
Abstract
Osteocytes actively participate in almost every phase of mineral handling by bone. They regulate the mineralisation of osteoid during bone formation, and they are also a major RANKL-producing cell. Osteocytes are thus able to liberate bone mineral by regulating osteoclast differentiation and activity in response to a range of stimuli, including bone matrix damage, bone disuse and mechanical unloading, oestrogen deficiency, high-dose glucocorticoid and chemotherapeutic agents. At least some of these activities may be regulated by the osteocyte-secreted product, sclerostin. There is also mounting evidence that in addition to regulating phosphate homeostasis systemically, osteocytes contribute directly to calcium homeostasis in the mature skeleton. Osteocyte cell death and the local loss of control of bone mineralisation may be the cause of focal hypermineralisation of bone and osteopetrosis, as seen in aging and pathology. The sheer number of osteocytes in bone means that ‘a little give and take’ in terms of regulation of bone mineral content translates into a powerful whole organism effect.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteocytes (OY) are the predominant bone cell type, comprising 90–95% of all bone cells and are also the most long lived, with a life span of up to 25 years (reviewed in Ref. [1]). Each osteocyte possesses numerous, up to 50, long and branched cellular processes that extend throughout the bone by virtue of a network of interconnecting canaliculi (Fig. 1). OY processes contact each other, and possibly other cell types, by means of specialised nodal structures termed gap junctions, comprising connexins (mainly connexin 43) and integrins, which facilitate the intercellular transport of small signalling molecules such as prostaglandins and nitric oxide [2]. In cross-sections of human cortical bone, the distance between canaliculi is typically around 4 μm [3] and OY processes, if not OY cell bodies, are never far from the other bone cell types including lining cells, osteoblasts, bone endothelial cells and bone resorbing osteoclasts. Indeed, the number of canaliculi contacting the under surface of each lining osteoblast was calculated in rabbit bone to be between 8 and 20 per cell, the number of contacts increasing with the age of the animal [4]. Kamioka et al. observed in mouse calvariae that more mature OY were extensively connected to immature or osteoid osteocytes, OY most recently embedded in mineralising osteoid, and it was these cells that retained contact via their processes to the surface osteoblasts [5]. In that study, a limited number of OY processes (about 3%) were also observed to extend through to the vascular side of the lining cell layer providing the potential to ‘talk to’ mesenchymal and haemopoietic cells resident in the marrow [5]. The OY syncytium is therefore ideally placed to both sense and communicate requirements for mineral homeostasis, skeletal loading, as well as insults inflicted upon it.

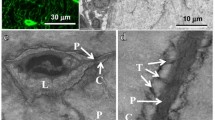
Osteocyte connectivity. SEM image of resin-embedded and acid-etched rat tibial bone, demonstrating the elongated osteocyte lacunae and the associated network of highly branched canaliculi providing many potential channels for communication between OY and other bone cell types. Image generated originally by K. McKenzie, University of Aberdeen and is downloadable from http://www.flickr.com/photos/wellcomeimages/5814814672/
Evidence now suggests that OY participate in almost every phase of mineral handling by bone. In an anabolic sense, OY activities result in mineralisation of osteoid laid down by osteoblasts, and the rate and extent of this mineralisation is also regulated by OY. In a catabolic sense, OY are a major RANKL-producing cell in bone and are able to liberate bone mineral by inducing osteoclast differentiation and activity in response to a range of stimuli, including bone matrix damage, bone disuse and unloading, oestrogen deficiency, high-dose glucocorticoid and chemotherapeutic agents. OY are a major cellular source of the hormone fibroblast growth factor (FGF) 23, which acts on the kidney (via co-receptors FGF-R1 and klotho) to regulate phosphate homeostasis; this activity of OY has been reviewed elsewhere [6] and is not considered further here. In addition, however, there is mounting evidence that OY manage calcium homeostasis in the mature skeleton by extracting calcium from the peri-lacuna space to meet a calcium challenge and replacing it when circulating calcium demands are met. Loss of OY viability in addition to stimulating osteoclastic resorption may also be the cause of hypermineralisation of bone and, as seen in aging and pathology, OY and their lacunae being replaced by mineral. This review aims to summarise these diverse actions of OY in terms of the regulation of bone mineral homeostasis.
Osteocytes organise bone mineral deposition
The process of mineralisation of osteoblast synthesised osteoid is intimately linked with OY differentiation from osteoblasts [1, 7]. Agents that promote the transition of osteoblasts to OY, such as vitamin K [8], vitamin D [9] or strontium [10] also increase the mineralisation of osteoblasts in culture. Franz-Odendaal et al. [1] comprehensively reviewed current knowledge of the process by which OY become embedded into osteoid and subsequently surrounded by mineralised bone matrix. Although the process is only partly understood, and its regulation remains to be elucidated, these authors describe a remarkable change in morphology and gene expression that occurs during the osteoblast-OY transition. The details of this transition are likely dependent on the mode of ossification - intramembranous, perichondral or endochondral - and on the type of bone being generated - woven or lamella bone [1]. The mineralisation of lamella bone has been difficult to study but it is becoming clear that mineralisation accompanies this transition of osteoblast to OY. A study by Barragan-Adjemian et al. [7] utilised the MLO-A5 cell line, described as late osteoblasts/early osteocytes, and which mineralise as they differentiate in culture, to explore the process. They found that mineral was laid down along and within type I collagen fibres surrounding these cells, in small mineralised spherical structures, similar to those described at the mineralisation front in bone, and termed ‘calcospherulites’ [11, 12]. Calcospherulites are thought be represent clusters of mineral-containing matrix vesicles [11]. These mineralised structures are initially associated with the dendritic processes of the developing OY-like cells and eventually increase in size, coalesce and surround the mineralising cells [7]. The authors noted that these events were consistent with those they observed at the mineralisation front in murine lamella bone.
As discussed by Murshed et al. [13], mineralisation is prevented in all non-mineralising tissues by the ubiquitous expression of the powerful mineralisation inhibitor, pyrophosphate (PPi). These authors argued that bone is unique in expressing both type I collagen and tissue non-specific alkaline phosphatase (TNAP), which hydrolyses PPi and provides the inorganic phosphate (Pi) for hydroxyapatite-like (calcium and phosphate) mineral formation [14]. Murshed et al. further showed that ectopic extra-cellular matrix (ECM) mineralisation could be induced by engineering the expression of TNAP in a type I collagen rich tissue, the skin [13]. While adequate levels of Pi, together with type I collagen, may be necessary for ECM mineralisation, they appear not to be sufficient for mineralisation in bone. The molecular regulation of mineralisation in bone is complex and seems to involve a large number of molecules, which function to regulate the availability of calcium and phosphate to precipitate in hydroxyapatite crystals, and to regulate crystal growth. For example, Midura et al. [15] have pointed to an essential role for bone acidic protein-75 (BAG-75) and bone sialoprotein-1 (BSP-1) in mineralisation. BAG-75 and BSP-1 are enriched within the calcospherulites in mineralising bone and it appears that enzymically cleaved products of these proteins are required for mineralisation, since inhibition of the enzyme, SKI-1, which produces this cleavage, also blocks mineralisation in cultures of UMR106-01 osteoblastic cells [16]. Mineralisation is also controlled by a number of factors produced by osteoblasts and osteocytes. As reviewed [17], among these are a group of extracellular matrix proteins, small integrin-binding ligand, N-linked glycoproteins (SIBLINGs), which include matrix extracellular phosphoglycoprotein (MEPE), osteopontin (OPN), dentin matrix protein 1 (DMP1) and bone sialoprotein. MEPE and several other SIBLINGs, including OPN [18] contain an acidic serine aspartate-rich MEPE-associated motif (ASARM) [19]. In the cases of MEPE and OPN, this motif, when enzymically released in the form of a phosphorylated peptide, physically associates with mineral crystals and negatively affects mineralization [17–20]. The protein phosphate-regulating gene with homologies to endopeptidases on the X-chromosome (PHEX) is made abundantly by OY and promotes mineralisation, firstly by binding to MEPE and preventing ASARM peptide generation [21] and secondly, by cleaving the otherwise protease-resistant MEPE-ASARM peptides, releasing their inhibitory effect on mineralisation [21–24]. It is noteworthy that ASARM peptides are chiefly responsible for the in vitro mineralization defect in HYP mice [24], a murine equivalent of human X-linked hypophosphatemia, both conditions arising because of a defect in the PEX/PHEX locus [24, 25]. A further detailed description of biomineralisation is beyond the scope of this review, however, some of the other factors with proposed roles in the promotion or inhibition of bone mineralisation are listed in Table 1 and are the subject of a comprehensive review by Gorski [26]. The point that we wish to make here is, as also proposed by Irie et al. [27], that the differentiation of OY and the process of mineralisation are inseparably linked so that mature OY only appear in association with mineralisation and mineralisation is dependent on the maturation of OY. After the initial phase of bone formation and mineralisation, the process of ‘secondary mineralisation’, which more slowly adds an additional 10% or so of mineral, takes place [28]. The mechanism of this latter process is not well understood.
Osteocytes regulate bone formation, including mineral deposition
Bone formation, the laying down and mineralisation of osteoid, occurs either during new bone formation (modelling) or following a resorptive episode (remodelling). Many factors that promote bone formation in situ have been recognised, and the Bone Morphogenetic Protein (BMP) (reviewed in Ref. [29]) and wingless integration (Wnt) (reviewed in Ref. [30]) signalling pathways are major contributors here. It is equally important to understand the negative regulation of this process, which is required to produce the correct amount of bone. Thus, negative regulators of BMP signalling (inhibitors include Noggin, Follistatin, Chordin family, Twisted gastrulation, Dan/Cerberus family, Gremlin) [29, 31] and the Wnt family of molecules (inhibitors include secreted frizzled-related proteins (sFRPs 1, 2, 3, 4, 5 and FrzB), Wnt inhibitory factor 1, cerberus, Dickkopf 1 and sclerostin) [32], have been identified. In particular, sclerostin, the SOST gene product, has gained attention because of its strong association with bone formation and bone mass. Human conditions, in which loss of function of SOST occurs (sclerosteosis [33]) or SOST is under-expressed due to a mutation in a distal enhancer region (Van Buchem’s disease [34–36]) are characterised by high bone mass. Genetically engineered SOST deficiency in mice also results in a high bone mass phenotype [37]. Sclerostin is of particular relevance here as it is made almost exclusively by OY in bone [38–40]. Sclerostin is also produced by hypertrophic chondrocytes in the growth plate [41] and by articular chondrocytes [42]. The above findings indicate an important role for sclerostin in the regulation of bone mass and sclerostin has now been identified as a target for anabolic treatment of bone, with neutralizing antibody and small molecule inhibitor approaches being pursued [43]. Accumulating evidence [38, 44–47] suggests that sclerostin acts by inhibiting canonical Wnt signalling by binding to the Wnt co-receptors, low-density lipoprotein receptors (LRPs) 5 and 6 [48, 49], and probably LRP 4 [50, 51]. Leupin and colleagues reported in abstract [52] that in addition to LRPs 4, 5 and 6, other molecules including glypicans may also have sclerostin receptor properties. Sclerostin also appears to inhibit bone morphogenetic protein signalling [38, 53], and we have reported that recombinant human sclerostin (rhSCL) stimulated a p42/p44 mitogen-activated protein kinase response in human primary osteoblasts [54], suggesting an additional pathway(s) of action.
It is likely that sclerostin has multiple activities (see below). However, to identify the cell type responsive to sclerostin during bone formation, we have tested the hypothesis that sclerostin regulates the behaviour of cells actively involved in mineralization in adult bone, namely cells in the osteoblast–osteocyte transition. Differentiating cultures of human primary osteoblasts under mineralising conditions were found to be exquisitely sensitive to rhSCL from the beginning of in vitro mineralization [17]. Treatment of human osteoblasts differentiated to a pre-OY stage with rhSCL markedly increased their expression of the pre-osteocyte marker, E11 and suppressed expression of the mature OY markers, DMP1 and SOST, implying inhibition of osteoblast-OY transition. Concomitantly, rhSCL increased MEPE expression and decreased PHEX expression, suggesting regulation of mineralisation by sclerostin through the PHEX/MEPE-ASARM axis [24]. Indeed, we found that mineralization by human osteoblasts was strongly inhibited by synthetic tri-phosphorylated ASARM peptides. Consistent with MEPE-ASARM being important in mineralisation and in the mechanism of sclerostin action, antibody-mediated neutralization of endogenous MEPE-ASARM or co-incubation with a PHEX peptide, SPR4, that effectively neutralises MEPE [24], both antagonised the effect of rhSCL on mineralization [17]. These results suggest that sclerostin acts through regulation of the PHEX/MEPE axis, at least at the early osteocyte stage, and serves as a ‘master’ regulator of physiological bone mineralization.
Osteocytes essential in osteoclastic breakdown of bone mineral
Osteoclast resorption of bone in the post-developmental skeleton appears to be largely controlled by OY. This is exemplified by two recent studies, in which the ability of OY to support osteoclastic activity was genetically inhibited, by conditionally deleting RANKL expression under control of the Dmp1 promoter [55, 56]. This is discussed further below. OY regulate osteoclast activity in both physiological and pathological resorption, although resorption in some non-physiological situations, such as that of non-vital bone graft [57], proceed via mechanisms that are still unclear. Osteoclastic resorption is essential for bone growth and morphogenesis and tooth eruption during development although OY may not play a significant role in these processes [55, 56]. In young animals, osteoclasts also appear to have an important role in calcium homeostasis, which is probably reduced to a minor role in the mature skeleton. This is exemplified by the fact that calcitonin, a potent inhibitor of osteoclastic resorption, is a regulator of extracellular fluid calcium in young and growing animals, in which rapid bone modelling and remodelling are required for development of the skeleton while its calcium-lowering effect is less marked with increasing age [58]. In the post-developmental skeleton, osteoclast activity is essential for the repair of bone matrix damage, and to remove fracture callus. For each of these osteoclastic roles, the factors that determine at which skeletal sites osteoclast recruitment and activation will take place, representing the molecular ‘area code’ proposed by Parfitt [59], are not fully understood. However, for a number of skeletal influences that result in increased local or systemic resorption, OY appear to have a central mediating role (Fig. 2), as will be discussed below.

The central role of the osteocyte in both bone anabolism and catabolism
A link between osteocyte apoptosis and bone resorption induced by bone microdamage
The first such influence is bone microdamage, which acts as a signal for removal and renewal of damaged bone matrix. That microdamage within the bone matrix would disrupt OY canaliculi, causing OY apoptosis, is intuitively obvious, given the high density of OY in both cortical and cancellous bone [60]. However, this link has been formally demonstrated by numbers of investigators [61, 62]. Franssen et al. [63] showed that the orthopaedic procedure of K-wire drilling resulted in empty OY lacunae, as a function of the distance from the drill hole. It has been further shown that experimentally induced microdamage resulted in a transient burst of OY apoptosis, which was followed several days later by co-localised osteoclastic invasion [64]. Osteotomy in chicken radii resulted in rapid OY apoptosis, which temporally preceded an increase in osteoclast presence at the same sites [65]. Experiments, in which microdamage was induced by fatigue loading of rat ulnae, showed that OY apoptosis and the associated intra-cortical bone remodelling were largely a function of linear cracks in the bone matrix, rather than diffuse microdamage, the latter having little effect on OY viability [66]. Strong evidence that OY apoptosis is causal of microdamage-induced bone remodelling has been provided by experiments, in which induction of microdamage in rats was preceded by administration of a pan-caspase inhibitor [67]. Continuous exposure to the inhibitor completely blocked both OY apoptosis and activation of bone resorption. Verborgt et al. [68] investigated the mechanisms, by which OY apoptosis occurs in highly specific spatial association with microdamage and subsequent osteoclastic remodelling. They showed that apoptosing OY close to the site of microdamage express the pro-apoptotic protein Bax, while the anti-apoptotic protein Bcl-2 is expressed by adjacent OY further from the damaged zone. The authors proposed that these mechanisms serve to confine OY apoptosis to sites of microdamage and to also provide spatial guidance for the resorption processes that occur in association with OY apoptosis after episodes of microdamage in bone.
A link between osteocyte apoptosis and osteoclastic resorption due to unloading of bone
The second influence coupling OY apoptosis to osteoclast activation is the lack of sufficient loading of the bone. As reviewed by Noble [62], OY are exquisitely sensitive to loading, and respond both in vitro and in vivo by dramatically changing their expression of a profile of molecules. However, it is also the case that OY viability is dependent on bone loading, probably because loading provides the motive force for interstitial fluid movement along OY canaliculi. Cyclical mechanical stimulation of bone in a bioreactor ex vivo maintained OY viability compared with unloaded bone samples [69]. Noble and co-workers [64] demonstrated a biphasic OY response to loading, with the results dependent on the strain magnitudes achieved in the loading. Lower strains were protective of OY, while higher magnitude strains produced matrix damage and OY apoptosis. Depriving turkey ulnae of mechanical loading greatly reduced diffusion of interstitial fluid, and rapidly induced osteocyte hypoxia [70]. In a model of weightlessness induced in mice by tail suspension there was an increased prevalence of apoptotic OY in both cortical and cancellous bone [71]. This was followed by reduced BMD due to increased osteoclast number and activity. These data strongly suggested that OY apoptosis was spatially associated with the induction of osteoclastic resorption by unloading. This link between OY apoptosis and osteoclast-mediated resorption was supported by experiments in transgenic mice, in which OY can be ablated in an inducible manner post-natally: the increase in osteoclast number seen in control mice during mechanical unloading was suppressed in OY-ablated mice [72].
A link between osteocyte apoptosis and osteoclastic resorption due to oestrogen deficiency
Oestrogen acts through numerous mechanisms to maintain bone health and bone loss after the menopause is contributed to by oestrogen deficiency. Similarly, oestrogen deficiency is an important driver of osteoporosis induced by ovariectomy in numerous animal studies. It has been reported that oestrogen withdrawal in pre-menopausal women, induced by analogues of gonadotropin-releasing hormone, was accompanied by OY apoptosis [73]. To investigate the spatial and temporal links between OY apoptosis produced by oestrogen deficiency and the subsequent bone resorption, experiments were performed in ovariectomised mice, described by Emerton et al. [74]. Their work showed that ovariectomy caused site-specific OY apoptosis, as measured by caspase-3 activation in the femoral cortex, with substantial OY apoptosis limited in the femoral mid-diaphyseal cortex to the posterior of the bone. Morphology of OY at these sites was severely deranged. Whereas OY apoptosis was maximally seen by 3 days after ovariectomy, endocortical resorption, which occurred solely at surfaces adjacent to regions of OY apoptosis, did not occur until 14 days post-ovariectomy. A clear link between OY apoptosis and osteoclast resorption was demonstrated by co-treating animals with a pan-caspase inhibitor, which suppressed OY apoptosis and completely prevented the ovariectomy-induced increase in osteoclastic activity. Similar links between OY death and osteoclast activity have been described in other scenarios, which include chemotherapy-induced OY apoptosis [75] and ischaemia (reviewed in Ref. [76]). However, osteocyte cell death due to glucocorticoid excess does not appear to provoke an osteoclastogenic response [77, 78].
Mechanisms for osteocyte apoptosis-mediated osteoclastic resorption: RANKL
From the above, it can be concluded that pathophysiological resorption is likely to be largely stimulated by OY apoptosis, although it remains unclear whether initiation of physiological resorption is predominantly stochastic or also determined by sites of OY death. Evidence for the latter explanation is the association of apoptotic OY with osteoclasts seen in normal bone tissue. Bronckers et al. [79], in an excellent and insightful paper, described OY death in deeper layers of normal hamster jaw bone at sites of intense resorption. Although viable OY were plentiful around osteoclasts, proportionately more apoptotic OY, as defined by fragmented DNA, were in contact with osteoclasts. The authors speculated that ‘it is conceivable that osteocytes, if undergoing programmed cell death, transmit signals through the canaliculi to the endosteal surface to recruit or activate osteoclasts. Alternatively, cell death may interrupt the secretion of factors made by the osteocytes to modulate osteoclast activity’. Since the time of that report, the central molecule that causes differentiation and activation of osteoclast precursors has been identified as RANKL (reviewed in Ref. [80]). RANKL binds to its cognate receptor, RANK, on myeloid osteoclast precursors and thereby promotes osteoclast differentiation and bone resorbing activity [81]. OPG, a secreted member of the TNF receptor superfamily, is a natural potent antagonist of RANKL [82]. While it is clear that both osteoblasts and osteocytes, as well as other cell types, express RANKL [80], until recently, it was unclear which cells of the osteoblastic lineage in bone express RANKL in a manner that leads to osteoclastic resorption. However, two groups have now independently shown that conditional deletion of the RANKL gene selectively in OY, under the control of the Dmp1 promoter, prevents bone resorption leading to osteopetrosis [55, 56]. Nakashima et al. [55] showed that isolated populations of OY express much more RANKL than osteoblasts and have a much greater capacity to induce osteoclast formation from precursor cells. Xiong et al. [56] showed that RANKL deletion in OY prevents bone resorption due to mechanical unloading. These data are consistent with observations in OY-ablated mice, in which massive bone resorption was seen several days after induction of OY ablation, in association with dramatically increased RANKL expression in the bone [70, 72]. That OY death is associated with increased (relative) RANKL expression and increased osteoclast formation is supported by studies where the OY-like cell line MLO-Y4 was damaged by mechanical means [83] or induced to undergo apoptosis by serum starvation [84]. Kogianni et al. [85] reported that apoptotic bodies derived from MLO-Y4 cells provided stimulus for osteoclast differentiation independently of RANKL, however the responsible mechanism was not described. It is important to note that living OY also are capable of supporting osteoclastogenesis. For example, we have shown that exposure of human primary OY-like cells to polyethylene particles, a known stimulus for periprosthetic osteolysis, produced a catabolic phenotype in the cells, characterised by increased expression of RANKL (which also occurred in MLO-Y4 cells exposed to PE particles), M-CSF and IL-8 [86]. Further, as discussed below, we have also found that sclerostin increases OY support of osteoclast formation and activity, in the absence of increased rates of apoptosis [87]. However, it does appear that OY in the path of a resorbing osteoclast may be engulfed and degraded by the osteoclast [79, 88], although the extent and nature of the signalling between these two cell types is currently under-studied. It is also important to remember that OY apoptosis triggers bone remodelling, so in addition to being important harbingers of resorption, the extent of OY apoptosis may be indicative of bone formation to follow. Indicative that this may be the case is shown in a study by Zarrinkalam et al. [89], who reported that while the histomorphometric parameters of bone formation BV/TV, Tb.Th, OS/BS, BFR and MAR correlated positively with the total number of occupied (viable) OY lacunae in sheep bone, they equally strongly correlated negatively with the numbers of empty lacunae, which were presumably representative of apoptotic OY.
Osteoclast activation by osteocytes: sclerostin
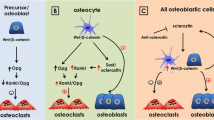
As discussed earlier, sclerostin was first identified as an anti-anabolic agent in bone [38], however, accumulating evidence also suggests a catabolic role. For example, ovariectomised rats treated with neutralizing antibody to SCL showed protection against bone loss and this was associated with a marked decrease in osteoclast surface, to below the level seen in sham-operated animals [90]. Also, in 10-month-old intact female rats, anti-SCL treatment was associated with a dramatic inhibition of osteoclastic activity [91]. Perhaps more strikingly, a recent report by Padhi and colleagues [43] of a phase-I clinical trial in healthy human subjects showed that a single subcutaneous or intravenous dose of a neutralizing antibody to sclerostin (AMG 785) resulted in a rapid and significant reduction of the serum resorption marker, serum C-telopeptide of collagen (sCTx). The initial decrease in sCTX in that study appeared to precede observed AMG 785-induced increases in the serum markers of bone formation (N-terminal propeptide of type 1 procollagen and bone alkaline phosphatase) [43]. Together, these findings are consistent with the reported activity of sclerostin as a Wnt inhibitor [53] and with OPG being expressed in response to canonical [92] and potentially non-canonical [93] Wnt signalling. However, it is possible that sclerostin has a more direct effect on OY support of osteoclastic activity. Our recent findings suggest that sclerostin has paracrine and/or autocrine actions on OY [17, 87]. Recombinant human SCL dose-dependently up-regulated the expression of RANKL mRNA and to a lesser extent, down-regulated that of OPG, causing a primarily RANKL-driven increase in the RANKL/OPG mRNA ratio in both human bone-derived OY-like cells and in MLO-Y4 cells [87]. Moreover, rhSCL-treated MLO-Y4 cells were better able to support the formation and, to a greater extent (up to sevenfold), the resorptive activity of osteoclasts derived from either mouse splenocytes or human peripheral blood mononuclear cell precursors. The effects of rhSCL on both osteoclast formation and activity were completely abolished by co-addition of recombinant OPG, demonstrating that RANKL signalling was essential for the sclerostin effect. It is noteworthy that rhSCL did not induce apoptosis of MLO-Y4 cells, as determined by caspase activity assays, demonstrating that the osteoclastic response was in this case not driven by apoptosing osteocytes [87]. If further substantiated, these results create a new paradigm because they suggest that in situations where sclerostin is increased, for example due to mechanical unloading [94] or perhaps in response to pro-inflammatory cytokines, TWEAK and TNF-α [54], a catabolic response may occur through OY promotion of osteoclast formation and activity. The link between sclerostin and RANKL expression by OY and the potential effects on osteoclast formation and activity are depicted schematically in Fig. 3.

Cartoon representing the potential role of osteocyte-derived sclerostin in regulating the expression of OY RANKL and subsequent control of osteoclast formation and/or the control of osteoclast activity, for example during remodelling of cortical bone. In this model, sclerostin expressed by mature OY in response to a catabolic stimulus such as mechanical unloading may act in an autocrine manner (curved dotted arrows), increasing the local expression of RANKL relative to OPG, or in a paracrine (straight dotted arrows) manner by inducing such expression in sclerostin-negative OY, including immature OY immediately below the osteoblastic lining cells and in the lining cells themselves. An effect of sclerostin and OY-derived RANKL on both endosteal and intra-cortical remodelling may occur either at the stage of osteoclast precursor recruitment/chemotaxis, proliferation/differentiation and/or on the activity of mature osteoclasts. Osteocytes may also ‘talk to’ cells in the bone marrow via a limited number of cell processes extending into the marrow space [5]. This may result in effects on bone marrow stromal cell expression of RANKL and OPG and subsequent regulation of osteoclastogenesis by these cells. Note that a role for apoptosing OY is not ruled out here as the initial stimulus for sclerostin expression, perhaps secondarily to microdamage and local bone unloading, or that the resorbing osteoclast is exposed to higher RANKL levels during engulfment of the expressing OY. The rationale for this cartoon is derived from our recent publication [87] and from the recent work of Nakashima et al. [55] and Xiong et al. [56]
Osteocytic osteolysis
There is good evidence that OY can contribute directly to calcium and phosphate mobilization in a process termed osteocytic osteolysis [95]. Some of this evidence is summarised in recent reviews [96, 97]. In addition, Talmage and co-workers produced a series of papers, reviewed in Ref. [98], in which they argued that the minute to minute control of the extracellular calcium concentration is largely governed by PTH, acting on cells at the bone matrix interface. Clearly, a substantial component (around 90%) of this interface comprises osteocytic lacunae and canaliculi. Haller and Zimny showed expanded OY lacunae, altered OY morphology and irregular OY lacunar borders in alveolar bone of hibernating ground squirrels [99]. Commenting on their findings, the authors noted that ‘these changes suggest that minerals are mobilised from bone during hibernation for utilization elsewhere in the body in order to maintain the minimal level of metabolism necessary for survival’. This insightful study suggests that to an extent yet to be determined, calcium demands, at least in the mature animal, are addressed by osteocytic release of bone mineral. There have been few studies to investigate this possibility in mammals [96], and more work needs to be done, especially using newly available approaches to measure mineral around OY three dimensionally. However, since calcium homeostasis is regulated by PTH, observations by a number of investigators (reviewed by Tazawa et al. [100] and more recently by Qing and Bonewald [96]), in rats, are consistent with the notion that calcium mobilisation can be mediated by OY. Tazawa and co-workers [100] administered human PTH to 8-month-old rats continuously for 4 weeks, and observed enlarged OY lacunae in the PTH group (137.0 μm2 in the PTH group versus 93.9 μm2 in the controls). Recent work supports those studies and further suggests a role for OY in calcium homeostasis. Mice, in which the PTH receptor was ablated specifically in OY, had lower serum calcium levels than littermate controls in the face of a low calcium diet [101]. The mechanism, by which OY might participate in releasing exchangeable calcium from bone into the extracellular fluid, is not understood. A number of early studies, as noted by Parfitt [102], identified the OY expression of tartrate resistant acid phosphatase (TRAP). Interestingly, Tazawa et al. also found TRAP expression by some OY in their PTH treated animals [100]. Tashjian and co-workers reported that cells of the osteoblast lineage (they did not investigate OY per se), responded to PTH, both in vitro and in calvarial explants, by rapidly acidifying their extracellular environment [103, 104]. Interestingly, ATP6V0b, a subunit of the lysosomal proton pump V0 which is responsible for acid extrusion in osteoclasts, was found to be produced and differentially expressed in MLO-Y4 cells [105]. It has been reported in abstract that osteocytic peri-lacunar remodelling may provide calcium for milk production during lactation [106]. In this study, OY in bone from lactating mice expressed TRAP, which was rapidly lost after weaning. Nakano et al. [107] found expression of both TRAP protein and mRNA in OY located close to osteoclast resorbing surfaces in rat and canine bones. There is some evidence that mineral removed from OY lacunae can be replaced [96], for example as seen by calcein labelling in patients with secondary hyperparathyroidism following parathyroidectomy [108].
The above data suggest that some features of OY osteolysis may be similar to the machinery used by osteoclasts to resorb bone mineral, with perhaps the difference that OY ‘resorption’ appears to be self-limiting, compared with the relatively extensive removal of bone performed by osteoclasts. This intriguing question requires to be further explored because it potentially has therapeutic implications. If OY can indeed remove bone mineral physiologically, another question is that of the relative contributions to calcium homeostasis of OY and osteoclasts, as well as other, perhaps cell-independent mechanisms. In relation to this question, Parfitt [28] referred to experiments using radiolabelled calcium, which showed ‘immediate rapid uptake at all bone surfaces accessible to the circulation, regardless of their cellular activity or degree of mineralisation’. He described a pool of calcium that is exchangeable with these surfaces, and which is much greater than can be accounted for by remodelling (osteoclastic activity) and larger than the component that is susceptible to regulation in the kidney by PTH. He states that, while PTH maintains the plasma calcium ‘set point’, this cannot be achieved by regulating (osteoclastic) resorption (because this would be too slow and because resorption blockers, such as alendronate, do not interfere with long-term calcium homeostasis). He therefore declared it a ‘mystery’ as to how PTH regulates the bone-blood calcium equilibrium ‘set point’. We may therefore conclude that calcium exchange between blood and bone may occur both spontaneously, possibly simply along physico-chemical gradients, as well as what has now been experimentally demonstrated, an OY-mediated mechanism driven by PTH [28]. A related concept also discussed by Parfitt [102], and one not well recognised, is that osteocytes and osteoclasts may resorb bone sympathetically. For example, Baylink et al. [109] demonstrated an increase in the volume of the mean osteocyte lacuno-canalicular size as well as an increase in osteoclastic resorption volume in response to vitamin D2, and suggested that the two cell types work together as a ‘resorbing unit’ to contribute to the vitamin D-induced hypercalcaemia.
Osteocyte lacuna hypermineralisation
As reviewed by Roschger et al. [110], bone is formed in separate ‘bone packets’, which are produced at different times during discreet modelling and remodelling cycles. Therefore, bone material is heterogeneously mineralised, with different packets having a characteristic bone mineralisation density distribution (BMDD), in turn reflecting bone turnover, mineralisation kinetics and bone matrix age. Kingsmill and Boyde [111] found that mineral density in the mandible was lowest at sites known to undergo the greatest net resorption. Interestingly, regions of highest density corresponded to those predicted to incur the highest principal strains, suggesting some ability to regulate this parameter. BMDD changes across various skeletal conditions and disease states, such as osteoporosis, hyperparathyroidism and osteogenesis imperfecta, and anti-resorptive treatment for these, and appears to relate to the rate of bone turnover [110]. In a model of ischaemic osteonecrosis, necrotic subchondral bone in the femoral head had increased mean BMDD [112]. This study raised the question of the mechanism of the increased mineral content and how cell death might contribute to this. Part of the answer to this might be the observation of OY lacunae filled with hypermineralised material [113, 114]. These hypermineralised OY (Frost coined the term ‘micropetrosis’ [115]) may be the reason for the reported age-related decrease in total OY lacunae number per bone area, since there is a dramatic increase in mineralised lacunae in older individuals [113]. The cause of the hypermineralised lacunae is not known but it seems reasonable that OY death is a major contributor. In an interesting study by Weinstein et al. [78], it was shown that glucocorticoid administration to mice caused OY apoptosis, which could be rescued by co-treatment with OPG-Fc. Intriguingly, the decreased OY apoptosis due to OPG-Fc was correlated with preservation of the bone interstitial fluid flow (shown by the ability of the dye procion red to travel through the bone) and BMD, suggesting that the two changes were related. This suggests that any cause of loss of adequate fluid flow through OY canaliculi, such as glucocorticoid, vascular disease or bone matrix damage, might lead to OY apoptosis and subsequently to mineral replacing the cells. A vascular mechanism is also suggested by hypermineralised OY being preferentially found in clusters in a bone packet. However, it remains unclear as to why OY apoptosis might sometimes lead to osteoclastic resorption and sometimes to micropetrosis and increased mineralisation of the surrounding bone. Bell et al. [114] found that mineralised OY lacunae, if carefully demineralised, revealed the remains of the OY cell skeleton, suggesting that the cells are mineralised during or as a consequence of cell death. It is noteworthy that healthy OY maintain an unmineralised peri-lacunar space, suggesting active cell-mediated inhibition of mineralisation in this zone. For example, mineralisation inhibitory ASARM peptides derived from MEPE or OPN are present in the lacunae of both osteoid and mature osteocytes [18, 23, 116]. It is possible that upon OY death, the local concentration of inhibitory ASARM peptides decreases, possibly due to the unrestrained activity of the enzyme, PHEX, resulting in the unregulated mineralisation of the remaining peri-lacunar matrix. A further explanation may lie in the cessation of production upon OY death of sclerostin, which we have shown inhibits mineralisation through the production of MEPE and MEPE-ASARM peptides [17]. Alternatively, Busse et al. [113] suggested a pathway leading to hypermineralised OY lacunae, whereby OY death might lead to a deterioration of bone fluid flow and a decreased ability to detect microdamage, leading to insufficient and/or delayed repair of the bone matrix. They speculated that this would in turn result in an accumulation of microdamage in the bone matrix and increased bone fragility. Figure 4 summarises the ways in which OY appear to be able to regulate the peri-osteocyte mineral in bone.

Osteocyte regulation of lacuno-canalicular mineralization
Summary and future directions
We have attempted to summarise some of what is now a great deal of evidence for a central role for OY in all phases of mineral handling in the bone. Rather than OY being inert within the bone, their development is intimately associated with the formation of the mineralised bone matrix. Bone mineral formation is also regulated by OY, by their production of the inhibitor, sclerostin. Sclerostin may also have a catabolic role in promoting osteoclastic release of bone mineral, in a manner that does not require initial OY apoptosis. However, osteoclast development and activation may also be driven by OY cell death. Finally, there appears to be a mode of OY death that leads to hypermineralisation of OY lacunae. In order to gain a comprehensive understanding about the role of OY in the processes discussed here and those not covered, for example the OY syncytium as an endocrine organ, it will be necessary to examine more closely the possible involvement of OY both in in vivo and in vitro models. In vivo studies are necessarily painstaking and require high-resolution histological assessment and lacunae imaging. The study of OY cell biology to date has been facilitated to a large degree by the study of a single murine cell line, MLO-Y4, established in the laboratories of Bonewald [117]. Like all models, this cell line has some limitations, for example it expresses extremely low levels of the mature osteocyte gene, Sost, and does not express the osteocyte product FGF23. Having said that, it is undoubtedly an extremely useful model, and in the majority of cases, findings using this cell line are consistent with findings in either primary cells or with in vivo findings and observations. A more recent osteocyte cell line model, IDG-SW3 [118], will no doubt impact greatly on future research. Ultimately though, we need to refer to human and/or large mammal models of these cells, as well as models that conserve or recapitulate the native three-dimensional conformation of the OY under study. We also need to bear in mind that osteocytes in the bones of large animals, unlike in rodents, are organised into osteons and it may well be that this additional degree of hierarchy and positioning imparts further complexity to the role of these important cells.
References
Franz-Odendaal TA, Hall BK, Witten PE (2006) Buried alive: how osteoblasts become osteocytes. Dev Dyn 235:176–190
Batra N, Kar R, Jiang JX (2012) Gap junctions and hemichannels in signal transmission, function and development of bone. Biochim Biophys Acta (in press)
Marotti G, Ferretti M, Remaggi F, Palumbo C (1995) Quantitative evaluation on osteocyte canalicular density in human secondary osteons. Bone 16:125–128
Marotti G, Ferretti M, Muglia MA, Palumbo C, Palazzini S (1992) A quantitative evaluation of osteoblast–osteocyte relationships on growing endosteal surface of rabbit tibiae. Bone 13:363–368
Kamioka H, Honjo T, Takano-Yamamoto T (2001) A three-dimensional distribution of osteocyte processes revealed by the combination of confocal laser scanning microscopy and differential interference contrast microscopy. Bone 28:145–149
Bergwitz C, Juppner H (2010) Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med 61:91–104
Barragan-Adjemian C, Nicolella D, Dusevich V, Dallas MR, Eick JD, Bonewald LF (2006) Mechanism by which MLO-A5 late osteoblasts/early osteocytes mineralize in culture: similarities with mineralization of lamellar bone. Calcif Tissue Int 79:340–353
Atkins GJ, Welldon KJ, Wijenayaka AR, Bonewald LF, Findlay DM (2009) Vitamin K promotes mineralization, osteoblast-to-osteocyte transition, and an anticatabolic phenotype by {gamma}-carboxylation-dependent and -independent mechanisms. Am J Physiol Cell Physiol 297:C1358–C1367
Atkins GJ, Anderson PH, Findlay DM, Welldon KJ, Vincent C, Zannettino AC, O’Loughlin PD, Morris HA (2007) Metabolism of vitamin D3 in human osteoblasts: evidence for autocrine and paracrine activities of 1 alpha,25-dihydroxyvitamin D3. Bone 40:1517–1528
Atkins GJ, Welldon KJ, Halbout P, Findlay DM (2009) Strontium ranelate treatment of human primary osteoblasts promotes an osteocyte-like phenotype while eliciting an osteoprotegerin response. Osteoporos Int 20:653–664
Boyde A, Sela J (1978) Scanning electron microscope study of separated calcospherites from the matrices of different mineralizing systems. Calcif Tissue Res 26:47–49
Midura RJ, Vasanji A, Su X, Wang A, Midura SB, Gorski JP (2007) Calcospherulites isolated from the mineralization front of bone induce the mineralization of type I collagen. Bone 41:1005–1016
Murshed M, Harmey D, Millan JL, McKee MD, Karsenty G (2005) Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev 19:1093–1104
Orimo H (2010) The mechanism of mineralization and the role of alkaline phosphatase in health and disease. J Nihon Med Sch 77:4–12
Midura RJ, Wang A, Lovitch D, Law D, Powell K, Gorski JP (2004) Bone acidic glycoprotein-75 delineates the extracellular sites of future bone sialoprotein accumulation and apatite nucleation in osteoblastic cultures. J Biol Chem 279:25464–25473
Gorski JP, Huffman NT, Chittur S, Midura RJ, Black C, Oxford J, Seidah NG (2011) Inhibition of proprotein convertase SKI-1 blocks transcription of key extracellular matrix genes regulating osteoblastic mineralization. J Biol Chem 286:1836–1849
Atkins GJ, Rowe PS, Lim HP, Welldon KJ, Ormsby R, Wijenayaka AR, Zelenchuk L, Evdokiou A, Findlay DM (2011) Sclerostin is a locally acting regulator of late-osteoblast/preosteocyte differentiation and regulates mineralization through a MEPE-ASARM-dependent mechanism. J Bone Miner Res 26:1425–1436
Addison WN, Masica DL, Gray JJ, McKee MD (2010) Phosphorylation-dependent inhibition of mineralization by osteopontin ASARM peptides is regulated by PHEX cleavage. J Bone Miner Res 25:695–705
Rowe PS (2004) The wrickkened pathways of FGF23, MEPE and PHEX. Crit Rev Oral Biol Med 15:264–281
Rowe PS, Kumagai Y, Gutierrez G, Garrett IR, Blacher R, Rosen D, Cundy J, Navvab S, Chen D, Drezner MK, Quarles LD, Mundy GR (2004) MEPE has the properties of an osteoblastic phosphatonin and minhibin. Bone 34:303–319
Guo R, Rowe PS, Liu S, Simpson LG, Xiao ZS, Quarles LD (2002) Inhibition of MEPE cleavage by PHEX. Biochem Biophys Res Commun 297:38–45
Rowe PS, Garrett IR, Schwarz PM, Carnes DL, Lafer EM, Mundy GR, Gutierrez GE (2005) Surface plasmon resonance (SPR) confirms that MEPE binds to PHEX via the MEPE-ASARM motif: a model for impaired mineralization in X-linked rickets (HYP). Bone 36:33–46
Addison WN, Nakano Y, Loisel T, Crine P, McKee MD (2008) MEPE-ASARM peptides control extracellular matrix mineralization by binding to hydroxyapatite: an inhibition regulated by PHEX cleavage of ASARM. J Bone Miner Res 23:1638–1649
Martin A, David V, Laurence JS, Schwarz PM, Lafer EM, Hedge AM, Rowe PS (2008) Degradation of MEPE, DMP1, and release of SIBLING ASARM-peptides (minhibins): ASARM-peptide(s) are directly responsible for defective mineralization in HYP. Endocrinology 149:1757–1772
Yuan B, Takaiwa M, Clemens TL, Feng JQ, Kumar R, Rowe PS, Xie Y, Drezner MK (2008) Aberrant PHEX function in osteoblasts and osteocytes alone underlies murine X-linked hypophosphatemia. J Clin Invest 118:722–734
Gorski JP (2011) Biomineralization of bone: a fresh view of the roles of non-collagenous proteins. Front Biosci 17:2598–2621
Irie K, Ejiri S, Sakakura Y, Shibui T, Yajima T (2008) Matrix mineralization as a trigger for osteocyte maturation. J Histochem Cytochem 56:561–567
Parfitt AM (2003) Misconceptions (3): calcium leaves bone only by resorption and enters only by formation. Bone 33:259–263
Canalis E, Economides AN, Gazzerro E (2003) Bone morphogenetic proteins, their antagonists, and the skeleton. Endocr Rev 24:218–235
Bonewald LF (2011) The amazing osteocyte. J Bone Miner Res 26:229–238
Chen D, Zhao M, Mundy GR (2004) Bone morphogenetic proteins. Growth Factors 22:233–241
Canalis E (2009) Growth factor control of bone mass. J Cell Biochem 108:769–777
Baron R, Rawadi G, Roman-Roman S (2006) Wnt signaling: a key regulator of bone mass. Curr Top Dev Biol 76:103–127
ten Dijke P, Krause C, de Gorter DJ, Lowik CW, van Bezooijen RL (2008) Osteocyte-derived sclerostin inhibits bone formation: its role in bone morphogenetic protein and Wnt signaling. J Bone Joint Surg Am 90(Suppl 1):31–35
Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, Dioszegi M, Dikkers FG, Hildering P, Willems PJ, Verheij JB, Lindpaintner K, Vickery B, Foernzler D, Van Hul W (2002) Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet 39:91–97
Staehling-Hampton K, Proll S, Paeper BW, Zhao L, Charmley P, Brown A, Gardner JC, Galas D, Schatzman RC, Beighton P, Papapoulos S, Hamersma H, Brunkow ME (2002) A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12–q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet 110:144–152
Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D’Agostin D, Kurahara C, Gao Y, Cao J, Gong J, Asuncion F, Barrero M, Warmington K, Dwyer D, Stolina M, Morony S, Sarosi I, Kostenuik PJ, Lacey DL, Simonet WS, Ke HZ, Paszty C (2008) Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res 23:860–869
Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K, Appleby M, Brunkow ME, Latham JA (2003) Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J 22:6267–6276
Winkler DG, Yu C, Geoghegan JC, Ojala EW, Skonier JE, Shpektor D, Sutherland MK, Latham JA (2004) Noggin and sclerostin bone morphogenetic protein antagonists form a mutually inhibitory complex. J Biol Chem 279:36293–36298
Poole KE, van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Lowik CW, Reeve J (2005) Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J 19:1842–1844
van Bezooijen RL, Bronckers AL, Gortzak RA, Hogendoorn PC, van der Wee-Pals L, Balemans W, Oostenbroek HJ, Van Hul W, Hamersma H, Dikkers FG, Hamdy NA, Papapoulos SE, Lowik CW (2009) Sclerostin in mineralized matrices and van Buchem disease. J Dent Res 88:569–574
Chan BY, Fuller ES, Russell AK, Smith SM, Smith MM, Jackson MT, Cake MA, Read RA, Bateman JF, Sambrook PN, Little CB (2011) Increased chondrocyte sclerostin may protect against cartilage degradation in osteoarthritis. Osteoarthr Cartil 19:874–885
Padhi D, Jang G, Stouch B, Fang L, Posvar E (2011) Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res 26:19–26
Kusu N, Laurikkala J, Imanishi M, Usui H, Konishi M, Miyake A, Thesleff I, Itoh N (2003) Sclerostin is a novel secreted osteoclast-derived bone morphogenetic protein antagonist with unique ligand specificity. J Biol Chem 278:24113–24117
Sutherland MK, Geoghegan JC, Yu C, Winkler DG, Latham JA (2004) Unique regulation of SOST, the sclerosteosis gene, by BMPs and steroid hormones in human osteoblasts. Bone 35:448–454
van Bezooijen RL, Roelen BA, Visser A, van der Wee-Pals L, de Wilt E, Karperien M, Hamersma H, Papapoulos SE, ten Dijke P, Lowik CW (2004) Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med 199:805–814
Winkler DG, Sutherland MS, Ojala E, Turcott E, Geoghegan JC, Shpektor D, Skonier JE, Yu C, Latham JA (2005) Sclerostin inhibition of Wnt-3a-induced C3H10T1/2 cell differentiation is indirect and mediated by bone morphogenetic proteins. J Biol Chem 280:2498–2502
Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, Harris SE, Wu D (2005) Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem 280:19883–19887
van Bezooijen RL, Svensson JP, Eefting D, Visser A, van der Horst G, Karperien M, Quax PH, Vrieling H, Papapoulos SE, ten Dijke P, Lowik CW (2007) Wnt but not BMP signaling is involved in the inhibitory action of sclerostin on BMP-stimulated bone formation. J Bone Miner Res 22:19–28
Choi HY, Dieckmann M, Herz J, Niemeier A (2009) Lrp4, a novel receptor for Dickkopf 1 and sclerostin, is expressed by osteoblasts and regulates bone growth and turnover in vivo. PLoS One 4:e7930
Leupin O, Piters E, Halleux C, Hu S, Kramer I, Morvan F, Bouwmeester T, Schirle M, Bueno-Lozano M, Ramos Fuentes FJ, Itin PH, Boudin E, De Freitas F, Jennes K, Brannetti B, Charara N, Ebersbach H, Geisse S, Lu CX, Bauer A, Van Hul W, Kneissel M (2011) Bone overgrowth-associated mutations in LRP4 impair sclerostin-facilitator function. J Biol Chem 286:19489–19500
Leupin O, Halleux C, Morvan F, Hu S, Lu C, Bauer A, Kneissel M (2009) LRP4 is a novel osteoblast and osteocyte expressed specific facilitator of SOST-mediated inhibition of in vitro bone formation. J Bone Miner Res 24(Suppl 1). Available at: http://www.asbmr.org/Meetings/AnnualMeeting/AbstractDetail.aspx?aid=c8cba0d3-a82c-4036-b4d8-29979e61e7a2
Krause C, Korchynskyi O, de Rooij K, Weidauer SE, de Gorter DJ, van Bezooijen RL, Hatsell S, Economides AN, Mueller TD, Lowik CW, Ten Dijke P (2010) Distinct modes of inhibition by sclerostin on bone morphogenetic protein and Wnt signaling pathways. J Biol Chem 285:41614–41626
Vincent C, Findlay DM, Welldon KJ, Wijenayaka AR, Zheng TS, Haynes DR, Fazzalari NL, Evdokiou A, Atkins GJ (2009) Pro-inflammatory cytokines TNF-related weak inducer of apoptosis (TWEAK) and TNFalpha induce the mitogen-activated protein kinase (MAPK)-dependent expression of sclerostin in human osteoblasts. J Bone Miner Res 24:1434–1449
Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, Bonewald LF, Kodama T, Wutz A, Wagner EF, Penninger JM, Takayanagi H (2011) Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med 17:1231–1234
Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA (2011) Matrix-embedded cells control osteoclast formation. Nat Med 17:1235–1241
McGee MA, Findlay DM, Howie DW, Carbone A, Ward P, Stamenkov R, Page TT, Bruce WJ, Wildenauer CI, Toth C (2004) The use of OP-1 in femoral impaction grafting in a sheep model. J Orthop Res 22:1008–1015
Cooper CW, Hirsch PF, Toverud SU, Munson PL (1967) An improved method for the biological assay of thyrocalcitonin. Endocrinology 81:610–616
Parfitt AM (1998) Osteoclast precursors as leukocytes: importance of the area code. Bone 23:491–494
Ejiri S, Ozawa H (1982) Scanning electron microscopic observations of rat tibia using the HCl-collagenase method. Arch Histol Jpn 45:399–404
Colopy SA, Benz-Dean J, Barrett JG, Sample SJ, Lu Y, Danova NA, Kalscheur VL, Vanderby R Jr, Markel MD, Muir P (2004) Response of the osteocyte syncytium adjacent to and distant from linear microcracks during adaptation to cyclic fatigue loading. Bone 35:881–891
Noble B (2003) Bone microdamage and cell apoptosis. Eur Cell Mater 6:46–55, discusssion 55
Franssen BB, van Diest PJ, Schuurman AH, Kon M (2008) Drilling K-wires, what about the osteocytes? An experimental study in rabbits. Arch Orthop Trauma Surg 128:83–87
Noble BS, Peet N, Stevens HY, Brabbs A, Mosley JR, Reilly GC, Reeve J, Skerry TM, Lanyon LE (2003) Mechanical loading: biphasic osteocyte survival and targeting of osteoclasts for bone destruction in rat cortical bone. Am J Physiol Cell Physiol 284:C934–C943
Clark WD, Smith EL, Linn KA, Paul-Murphy JR, Muir P, Cook ME (2005) Osteocyte apoptosis and osteoclast presence in chicken radii 0–4 days following osteotomy. Calcif Tissue Int 77:327–336
Herman BC, Cardoso L, Majeska RJ, Jepsen KJ, Schaffler MB (2010) Activation of bone remodeling after fatigue: differential response to linear microcracks and diffuse damage. Bone 47:766–772
Cardoso L, Herman BC, Verborgt O, Laudier D, Majeska RJ, Schaffler MB (2009) Osteocyte apoptosis controls activation of intracortical resorption in response to bone fatigue. J Bone Miner Res 24:597–605
Verborgt O, Tatton NA, Majeska RJ, Schaffler MB (2002) Spatial distribution of Bax and Bcl-2 in osteocytes after bone fatigue: complementary roles in bone remodeling regulation? J Bone Miner Res 17:907–914
Mann V, Huber C, Kogianni G, Jones D, Noble B (2006) The influence of mechanical stimulation on osteocyte apoptosis and bone viability in human trabecular bone. J Musculoskelet Neuronal Interact 6:408–417
Dodd JS, Raleigh JA, Gross TS (1999) Osteocyte hypoxia: a novel mechanotransduction pathway. Am J Physiol 277:C598–C602
Aguirre JI, Plotkin LI, Stewart SA, Weinstein RS, Parfitt AM, Manolagas SC, Bellido T (2006) Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J Bone Miner Res 21:605–615
Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, Ito M, Takeshita S, Ikeda K (2007) Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab 5:464–475
Tomkinson A, Reeve J, Shaw RW, Noble BS (1997) The death of osteocytes via apoptosis accompanies estrogen withdrawal in human bone. J Clin Endocrinol Metab 82:3128–3135
Emerton KB, Hu B, Woo AA, Sinofsky A, Hernandez C, Majeska RJ, Jepsen KJ, Schaffler MB (2010) Osteocyte apoptosis and control of bone resorption following ovariectomy in mice. Bone 46:577–583
Shandala T, Ng YS, Hopwood B, Yip YC, Foster BK, Xian CJ (2012) The role of osteocyte apoptosis in cancer chemotherapy-induced bone loss. J Cell Physiol (in press)
Findlay DM (2007) Vascular pathology and osteoarthritis. Rheumatology (Oxford) 46:1763–1768
Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC (1998) Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest 102:274–282
Weinstein RS, O’Brien CA, Almeida M, Zhao H, Roberson PK, Jilka RL, Manolagas SC (2011) Osteoprotegerin prevents glucocorticoid-induced osteocyte apoptosis in mice. Endocrinology 152:3323–3331
Bronckers AL, Goei W, Luo G, Karsenty G, D’Souza RN, Lyaruu DM, Burger EH (1996) DNA fragmentation during bone formation in neonatal rodents assessed by transferase-mediated end labeling. J Bone Miner Res 11:1281–1291
Findlay DM, Atkins GJ (2011) Relationship between serum RANKL and RANKL in bone. Osteoporos Int 22:2597–2602
Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T (1998) Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A 95:3597–3602
Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, Fujise N, Sato Y, Goto M, Yamaguchi K, Kuriyama M, Kanno T, Murakami A, Tsuda E, Morinaga T, Higashio K (1998) Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology 139:1329–1337
Kurata K, Heino TJ, Higaki H, Vaananen HK (2006) Bone marrow cell differentiation induced by mechanically damaged osteocytes in 3D gel-embedded culture. J Bone Miner Res 21:616–625
Al-Dujaili SA, Lau E, Al-Dujaili H, Tsang K, Guenther A, You L (2011) Apoptotic osteocytes regulate osteoclast precursor recruitment and differentiation in vitro. J Cell Biochem 112:2412–2423
Kogianni G, Mann V, Noble BS (2008) Apoptotic bodies convey activity capable of initiating osteoclastogenesis and localized bone destruction. J Bone Miner Res 23:915–927
Atkins GJ, Welldon KJ, Holding CA, Haynes DR, Howie DW, Findlay DM (2009) The induction of a catabolic phenotype in human primary osteoblasts and osteocytes by polyethylene particles. Biomaterials 30:3672–3681
Wijenayaka AR, Kogawa M, Lim HP, Bonewald LF, Findlay DM, Atkins GJ (2011) Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS One 6:e25900
Palumbo C, Ferretti M, Ardizzoni A, Zaffe D, Marotti G (2001) Osteocyte-osteoclast morphological relationships and the putative role of osteocytes in bone remodeling. J Musculoskelet Neuronal Interact 1:327–332
Zarrinkalam MR, Mulaibrahimovic A, Atkins GJ, Moore RJ (2012) Changes in osteocyte density correspond with changes in osteoblast and osteoclast activity in an osteoporotic sheep model. Osteoporos Int (in press)
Li X, Ominsky MS, Warmington KS, Morony S, Gong J, Cao J, Gao Y, Shalhoub V, Tipton B, Haldankar R, Chen Q, Winters A, Boone T, Geng Z, Niu QT, Ke HZ, Kostenuik PJ, Simonet WS, Lacey DL, Paszty C (2009) Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res 24:578–588
Tian X, Setterberg RB, Li X, Paszty C, Ke HZ, Jee WS (2010) Treatment with a sclerostin antibody increases cancellous bone formation and bone mass regardless of marrow composition in adult female rats. Bone 47:529–533
Glass DA 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, Karsenty G (2005) Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell 8:751–764
Kaneuji T, Ariyoshi W, Okinaga T, Toshinaga A, Takahashi T, Nishihara T (2011) Mechanisms involved in regulation of osteoclastic differentiation by mechanical stress-loaded osteoblasts. Biochem Biophys Res Commun 408:103–109
Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, Mantila SM, Gluhak-Heinrich J, Bellido TM, Harris SE, Turner CH (2008) Mechanical stimulation of bone in vivo reduces osteocyte expression of sost/sclerostin. J Biol Chem 283:5866–5875
Belanger LF, Belanger C, Semba T (1967) Technical approaches leading to the concept of osteocytic osteolysis. Clin Orthop Relat Res 54:187–196
Qing H, Bonewald LF (2009) Osteocyte remodeling of the perilacunar and pericanalicular matrix. Int J Oral Sci 1:59–65
Teti A, Zallone A (2009) Do osteocytes contribute to bone mineral homeostasis? Osteocytic osteolysis revisited. Bone 44:11–16
Talmage RV, Mobley HT (2008) Calcium homeostasis: reassessment of the actions of parathyroid hormone. Gen Comp Endocrinol 156:1–8
Haller AC, Zimny ML (1977) Effects of hibernation on interradicular alveolar bone. J Dent Res 56:1552–1557
Tazawa K, Hoshi K, Kawamoto S, Tanaka M, Ejiri S, Ozawa H (2004) Osteocytic osteolysis observed in rats to which parathyroid hormone was continuously administered. J Bone Miner Metab 22:524–529
Powell WF Jr, Barry KJ, Tulum I, Kobayashi T, Harris SE, Bringhurst FR, Pajevic PD (2011) Targeted ablation of the PTH/PTHrP receptor in osteocytes impairs bone structure and homeostatic calcemic responses. J Endocrinol 209:21–32
Parfitt AM (1977) The cellular basis of bone turnover and bone loss: a rebuttal of the osteocytic resorption—bone flow theory. Clin Orthop Relat Res 127:236–247
Barrett MG, Belinsky GS, Tashjian AH Jr (1997) A new action of parathyroid hormone. receptor-mediated stimulation of extracellular acidification in human osteoblast-like SaOS-2 cells. J Biol Chem 272:26346–26353
Belinsky GS, Tashjian AH Jr (2000) Direct measurement of hormone-induced acidification in intact bone. J Bone Miner Res 15:550–556
Bivi N, Bereszczak JZ, Romanello M, Zeef LA, Delneri D, Quadrifoglio F, Moro L, Brancia FL, Tell G (2009) Transcriptome and proteome analysis of osteocytes treated with nitrogen-containing bisphosphonates. J Proteome Res 8:1131–1142
Qing H, Ardeshirour L, Dusevich V, Dallas M, Wysolmerski JJ, Bonewald LF (2008) Osteocytic perilacunar remodelling as a significant source of calcium during lactation. J Bone Miner Res 23:s401
Nakano Y, Toyosawa S, Takano Y (2004) Eccentric localization of osteocytes expressing enzymatic activities, protein, and mRNA signals for type 5 tartrate-resistant acid phosphatase (TRAP). J Histochem Cytochem 52:1475–1482
Yajima A, Inaba M, Tominaga Y, Nishizawa Y, Ikeda K, Ito A (2010) Increased osteocyte death and mineralization inside bone after parathyroidectomy in patients with secondary hyperparathyroidism. J Bone Miner Res 25:2374–2381
Baylink D, Sipe J, Wergedal J, Whittemore OJ (1973) Vitamin D-enhanced osteocytic and osteoclastic bone resorption. Am J Physiol 224:1345–1357
Roschger P, Paschalis EP, Fratzl P, Klaushofer K (2008) Bone mineralization density distribution in health and disease. Bone 42:456–466
Kingsmill VJ, Boyde A (1998) Mineralisation density of human mandibular bone: quantitative backscattered electron image analysis. J Anat 192(Pt 2):245–256
Hofstaetter JG, Roschger P, Klaushofer K, Kim HK (2010) Increased matrix mineralization in the immature femoral head following ischemic osteonecrosis. Bone 46:379–385
Busse B, Djonic D, Milovanovic P, Hahn M, Puschel K, Ritchie RO, Djuric M, Amling M (2010) Decrease in the osteocyte lacunar density accompanied by hypermineralized lacunar occlusion reveals failure and delay of remodeling in aged human bone. Aging Cell 9:1065–1075
Bell LS, Kayser M, Jones C (2008) The mineralized osteocyte: a living fossil. Am J Phys Anthropol 137:449–456
Frost HM (1960) Micropetrosis. J Bone Joint Surg Am 42-A:144–150
Gaudin-Audrain C, Gallois Y, Pascaretti-Grizon F, Hubert L, Massin P, Basle MF, Chappard D (2008) Osteopontin is histochemically detected by the AgNOR acid-silver staining. Histol Histopathol 23:469–478
Kato Y, Windle JJ, Koop BA, Mundy GR, Bonewald LF (1997) Establishment of an osteocyte-like cell line, MLO-Y4. J Bone Miner Res 12:2014–2023
Woo SM, Rosser J, Dusevich V, Kalajzic I, Bonewald LF (2011) Cell line IDG-SW3 replicates osteoblast-to-late-osteocyte differentiation in vitro and accelerates bone formation in vivo. J Bone Miner Res 26:2634–2646
Wang W, Xu J, Du B, Kirsch T (2005) Role of the progressive ankylosis gene (ank) in cartilage mineralization. Mol Cell Biol 25:312–323
Golub EE (2009) Role of matrix vesicles in biomineralization. Biochim Biophys Acta 1790:1592–1598
Kalamajski S, Aspberg A, Lindblom K, Heinegard D, Oldberg A (2009) Asporin competes with decorin for collagen binding, binds calcium and promotes osteoblast collagen mineralization. Biochem J 423:53–59
Gorski JP, Wang A, Lovitch D, Law D, Powell K, Midura RJ (2004) Extracellular bone acidic glycoprotein-75 defines condensed mesenchyme regions to be mineralized and localizes with bone sialoprotein during intramembranous bone formation. J Biol Chem 279:25455–25463
Berendsen AD, Fisher LW, Kilts TM, Owens RT, Robey PG, Gutkind JS, Young MF (2011) Modulation of canonical Wnt signaling by the extracellular matrix component biglycan. Proc Natl Acad Sci U S A 108:17022–17027
Huffman NT, Keightley JA, Chaoying C, Midura RJ, Lovitch D, Veno PA, Dallas SL, Gorski JP (2007) Association of specific proteolytic processing of bone sialoprotein and bone acidic glycoprotein-75 with mineralization within biomineralization foci. J Biol Chem 282:26002–26013
Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE (2006) Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet 38:1310–1315
Yadav MC, Simao AM, Narisawa S, Huesa C, McKee MD, Farquharson C, Millan JL (2011) Loss of skeletal mineralization by the simultaneous ablation of PHOSPHO1 and alkaline phosphatase function: a unified model of the mechanisms of initiation of skeletal calcification. J Bone Miner Res 26:286–297
Yoshiko Y, Candeliere GA, Maeda N, Aubin JE (2007) Osteoblast autonomous Pi regulation via Pit1 plays a role in bone mineralization. Mol Cell Biol 27:4465–4474
Terkeltaub RA (2001) Inorganic pyrophosphate generation and disposition in pathophysiology. Am J Physiol Cell Physiol 281:C1–C11
Fedde KN, Blair L, Silverstein J, Coburn SP, Ryan LM, Weinstein RS, Waymire K, Narisawa S, Millan JL, MacGregor GR, Whyte MP (1999) Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia. J Bone Miner Res 14:2015–2026
Acknowledgements
The authors gratefully acknowledge Drs. Kamarul Ariffin Khalid and Nobuaki Ito for their critical comments.
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Atkins, G.J., Findlay, D.M. Osteocyte regulation of bone mineral: a little give and take. Osteoporos Int 23, 2067–2079 (2012). https://doi.org/10.1007/s00198-012-1915-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-012-1915-z