) and clumsy gait (family 7) in childhood [2, 3]. In spinal muscular atrophy (SMA), PLS3 expression restored the motor neuron and rescued neuromuscular junction defects [27]. Furthermore, knockdown of PLS3 in zebrafish caused muscle tissue deformities [1]. These findings highlighted the potential importance of assessing neuromuscular function in patients with PLS3 mutations. The mutations in family 2, 3, 7, and 9 affected more than one structural motif, however no obvious phenotypic differences were observed among patients with different motifs affected. Moreover, Lyon et al. found that EF-hand motifs of PLS3, but not actin-binding domains, were important to rescue motor axon outgrowth defects in SMN-knockdown zebrafish [27]. Therefore, we suspected that patients with PLS3 mutations involving EF-hand motifs might have more severe manifestations. However, the manifestations of family 2 and 9 were similar to those of the other families with mutations not affecting EF-hand motifs. Thus, the genotype-phenotype correlations were still needed to be investigated in patients with PLS3 mutations.
Due to its X-chromosomal location, mutations in PLS3 affected male patients in a more severe manner than female patients. Few female patients with PLS3 mutations had recurrent peripheral fractures and multiple vertebral compressed fractures [3]. The proband’s mother had only osteopenia and never suffered from bone fractures, which was in line with the penotypes of heterozygous females in other families with PLS3 mutations [1,2,3].
Bisphosphonates were widely used to treat bone fragility in children [28,29,30], and this treatment was given to almost all affected patients in family 2 to family 9, with the exception of family 8 and 10. Although some patients had long bone fractures during the treatment, bisphosphonates could increase BMD Z-score (or T-score). In our patient, zoledronic acid resulted in an apparent increase in BMD Z-score and reshape of compressed vertebrae. As we know, bone growth was quickly and BMD was obviously increased in puberty [31]. We could not confirm that the increase in BMD and its Z-score of our patient was the effects of zoledronic acid or skeletal growth, because we did not establish a control and our patient was at puberty. The effects of bisphosphonates on bone of patients with PLS3 mutation needed to be further evaluated in a large sample of patients. However, we deduced the reshape of compressed vertebrae was the effects of bisphosphonate treatment. Thirty-seven children with OI, who received intravenous bisphosphonates treatment before 5 years old, were followed for more than 10 years. Thirty-five percent of vertebrae were compressed at baseline, whereas only 6% of vertebrae were compressed at the last evaluation [32]. In addition, Land et al. found compressed vertebrae would become more flatten and more concave with age if they did not receive medical treatment [33]. Therefore, we thought that bisphosphonates treatment were beneficial to reshape the compressed vertebrae. The effectiveness of zoledronic acid stemmed from its capacity to suppress osteoclastic activity. Zoledronic acid might play roles through inhibiting the bone resorption of the patient with PLS3 mutations. However, PLS3 was indicated to modulate the dendrite function and mechanosense of osteocytes [2]. It was worth investigating if zoledronic acid also had subsequent effects on osteocytes.
In conclusion, this was the first case of X-linked early onset osteoporosis induced by a novel large fragment deletion from intron 9 to 3’UTR (E10-E16del) in PLS3. We elucidated the mechanism of the large fragment deletion causing by microhomology-mediated end joining through finding the precise breakpoint junctions. We also prospectively demonstrated that zoledronic acid could increase the BMD and its Z-score, and was beneficial to reshape the compressed vertebrae of the patient. Our findings could expand the pathogenic spectrum of X-linked early-onset osteoporosis and indicated the possible effects of bisphosphonates on this disease.
References
van Dijk FS, Zillikens MC, Micha D (2013) PLS3 mutations in X-linked osteoporosis with fractures. N Engl J Med 369:1529–1536
Fahiminiya S, Majewski J, Al-Jallad H (2014) Osteoporosis caused by mutations in PLS3: clinical and bone tissue characteristics. J Bone Miner Res 29:1805–1814
Laine CM, Wessman M, Toiviainen-Salo S (2015) A novel splice mutation in PLS3 causes X-linked early onset low-turnover osteoporosis. J Bone Miner Res 30:510–518
Nishi E, Masuda K, Arakawa M (2016) Exome sequencing-based identification of mutations in non-syndromic genes among individuals with apparently syndromic features. Am J Med Genet A 170:2889–2894
van de Laarschot DM, Zillikens MC (2016) Atypical femur fracture in an adolescent boy treated with bisphosphonates for X-linked osteoporosis based on PLS3 mutation. Bone 91:148–151
Delanote V, Vandekerckhove J, Gettemans J (2005) Plastins: versatile modulators of actin organization in (patho) physiological cellular processes. Acta Pharmacol Sin 26:769–779
Klein MG, Shi W, Ramagopal U (2004) Structure of the actin crosslinking core of fimbrin. Structure 12:999–1013
Tanaka-Kamioka K, Kamioka H, Ris H (1998) Osteocyte shape is dependent on actin filaments and osteocyte processes are unique actin-rich projections. J Bone Miner Res 13:1555–1568
Atkins GJ, Findlay DM (2012) Osteocyte regulation of bone mineral: a little give and take. Osteoporos Int 23:2067–2079
Khadilkar AV, Sanwalka NJ, Chiplonkar SA (2011) Normative data and percentile curves for Dual Energy X-ray Absorptiometry in healthy Indian girls and boys aged 5–17 years. Bone 48:810–819
Li H, Ji CY, Zong XN (2009) Height and weight standardized growth charts for Chinese children and adolescents aged 0 to 18 years. Zhonghua Er Ke Za Zhi 47:487–492 [Chinese]
Milner GR, Levick RK, Kay R (1986) Assessment of bone age: a comparison of the Greulich and Pyle, and the Tanner and Whitehouse methods. Clin Radiol 37:119–121
Makitie O, Doria AS, Henriques F (2005) Radiographic vertebral morphology: a diagnostic tool in pediatric osteoporosis. J Pediatr 146:395–401
Forlino A, Marini JC (2016) Osteogenesis imperfecta. Lancet 387:1657–1671
Fahiminiya S, Majewski J, Roughley P (2013) Whole-exome sequencing reveals a heterozygous LRP5 mutation in a 6-year-old boy with vertebral compression fractures and low trabecular bone density. Bone 57:41–46
Korvala J, Juppner H, Makitie O (2012) Mutations in LRP5 cause primary osteoporosis without features of OI by reducing Wnt signaling activity. BMC Med Genet 13:26
Lv F, Xu XJ, Wang JY (2016) Two novel mutations in TMEM38B result in rare autosomal recessive osteogenesis imperfecta. J Hum Genet 61:539–545
Cho SY, Law CY, Ng KL (2016) Novel large deletion in AVPR2 gene causing copy number variation in a patient with X-linked nephrogenic diabetes insipidus. Clin Chim Acta 455:84–86
Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27:573–580
Froyen G, Belet S, Martinez F (2012) Copy-number gains of huwe1 due to replication- and recombination-based rearrangements. Am J Hum Genet 91:252–264
Shaw CJ, Lupski JR (2004) Implications of human genome architecture for rearrangement-based disorders: the genomic basis of disease. Hum Mol Genet 13 Spec No 1:R57-64
Ottaviani D, LeCain M, Sheer D (2014) The role of microhomology in genomic structural variation. Trends Genet 30:85–94
Hastings PJ, Ira G, Lupski JR (2009) A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 5:e1000327
Li M, Lv F, Zhang Z (2016) Establishment of a normal reference value of parathyroid hormone in a large healthy Chinese population and evaluation of its relation to bone turnover and bone mineral density. Osteoporos Int 27:1907–1916
Rochefort GY, Pallu S, Benhamou CL (2010) Osteocyte: the unrecognized side of bone tissue. Osteoporos Int 21:1457–1469
Reddy R, Nguyen NM, Sarrabay G (2016) The genomic architecture of NLRP7 is Alu rich and predisposes to disease-associated large deletions. Eur J Hum Genet 24:1445–1452
Lyon AN, Pineda RH, Hao le T (2014) Calcium binding is essential for plastin 3 function in Smn-deficient motoneurons. Hum Mol Genet 23:1990–2004
Bishop N, Adami S, Ahmed SF (2013) Risedronate in children with osteogenesis imperfecta: a randomised, double-blind, placebo-controlled trial. Lancet 382:1424–1432
Baroncelli GI, Vierucci F, Bertelloni S (2013) Pamidronate treatment stimulates the onset of recovery phase reducing fracture rate and skeletal deformities in patients with idiopathic juvenile osteoporosis: comparison with untreated patients. J Bone Miner Metab 31:533–543
Dwan K, Phillipi CA, Steiner RD (2016) Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev 10:CD005088
Trejo P, Rauch F (2016) Osteogenesis imperfecta in children and adolescents—new developments in diagnosis and treatment. Osteoporos Int 27:3427–3437
Palomo T, Fassier F, Ouellet J (2015) Intravenous bisphosphonate therapy of young children with osteogenesis imperfecta: skeletal findings during follow up throughout the growing years. J Bone Miner Res 30:2150–2157
Land C, Rauch F, Munns CF (2006) Vertebral morphometry in children and adolescents with osteogenesis imperfecta: effect of intravenous pamidronate treatment. Bone 39:901–906
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No. 81570802), National Key Research and Development Program of China (No. 2016YFC0901501), and CAMS Initiative for Innovative Medicine (2016-I2M-3-003). We thank the staff in the department of radiology department for measurement of bone mineral density and interpretation of X-ray films. We also thank the boy with PLS3 mutation and his family members for participation in this research. We thank all unaffected, unrelated individuals for providing control DNA samples.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
None.
Rights and permissions
About this article

Cite this article
Lv, F., Ma, M., Liu, W. et al. A novel large fragment deletion in PLS3 causes rare X-linked early-onset osteoporosis and response to zoledronic acid. Osteoporos Int 28, 2691–2700 (2017). https://doi.org/10.1007/s00198-017-4094-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-017-4094-0