
Abstract
Vitamin D, a secosteroid hormone, affects multiple biological pathways via both genomic and nongenomic signalling. Several pathways have potential benefit to cardiovascular health, including effects on parathyroid hormone, the renin–angiotensin–aldosterone system, vascular endothelial growth factor and cytokine production, as well as direct effects on endothelial cell function and myocyte calcium influx. Observational data supports a link between low vitamin D metabolite levels and cardiovascular health. Cross-sectional data shows associations between low 25-hydroxyvitamin D levels and stroke, myocardial infarction, diabetes mellitus, hypertension, and heart failure. Longitudinal data also suggests a relationship with incident hypertension and new cardiovascular events. However, these associations are potentially confounded by reverse causality and by the effects that other cardiovascular risk factors have on vitamin D metabolite levels. Intervention studies to date suggest a modest antihypertensive effect of vitamin D, no effect on serum lipids, a small positive effect on insulin resistance and fasting glucose, and equivocal actions on arterial stiffness and endothelial function. Analysis of cardiovascular event data collected from osteoporosis trials does not currently show a clear signal for reduced cardiovascular events with vitamin D supplementation, but results may be confounded by the coadministration of calcium, and by the secondary nature of the analyses. Despite mechanistic and observational data that suggest a protective role for vitamin D in cardiovascular disease, intervention studies to date are less promising. Large trials using cardiovascular events as a primary outcome are needed before vitamin D can be recommended as a therapy for cardiovascular disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Following the discovery of vitamin D in the early twentieth century, decades of research have uncovered a myriad of potential biological effects. The traditionally recognised actions on calcium and bone metabolism are well documented, but more recent work has uncovered its potential role in the pathophysiology of cardiovascular disease. Furthermore, longitudinal and cross-sectional studies have shown associations between vitamin D deficiency and a variety of cardiovascular disease states and risk factors. Interventional trials in a variety of cardiometabolic conditions have sought to identify the possible role of vitamin D as a therapeutic agent in the management of cardiovascular risk factors. This review aims to highlight the vascular effects of vitamin D, provide an overview of existing observational data, critically review interventional trials of vitamin D in vascular disease to date and detail future directions in this field.
What is vitamin D?
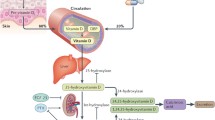
Vitamin D is a secosteroid hormone, mainly produced from the action of sunlight on skin. 7-Dehydrocholesterol in skin is converted to vitamin D3 (cholecalciferol) by the action of ultraviolet light. Small amounts of vitamin D3 can be gained from dietary sources and vitamin D2 from plant sources. Vitamins D3 and D2 need to be converted to their active form by the process of hydroxylation. Cholecalciferol is converted to 1α,25-dihydroxyvitamin D3 (1,25OHD, calcitriol) by first undergoing 25-hydroxylation in the liver to form 25-hydroxyvitamin D (25OHD), and then 1α hydroxylation in the kidney. Both processes are dependent on cytochrome CYP450 containing enzymes [1]. 1α Hydroxylase has also been found in many cells throughout the body, e.g., macrophages, thus conversion to the active form may occur in an autocrine or paracrine manner, rather than occurring only in the kidney as previously thought [2, 3]. 25OHD is the major circulating form of vitamin D bound to vitamin D-binding protein [4], and given its long half-life (several weeks), is the form most commonly measured to ascertain vitamin D repletion status. Controversy exists as to what levels of 25OHD should define repletion, insufficiency, and deficiency. For the purposes of this review, deficiency is used to denote levels below 25 nmol/L (a level associated with an increasing risk of rickets and osteomalacia), and insufficiency for levels below 75 nmol/L, a level postulated to associate with better health outcomes by some authors based on observational data [5].
Vitamin D receptors
1,25OHD binds to the vitamin D receptor (VDR). VDR is a nuclear steroid receptor expressed on at least 36 different tissues including cardiac muscle, vascular smooth muscle, endothelium, and lymphocytes [4]. Vitamin D receptors have similar features to other ligand-activated receptors such as the retinoic acid receptor and thyroid hormone receptor [1]. Indeed, the vitamin D receptor forms a heterodimer with the retinoic acid receptor, and it is this heterodimeric complex that binds to the vitamin D response element to mediate effects on gene transcription [6]. Vitamin D-responsive elements are found at the start of a large number of target genes [7]. Intriguingly, recent work suggests that vitamin D metabolites may also act via alternative nongenomic pathways, using an alternative binding site on the vitamin D receptor, rapid activation of cell signal pathways at the plasma membrane appear to mediate some effects [4, 6].
The major role of 1,25OHD is in calcium homeostasis. Circulating calcium levels are tightly regulated via the effects of calciotropic hormone systems including vitamin D and parathyroid hormone (PTH). 1,25OHD enhances production and activity of the TRPV6 ion channel and calbindin calcium binding protein in the intestinal epithelium to promote gut absorption of calcium. In the presence of low blood calcium, 1,25OHD and PTH act together to mobilise calcium from the skeleton through stimulating osteoclastogenesis and both act together to increase distal renal tubule reabsorption of calcium [7]. Vitamin D also plays a role in bone remodelling [1]. Calcium sensors in the parathyroid gland closely monitor serum calcium concentrations and PTH stimulates renal production of 1-alpha hydroxylase to increase 1,25OHD production.
Low vitamin D levels are common, especially in countries at latitudes >40° north or south, where insufficient ultraviolet light reaches Earth’s surface during winter to allow vitamin D synthesis. Indoor living, extensive skin cover by clothing, old age, obesity, dark skin, and genetic variation in metabolic pathways are all recognised to contribute to low 25OHD levels. Basic science investigation over the last 20 years has elucidated a number of pathways by which vitamin D metabolites might influence cardiovascular health, which are discussed below. We then review the observational data linking low 25OHD levels with cardiovascular disease before examining whether supplementation can ameliorate any deleterious effects on cardiovascular health.
Vitamin D and vascular biology
Effects on the renin–angiotensin–aldosterone system
Both animal and human studies have shown that vitamin D acts as a negative regulator of the renin–angiotensin–aldosterone system (RAAS) [8–10]. The RAAS is involved in the maintenance of electrolyte homoeostasis, blood pressure and intravascular volume. Vitamin D receptor knockout mice have markedly increased renin and hence angiotensin II activity [8]. This overproduction leads to hypertension, cardiac hypertrophy, and increased water intake and sodium absorption. Studies in humans also suggest effects of vitamin D on the RAAS, although this may be mediated via more complex effects on angiotensin II production rather than a direct effect on reducing renin levels [10]. Large prospective human studies have also identified a relationship between low vitamin D levels and increased activity of RAAS [9]. Correction of vitamin D insufficiency appears to have similar effects on tissue response to angiotensin II infusion in humans as does administration of captopril, suggesting a mechanism of action for vitamin D in humans that is similar to ACE inhibition [11].
Direct effects on calcium flux
1,25OHD can act through both genomic and nongenomic mechanisms at cardiac myocytes. Nongenomic pathways are mediated through the action of 1,25OHD at voltage-gated calcium channels in cardiac myocytes, providing a rapid influx of calcium into cells [12]. Animal studies have demonstrated that 1,25OHD directly alters myocyte contractility with accelerated relaxation which may be important for normal diastolic function. This mechanism was shown to occur through phosphorylation of protein kinase C [13]. 1,25OHD can also alter calcium influx into the cell via beta adrenergic mediated adenylate cyclase and cAMP pathways in animal models [12], potentially again enhancing myocyte contractility.
Vascular calcification
Calcium deposition in atherosclerotic plaques, vessel walls, and valves forms a major cause of cardiac morbidity and mortality. Arterial calcification is an additional risk factor for cardiovascular events independent of traditional risk factors [14]. Vitamin D has been associated with both increased vascular calcification and evidence conversely supports a protective effect. Observational studies in humans have shown an inverse correlation between vitamin D and coronary artery calcification [15]. Such studies are at variance with results in rat models, where the combination of nicotine and vitamin D can induce vascular calcification [16]. Recent evidence suggests that vascular calcification involves similar regulatory mechanisms and pathways to bone formation [17]. Several bone-associated proteins have been identified in the process of vascular calcification, including osteocalcin, matrix Gla protein, and osteoprotegerin. A number of bone matrix proteins have been identified which lead to osteoblast differentiation and subsequent mineral deposition in vascular endothelial cells [18]. Further research investigating the bone vascular axis is needed to outline the exact mechanisms linking vitamin D with vascular calcification.
Left ventricular hypertrophy and parathyroid hormone
VDR knockout mice display profound left ventricular hypertrophy at 12 months. VDR is present in myocytes and thus a direct effect on the regulation of myocyte growth is possible [12]. In addition, VDR are present in the parathyroid gland and 1,25OHD suppresses production of PTH and prevents proliferation of parathyroid glands [7]. As such, vitamin D helps to regulate parathyroid levels, and vitamin D insufficiency is associated with elevated PTH levels. Observational studies have suggested that elevated parathyroid hormone levels are associated with left ventricular hypertrophy [19] and patients with primary hyperparathyroidism have impaired endothelial function that can be improved by parathyroidectomy [20]. PTH levels were an independent risk factor for the development of heart failure in a large observational study [21], suggesting that such pathophysiological mechanisms may be clinically relevant.
Endothelial dysfunction
Endothelial dysfunction is the common end pathway for many cardiovascular risk factors, including smoking, hypertension, and hyperlipidaemia. It is thought to presage atherosclerosis and in turn leads to increased vascular tone. Vascular endothelial cells and smooth muscle cells possess VDR, and in vitro studies have shown that 1,25OHD increases nitric oxide synthase activity and nitric oxide production in human umbilical vein endothelial cells [22]. 1,25OHD also increases the angiogenic potential of endothelial cells; an effect that appears to be mediated in part by autocrine and paracrine effects of enhanced vascular endothelial growth factor (VEGF) production [23, 24]. Vitamin D also reduces the ability of endothelial cells to induce platelet aggregation [25]. Additional potential antithrombotic effects may be mediated by vitamin D-induced alterations in the production of proteins involved in regulation of thrombogenesis, e.g., thrombomodulin and antithrombin [26].
Insulin and glucose handling
Vitamin D may play a pathophysiological role in the development of both types 1 and 2 diabetes mellitus. Vitamin D receptors are found on islet cells and may stimulate insulin release by beta cells [27, 28]. Early animal studies demonstrated reduced secretion of insulin in response to glucose in vitamin D-deficient rats [29]. This finding may be driven in part by the elevated parathyroid hormone levels seen in vitamin D insufficiency [30]. In addition, the anti-inflammatory and immunomodulatory actions of vitamin D may ameliorate both the autoimmune pathology seen in type I diabetes mellitus and could ameliorate the chronic inflammation thought to contribute to insulin resistance in type 2 diabetes [31].
Immune/inflammatory modulation
Vitamin D receptors are present on a number of cells in the immune system including lymphocytes, monocytes, macrophages, and the thymus and its role in a number of immune-mediated diseases has been studied [1]. Indeed activated vitamin D preparations have long formed part of the therapeutic armamentarium used by dermatologists to treat immunologically-mediated skin disease. Immune cells play an important role in vascular disease via cytokine production and subsequent immunomodulation and inflammation; factors known to be important in the pathophysiology of heart failure and atherosclerosis. A demonstration of the potential role of vitamin D as an immunomodulator in vascular disease is the increase in interleukin 10 and decrease in tumour necrosis factor (TNF) seen in a randomised controlled trial of vitamin D3 and calcium supplementation in chronic heart failure [32]; both are cytokines thought to play a role in the pathophysiology of heart failure.
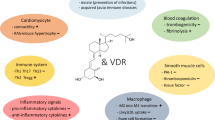
Vascular endothelial cells are known to express VDR. Studies have shown the ability of 1,25OHD to regulate innate immunity in endothelial cells when activated by lipopolysaccharides [33]. Jabalonski et al. [34] demonstrated a relationship between low vitamin D levels and increased vascular endothelial cell expression of NF-kappa B and interleukin 6. Such associations do not necessarily infer a causal relationship between low levels of vitamin D metabolites and inflammation, as some clinical studies suggest that the inflammatory response may lead to reduction of 25OHD levels [35]. Thus, a number of potentially plausible biological mechanisms exist that link vitamin D and its metabolites with effects on the cardiovascular system. These potential mechanisms are summarised in Fig. 1.

Potential biological pathways for effects of vitamin D on the cardiovascular system
Observational evidence linking vitamin D to vascular disease
Cross sectional and case–control data
A wealth of cross-sectional data has shown associations between a range of cardiometabolic risk factors and 25 hydroxyvitamin D levels. Cross-sectional data support inverse relationships between 25OHD levels and blood pressure [36, 37]. The degree of difference in blood pressure across the spectrum of 25OHD levels is small (3 mmHg for systolic blood pressure) but potentially significant at a population level. Similar relationships have been shown between higher 25OHD and higher HDL levels, and lower 25OHD and indices of impaired glucose homeostasis [37]. 25OHD levels are lower in patients with type 2 diabetes than in age- and sex-matched patients without diabetes [38] and participants in the 1958 British birth cohort with 25OHD levels in the highest tertile had only one third the prevalence of the metabolic syndrome compared to those with the lower tertile of 25OHD levels [39]. An association between low 25OHD levels and prevalent cardiometabolic risk factors extends even to adolescents [40]. Inverse associations were noted between 25OHD levels and blood pressure and insulin resistance, and higher 25OHD was associated with higher HDL in this population. These associations remained significant after correcting for obesity, age, sex, and ethnicity. Nontraditional risk factors for cardiovascular disease are also related to 25OHD levels in cross-sectional studies, worse endothelial function and arterial stiffness are correlated with lower 25OHD levels in healthy individuals [41], and lower 25OHD levels (<75 nmol/L) are related to worse endothelial function in patients with type 2 diabetes [42].
Patients with myocardial infarction, stroke, and heart failure [43–45] have 25OHD levels that are lower than age and sex matched controls. Patients with type 2 diabetes mellitus [46] with serum 25OHD levels of less than 50 nmol/L were 1.7 times as likely to have an existing diagnosis of cardiovascular disease, despite a similar prevalence of other cardiovascular risk factors. Cross-sectional data is however notoriously prone to confounding and this is a particular problem with vitamin D measurement. Almost any illness will lead to reduced activity, with consequent reduced sun exposure and lower 25OHD levels.
Longitudinal data
A considerable body of longitudinal data also exists supporting an association between 25OHD levels and a range of cardiovascular diseases. Some of the key data are outlined below, and a summary of relevant meta-analyses in this area is given in Table 1.
Hypertension
Lower baseline 25OHD has been associated with an increased risk of new incident hypertension [47, 48]. Studies followed patients after either direct measurement of 25OHD levels or after algorithm-based prediction of 25OHD levels for between 4 and 8 years. Meta-analysis of several cohort studies, all of which relied on self-reported hypertension, demonstrated that those with the lowest baseline 25OHD level have a 1.8 times higher risk of developing a new diagnosis of hypertension [49, 50]. However, more recent data from the Women’s Health Initiative trial found no relationship between baseline 25OHD level and the risk of either incident hypertension or of change in measured blood pressure over a 7-year follow up [51].
Metabolic syndrome and diabetes
Lower 25OHD levels are associated with a lower risk of progression to diabetes. This risk of progression in the Nurses’ Health study [52] was 48 % lower for those in the highest 25OHD quartile compared to those in the lowest quartile. Similar, though less striking, results were also seen in patients enrolled in the Diabetes Prevention Programme [53]. Meta-analysis of eight longitudinal studies confirmed a reduction of 43 % (95 % CI, 24–57 %) in incident cases of diabetes in those with serum 25OHD >62.5 nmol/L compared with those with serum 25OHD < 35 nmol/L [54]. Similar results have been seen with similar, updated meta-analyses [55, 56].
Cardiovascular events
In the Framingham study, participants without overt cardiovascular disease at baseline were at increased risk of cardiovascular events if their baseline 25OHD level was <37.5 nmol/L, risk of cardiovascular events was doubled at the lowest 25OHD concentrations [57]. The combination of hypertension and low 25OHD level dramatically increased the risk of future cardiovascular events. Interestingly, although progressively lower 25OHD concentrations below approximately 60 nmol/L were associated with a progressive increase in cardiovascular risk, no further reduction in risk was seen above this threshold.
Data from Austria examining the risk of cardiovascular events in patients referred for coronary angiography supports the above findings. In addition, low vitamin D levels in this cohort also predicted future heart failure. The relation between low 25OHD levels and future sudden death or heart failure appeared even stronger in the subgroup with metabolic syndrome at baseline, but interestingly, baseline 25OHD levels did not predict incident myocardial infarction in this subgroup [58]. A recent meta-analysis of studies examining the relationship between 25OHD level and incident stroke is also consistent with the above findings. Low 25OHD levels were associated with a 50 % increased risk of future stroke in a pooled analysis [59–61].
Systematic review of longitudinal data [49] suggests that a 25OHD level below 25 nmol/L is associated with a 1.2- to 2.5-fold increase in the risk of cardiovascular events compared to levels >75 nmol/L. Such associations remain robust after adjustment for age, sex, body mass index, smoking, and previous cardiovascular events and have been seen in other recent meta-analyses [56, 62–64] including one in patients with chronic kidney disease [65]. However, unmeasured confounders may still exist to explain the association between 25OHD levels and vascular disease.
Heart failure
Patients with heart failure have particularly low levels of 25OHD [44, 66]. This is almost certainly a reflection of the marked exercise intolerance that is a hallmark of heart failure. This limits physical activity and is thus likely to limit sun exposure; most heart failure patients are over the age of 75, and will therefore be less efficient at generating vitamin D in response to UV light.
Cross-sectional studies have confirmed a relationship between heart failure severity and lower 25OHD levels [67, 68]. Lower 25OHD levels correlate with higher levels of natriuretic peptides in patients with heart failure, lower peak exercise capacity, and worse disease-specific quality of life [44, 69]. Longitudinal data confirms that low 25OHD levels independently predict death in patients with heart failure; the relative risk of death was increased by 1.52 (95 % CI, 1.21–1.92) for heart failure patients with 25OHD levels <25 nmol/L compared to those with 25OHD levels of >75 nmol/L in a large Israeli population study [70].
Data on the relationship between 25OHD levels and incident heart failure are harder to interpret. PTH, but not 25OHD levels, was associated with incident heart failure in the Cardiovascular Health Study [21] despite the fact that 25OHD levels are known to have a major effect on PTH levels. The Intermountain study group [71] examined incidence of heart failure in routinely collected population data; although the incidence of decompensated heart failure episodes was higher in those with 25OHD levels <37.5 nmol/L, this group had a higher prevalence of previously diagnosed heart failure at baseline (19 vs 10 % for those with 25OHD > 75 nmol/L), follow up was short (mean, 1.3 years) and 25OHD sampling was based on clinical indication, rather than being a random population sample. Data from over 3,000 patients referred for coronary angiography [68] who had baseline 25OHD levels measured found that the risk of dying from heart failure was 2.5 times higher in those with baseline 25OHD <25 nmol/L. This result remained robust to multiple risk factor adjustments and benefitted from a longer follow up (median, 7.7 years)
Atrial fibrillation
Data on the relationship between 25OHD levels and atrial fibrillation are conflicting. Cross-sectional data from the Intermountain study shows a higher prevalence of atrial fibrillation in those with 25OHD levels <37.5 nmol/L [71], but longitudinal data from the same study showed no increase in incidence of new AF in those with baseline 25OHD <37.5 nmol/L compared to those with 25OHD >75 nmol/L. Longitudinal data from the Framingham study also suggests that 25OHD levels do not predict incident atrial fibrillation over and above the effect of traditional cardiovascular risk factors (adjusted hazard ratio 0.99 [95 % CI, 0.88–1.10] per SD increment in 25OHD) [72]. Interestingly, further data from the Intermountain study suggests that patients with very high (>250 nmol/L) 25OHD levels have a significantly increased risk of developing atrial fibrillation (adjusted hazard ratio, 2.5; p = 0.003) [73].
Problems with observational data
Any observational data, no matter how well prepared or controlled, is still potentially subject to residual confounding. This is a particular problem for observational studies of vitamin D and cardiovascular disease for several reasons. Firstly, any disease state is likely to reduce activity and therefore sun exposure, leading to reverse causality. This issue dogs all cross-sectional data and potentially confounds any longitudinal data with short follow-up times. Secondly, a number of risk factors for cardiovascular disease are also known to lead to low 25OHD levels. These include older age, obesity, smoking, and physical inactivity [38, 74]. Low 25OHD levels could therefore be a bystander (risk marker) due to the presence of these risk factors, but it is still possible that some of the deleterious effect of these risk factors on cardiovascular health is mediated via reduction of 25OHD levels. Dissecting out the independent effect of vitamin D from these other risk factors in observational studies is probably not possible without some residual confounding. Thirdly, it is entirely possible that other, poorly understood biological factors may confound any relationship. Recent work suggests that 25OHD is a negative acute-phase reactant, falling rapidly and significantly after a systemic inflammatory insult [35]. Therefore, the presence of low-grade chronic inflammation, known to be a pathophysiological contributor to cardiovascular disease and often present years before symptoms become overt, may drive both cardiovascular disease and low 25OHD levels. In a similar vein, ultraviolet light has other biological effects beyond vitamin D synthesis; UV light appears to have vascular effects independent of vitamin D, mediated by release of cutaneous nitric oxide stores [75]. Changes in external temperature with season may influence blood pressure [76], giving yet another biological mechanism for mediating cardiovascular risk that is correlated with, but not caused by, vitamin D. Finally, several different technologies exist for measuring 25OHD levels [77] and measurements have historically suffered from a lack of global standardisation (although this is now being addressed via a global program based on internal standards from the National Institute of Standards and Technology in the USA). Thus, comparison across studies and across sites is problematic, especially given the nonlinear response of some assays and the frequent reporting of results using cutoff values, which are highly sensitive to differences in assay technique. These potential confounding factors are summarised in Table 2.
Intervention studies
Effects of vitamin D on surrogate markers of vascular disease
Blood pressure
Some, but not all studies investigating the effect of vitamin D supplementation have seen improvements in blood pressure. Meta-analysis [78] confirmed a mean reduction of 3.6/3.1 mmHg with supplementation, but this effect was confined to studies with a mean baseline systolic BP > 140 mmHg. Studies examining normotensive individuals did not show any effect on blood pressure. This meta-analysis also suggested that vitamin D2 or D3 were more effective than 1-alpha hydroxylated versions of vitamin D. Similar small reductions in blood pressure were noted in another meta-analysis [79]. A summary of meta-analyses of intervention trials for blood pressure, other surrogate markers and cardiovascular event data is given in Table 3. Arterial stiffness, a key contributor to both hypertension and left ventricular hypertrophy via increased cardiac afterload, did not improve after 20 weeks of treatment with 3,000 IU of vitamin D3 per day [80] compared with placebo.
Left ventricular hypertrophy
Left ventricular hypertrophy (LVH) is a major independent risk factor for cardiovascular events and cardiovascular death. High blood pressure is a significant contributor to the development of LVH, but is by no means the only determinant. Few intervention studies to date have examined the effect of vitamin D supplementation on left ventricular mass; the Paricalcitol Capsule Benefits in Renal Failure-Induced Cardiac Morbidity (PRIMO) trial [81], examining the effect of paricalcitol on LV mass in patients with chronic kidney disease (CKD), showed no effect on LV mass after 48 weeks of therapy when compared to placebo. Results from other studies underway evaluating this question are awaited.
Cholesterol
No overall effect of vitamin D supplementation on cholesterol levels has been seen in studies to date [82]; however, there is an interesting, possibly two-way interaction between vitamin D and statin therapy, perhaps mediated by the cytochrome 3A4 pathway. Statin therapy has been shown to raise vitamin D levels slightly in some but not all studies [83–85]. Conversely, giving vitamin D to patients on atorvastatin with low vitamin D levels may lower cholesterol [86]. Such interactions have not been explored in detail in randomised-controlled trials, and represent an opportunity for future research.
B-type natriuretic peptide
B-type natriuretic peptide is a marker of future cardiovascular risk. Its utility is not confined to the diagnosis of heart failure. Ventricular wall stress and myocardial ischaemia both elevate levels of B-type natriuretic peptide (BNP), and a range of cardiac pathologies, including coronary artery disease, LVH, and atrial fibrillation can raise BNP levels.
A number of studies have examined the effect of vitamin D supplementation on BNP levels. One study on older heart failure patients [66] did demonstrate a reduction in BNP levels, but a second study in younger patients failed to demonstrate a fall despite a higher dose (2,000 IU per day of vitamin D3) being used [32]. No reduction was seen in patients supplemented with vitamin D after myocardial infarction [87] but bolus supplementation with either 100,000 or 200,000 IU of oral vitamin D3 lowered BNP levels relative to placebo in patients with type 2 diabetes mellitus [88]. One trial of vitamin D supplementation in patients with previous stroke [89] showed an increase in BNP levels in the intervention arm compared to placebo.
Endothelial function
Endothelial function denotes the final common pathway to vascular damage and is influenced by multiple vascular insults, including smoking, high cholesterol, elevated blood pressure, and pollution. It is an independent predictor of future cardiovascular events and is thought to represent the pathological lesion that leads to atherosclerotic change. A number of studies have now examined the effect of vitamin D supplementation on endothelial function across a range of diseases. Marked improvement in endothelial function was noted in an uncontrolled trial of supplementation performed in a healthy young Turkish population with 25OHD levels of <25 nmol/L [90]. Results from randomised controlled trials have been mixed: in patients with type 2 diabetes one study showed improvement in endothelial function after a single 100,000 IU oral dose of vitamin D2 [91] but another study showed no benefit after bolus supplementation with high dose vitamin D3 [88]. Bolus (60,000 IU per month) vitamin D3 supplementation also improved endothelial function in overweight African-Americans [92]. Transient improvements 8 weeks after supplementation were seen in patients with stroke, but no improvement was noted at 16 weeks [89]. Measurement of fingertip plethysmography (a correlate of coronary artery endothelial function) showed no improvement with supplementation in a group of patients with previous myocardial infarction [87]. It is possible that the lack of effect seen was due to the use of fingertip plethysmography as opposed to measurement of brachial artery function used in several other trials. Differences between trials may also be due to the differences in the proportion of patients already receiving therapies known to improve endothelial function, e.g., ACE inhibitors and statins.
A major caveat with most trials examining surrogate markers to date is that they have been of short duration, typically 6 months or less. It is therefore unclear if the effects of vitamin D on such surrogate markers (particularly blood pressure) are sustained in the longer term.
Cardiovascular outcome trials
To date, no large randomised controlled trials have been published that were designed specifically to test the effect of vitamin D supplementation on cardiovascular events. Some event data are available from existing trials, which were mostly conducted in patients with osteoporosis with the aim of reducing fractures or improving bone mineral density. Although there is a well-known association between osteoporosis and vascular disease, such trial results cannot reliably be generalised to wider populations, were not powered to detect differences in cardiovascular events, and were not designed to accurately classify, record, and adjudicate on the presence of cardiovascular events.
Nevertheless, a number of authors have attempted to analyse existing trials, either singly or in meta-analysis. The Women’s Health Initiative (WHI) study [93] randomised 36,000 women to receive 1g calcium plus 400 U vitamin D3 daily for 7 years. No difference was seen in either myocardial infarction or coronary heart disease death (HR, 1.04; 95 % CI, 0.92–1.18), stroke (HR, 0.95; 95 % CI, 0.82–1.10) or new-onset diabetes mellitus [94] (HR, 1.01; 95 % CI, 0.94–1.10). Trivedi et al. [95] randomised 2,286 older people to receive 100,000 IU oral vitamin D3 or placebo every 4 months for 5 years; the relative risk of both cardiovascular death (0.84) and overall mortality (0.88) was non‐significantly lower in the treatment arm.
A recent meta-analysis concluded that for the small number of eligible trials (n = 6 for myocardial infarction, n = 6 for stroke), no significant difference was detectable between the intervention and control groups for myocardial infarction (HR, 1.02; 95 % CI, 0.93–1.10) or stroke (HR, 1.05; 95 % CI, 0.88–1.25) [96]. Similar effects were seen in another recent meta-analysis [97]. However, these results were heavily influenced by the size of the included WHI trial relative to the other included trials. The dose of vitamin D used in the WHI trial was very small; extrapolation from other published data suggests that this dose was enough to increase circulating 25OHD levels by only 10 nmol/L [98]; smaller than might be needed to detect a reduction in cardiovascular events. Thus, the lack of effect in this trial (and by extension the aforementioned meta-analysis) may be due to inadequate dosing.
CKD
The effects of vitamin D supplementation in chronic kidney disease are even more complex than for other disease states. Impaired 1 alpha hydroxylation by the failing kidney has led to widespread use of 1-alpha hydroxylated vitamin D analogues and concern about the ability of vitamin D to increase serum calcium and hence calcium × phosphate product has led to development of analogues (e.g., paricalcitol) designed to have less effect on serum calcium. More recent data suggests that 1-alpha hydroxylation may still occur in the failing kidney, as well as occurring in other target tissues (e.g., macrophages) [2, 3]. Thus, it is biologically plausible that giving nonhydroxylated vitamin D2 or D3 to CKD patients could still have beneficial biological effects.
Intervention trials in CKD have shown mixed effects. No effect of 8 weeks of 40,000 IU/week supplementation with vitamin D3 was seen on blood pressure or arterial stiffness [99] despite an increase in 1,25OHD and a decrease in PTH levels in the treatment group. A recent meta-analysis [100] failed to find data on the rates of cardiovascular events from the small number (n = 5) of RCTs comparing vitamin D2 or D3 with placebo in patients with CKD; no effect on overall mortality was seen in another meta-analysis [101].
Recent trials of paricalcitol (1–2 μg per day for 24 weeks) have shown a reduction in proteinuria in patients with diabetic nephropathy [102], but the PRIMO trial failed to show regression of left ventricular hypertrophy in paricalcitol-treated patients, despite giving 2 μg per day for 48 weeks [81]. A meta-analysis of paricalcitol trials [103] showed no reduction in cardiovascular endpoints, and the effect of 1-alpha hydroxylated vitamin D on CKD progression and all-cause mortality remains unclear from existing trial data [101].
Diabetes/glycaemic control
Despite a considerable body of observational evidence, trial data examining the effect of vitamin D supplementation on glycaemic control in diabetes, or aiming to prevent onset of diabetes have been disappointing to date. A recent systematic review noted a small (−0.3 mmol/L) reduction in fasting glucose with vitamin D supplementation in patients with diabetes or impaired fasting glucose, no effect on glycosylated haemoglobin, and a modest improvement in insulin resistance [104]. Analyses of data from two osteoporosis trials (WHI and Randomised trial of vitamin D and calcium for the secondary prevention of osteoporosis‐related fractures in the elderly (RECORD)) failed to show any reduction in the rate of new diagnoses of type 2 diabetes mellitus after vitamin D treatment [94, 105].
The existing data pertinent to type 2 diabetes can be legitimately criticised as being derived from trials set up for other reasons. Imprecise definition of new onset diabetes diagnosis and lack of trials targeting groups at high risk of developing new onset type 2 diabetes are potential problems, although the existing evidence does not lend strong support for performing such trials. It is still possible that vitamin D could reduce microvascular or macrovascular adverse outcomes in patients with diabetes even without an effect on glycaemic control. Effects on VEGF system might mediate beneficial effects on microvascular function and improvement in blood pressure and endothelial function could still reduce macrovascular risk, which is less driven by glycaemic control. Trials are needed to test whether vitamin D can reduce the incidence of type 1 diabetes; intervention data are lacking in this area, and the different mechanisms postulated for benefits in type 1 diabetes (i.e., immunosuppression) support further work in this area.
Heart failure trials
Heart failure is a systemic disease; it affects not only cardiac function, but is accompanied by a syndrome of neurohormonal overactivation (particularly adrenergic and renin–angiotensin–aldosterone systems). Significant chronic inflammation is also a hallmark of heart failure, with elevated levels of proinflammatory cytokines, particularly TNF alpha. Anaemia of chronic disease is also part of the heart failure syndrome, but of even more note is the profound skeletal myopathy that accompanies heart failure. This myopathy affects predominantly type 1 muscle fibres, contributing in large part to the fatiguability and exercise intolerance that is the hallmark of chronic heart failure.
There are thus multiple potential targets on which vitamin D could potentially act in heart failure, given its potential effects on inflammation, muscle function, and endothelial function. Two randomised controlled trials have reported to date on the effects of vitamin D in heart failure. The first trial [32] compared 2,000 IU per day of oral vitamin D3 with placebo, given over a 9-month period. They found no effect of vitamin D on ejection fraction, exercise capacity or B-type natriuretic peptide levels, but did show a significant reduction in TNF alpha levels. The second trial [66] compared the effects of two 100,000 IU oral vitamin D2 doses given 10 weeks apart with placebo in older (mean age, 80 years) heart failure patients. This trial found no effect on exercise capacity, physical function, quality of life, or TNF alpha levels, although BNP levels were reduced in the intervention group.
Although no encouraging signals of effect in heart failure have yet been seen, it is possible that larger doses are needed and beneficial effects on hospitalisation and death may still be produced by vitamin D supplementation even in the absence of benefits on symptoms and physical function.
Cardiovascular safety
Most observational and interventional data collected to date does not suggest any deleterious effect of vitamin D on cardiovascular health, even with relatively long-term follow up. Animal models (particularly the rat) show that administration of vitamin D analogues may accelerate vascular calcification [16], but observational studies in humans suggest the opposite. Higher 25OHD levels were associated with lower coronary artery calcification scores [15] and no worsening of coronary artery calcification was seen in the WHI trial. One small study performed in rural Indian manual workers [106] suggested that those with very high 25OHD levels (>222 nmol/L) might have an increased risk of myocardial infarction. Such levels appear anomalously high, only being very rarely reached in most populations however, and the methodology of this study has been criticised. Data from the Framingham cohort suggest that the incidence of vascular events in a healthy population follows a U-shaped pattern, with a nadir at around 60 nmol/L [57], albeit with wide confidence intervals; a recent study of 250,000 community-dwelling people in Denmark also suggest a U-shaped pattern, with increasing risk of all-cause mortality above and below 50–60 nmol/L [107]. Other studies have found either linear relationships between 25OHD level and vascular risk or levels that flatten off above approximately 50 nmol/L [108].
The confounding effect of calcium
A key confounder in much of the interventional data collected to date (which originates from studies designed to improve bone health) is the coadministration of calcium. Recent meta-analyses have suggested that calcium, when compared to placebo, causes an increase in vascular death, particularly that due to myocardial infarction [109]. The interaction between calcium and vitamin D supplementation on cardiovascular disease is currently unclear. Reanalysis of the MRC RECORD trial suggests a trend to lower cardiovascular mortality in those given vitamin D alone vs placebo, higher mortality in those given calcium alone vs placebo, and a neutral effect of combined calcium and vitamin D [110]. Original analysis of the WHI trial data [93] suggested no effect of calcium plus vitamin D supplementation on either myocardial infarction or coronary heart disease death (HR, 1.04; 95 % CI, 0.92–1.18) or stroke (HR, 0.95; 95 % CI, 0.82–1.10), a result that was consistent with a meta-analysis of trials [96] that showed no effect of supplementation on either myocardial infarction or stroke. Reanalysis of the WHI dataset, omitting the 54 % of participants taking calcium supplements at baseline, suggested that calcium and vitamin D supplementation actually increased the risk of cardiovascular events by 15–20 % [111]. In contrast, one recent individual patient meta-analysis [112] including only large osteoporosis trials concluded that supplementation with vitamin D alone did not alter overall mortality (HR, 0.99; 95 % CI, 0.89–1.09) but combined calcium and vitamin D reduced overall mortality (HR, 0.91; 95 % CI, 0.85–1.00).
How then to interpret these conflicting data? The findings are certainly due in part to variation in the choice of trials to include in the different analyses. The WHI trial was performed on younger, healthier patients, and the vitamin D dose used was very low—possibly too low to produce a sufficient increment in vitamin D levels to affect cardiovascular health. It is therefore possible that the WHI results should be interpreted more as giving unopposed calcium, rather than calcium plus vitamin D. Further trials, rather than reanalysis of existing data, are required to resolve this controversy.
Future directions
Ultimately, the question of whether vitamin D supplementation improves vascular health can only be settled by performing large randomised controlled trials, specifically designed to answer this question. The concerns that have been raised over the possible deleterious effects of calcium should direct us to compare vitamin D with placebo, without coadministration of calcium. Debate continues as to what dose of vitamin D should be used in such trials; the current obsession with reaching prespecified levels of 25OHD has rather obscured any focus on dose–response relationships with vascular risk factors such as blood pressure and cholesterol. However, doses of vitamin D sufficient to reach mean 25OHD levels of around 75 nmol/L in trial populations will be needed to avoid criticism of insufficient dosing in the event that such trials are negative.
Trials will need to be powered to detect differences in cardiovascular event rates—in particular, cardiovascular death, new myocardial infarction and new stroke; incident heart failure is also an important outcome measure to record. Larger trials in populations with established heart failure are also needed, focusing on hospitalisation and death, but also on symptoms and quality of life. Further trials examining the effect of vitamin D supplementation on blood pressure are also needed, particularly focusing on populations with established hypertension, which have not been well studied to date. Such populations should be subdivided by existing treatment, and also by renin status, given the fact that vitamin D may reduce blood pressure via effects on the renin–angiotensin–aldosterone system. Existing data do not support trials of vitamin D specifically to prevent type 2 diabetes mellitus or to improve glycaemic control in this population, although the immunosuppressant actions of vitamin D still make type 1 diabetes a potential target for intervention trials, and it is still possible that vitamin D might be able to reduce micro and macrovascular complications in type 2 diabetes. Similarly, existing data do not support further trials using vitamin D as an adjunct to weight loss.
The current evidence does not therefore support routine supplementation with vitamin D to reduce cardiovascular risk at the population level. Supplementation for specific cardiovascular disease states cannot be recommended either, even for diseases such as heart failure which are associated with very low 25OHD levels. Until appropriate trial data are available, the balance between described risks (e.g., renal stones, costs, and tablet burden) and any potential benefits remains unclear, and extending the prescribing indications for vitamin D beyond its current use in osteomalacia, osteoporosis, and falls prevention in older patients in institutional care cannot be justified.
Conclusions
Vitamin D has a myriad of biological effects in addition to its traditionally ascribed roles in calcium metabolism and bone health. A number of biological pathways have been described that may mediate potentially beneficial effects on the cardiovascular system, and a large body of observational evidence supports an association between low 25OHD levels and both cardiovascular risk factors and cardiovascular events. However, such associations are prone to confounding, and existing trial data shows inconsistent effects on cardiovascular risk factors. Meta-analysis of cardiovascular data from osteoporosis trials has given inconsistent results, and large trials using cardiovascular events as a primary outcome are needed before vitamin D can be recommended as a therapy for cardiovascular disease.
References
Jones G, Strugnell SA, DeLuca HF (1998) Current understanding of the molecular actions of vitamin D. Physiol Rev 78:1193–1231
Jean G, Terrat JC, Vanel T et al (2008) Evidence for persistent vitamin D 1-alpha-hydroxylation in hemodialysis patients: evolution of serum 1,25-dihydroxycholecalciferol after 6 months of 25-hydroxycholecalciferol treatment. Nephron Clin Pract 110:c58–c65
Gottfried E, Rehli M, Hahn J et al (2006) Monocyte-derived cells express CYP27A1 and convert vitamin D3 into its active metabolite. Biochem Biophys Res Commun 349:209–213
Norman AW (2008) From vitamin D to hormone D: fundamentals of the vitamin D endocrine system essential for good health. Am J Clin Nutr 88:491S–499S
Bischoff-Ferrari HA, Giovannucci E, Willett WC et al (2006) Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr 84:18–28
Haussler MR, Jurutka PW, Mizwicki M et al (2011) Vitamin D receptor (VDR)-mediated actions of 1alpha,25(OH)(2)vitamin D(3): genomic and non-genomic mechanisms. Best Pract Res Clin Endocrinol Metab 25:543–559
DeLuca HF (2004) Overview of general physiologic features and functions of vitamin D. Am J Clin Nutr 80:1689S–1696S
Li YC, Kong J, Wei M et al (2002) 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest 110:229–238
Tomaschitz A, Pilz S, Ritz E et al (2010) Independent association between 1,25-dihydroxyvitamin D, 25-hydroxyvitamin D and the renin-angiotensin system: The Ludwigshafen Risk and Cardiovascular Health (LURIC) study. Clin Chim Acta 411:1354–1360
Forman JP, Williams JS, Fisher ND (2010) Plasma 25-hydroxyvitamin D and regulation of the renin–angiotensin system in humans. Hypertension 55:1283–1288
Vaidya A, Sun B, Larson C et al (2012) Vitamin D3 therapy corrects the tissue sensitivity to angiotensin ii akin to the action of a converting enzyme inhibitor in obese hypertensives: an interventional study. J Clin Endocrinol Metab 97:2456–2465
Simpson RU, Hershey SH, Nibbelink KA (2007) Characterization of heart size and blood pressure in the vitamin D receptor knockout mouse. J Steroid Biochem Mol Biol 103:521–524
Green JJ, Robinson DA, Wilson GE et al (2006) Calcitriol modulation of cardiac contractile performance via protein kinase C. J Mol Cell Cardiol 41:350–359
Lehto S, Niskanen L, Suhonen M et al (1996) Medial artery calcification. A neglected harbinger of cardiovascular complications in non-insulin-dependent diabetes mellitus. Arterioscler Thromb Vasc Biol 16:978–983
Watson KE, Abrolat ML, Malone LL et al (1997) Active serum vitamin D levels are inversely correlated with coronary calcification. Circulation 96:1755–1760
Niederhoffer N, Bobryshev YV, Lartaud-Idjouadiene I et al (1997) Aortic calcification produced by vitamin D3 plus nicotine. J Vasc Res 34:386–398
Demer LL, Tintut Y (2008) Vascular calcification: pathobiology of a multifaceted disease. Circulation 117:2938–2948
Hruska KA, Mathew S, Saab G (2005) Bone morphogenetic proteins in vascular calcification. Circ Res 97:105–114
Saleh FN, Schirmer H, Sundsfjord J et al (2003) Parathyroid hormone and left ventricular hypertrophy. Eur Heart J 24:2054–2060
Kosch M, Hausberg M, Vormbrock K et al (2000) Impaired flow-mediated vasodilation of the brachial artery in patients with primary hyperparathyroidism improves after parathyroidectomy. Cardiovasc Res 47:813–818
Kestenbaum B, Katz R, de Boer I et al (2011) Vitamin D, parathyroid hormone, and cardiovascular events among older adults. J Am Coll Cardiol 58:1433–1441
Molinari C, Uberti F, Grossini E et al (2011) 1 Alpha,25-dihydroxycholecalciferol induces nitric oxide production in cultured endothelial cells. Cell Physiol Biochem 27:661–668
Cardus A, Panizo S, Encinas M et al (2009) 1,25-Dihydroxyvitamin D3 regulates VEGF production through a vitamin D response element in the VEGF promoter. Atherosclerosis 204:85–89
Grundmann M, Haidar M, Placzko S et al (2012) Vitamin D improves the angiogenic properties of endothelial progenitor cells. Am J Physiol Cell Physiol. doi:10.1152/ajpcell.00030.2012
Stach K, Kalsch AI, Nguyen XD et al (2011) 1 Alpha,25-dihydroxyvitamin D3 attenuates platelet activation and the expression of VCAM-1 and MT1-MMP in human endothelial cells. Cardiology 118:107–115
Aihara K, Azuma H, Akaike M et al (2004) Disruption of nuclear vitamin D receptor gene causes enhanced thrombogenicity in mice. J Biol Chem 279:35798–35802
Pitocco D, Crino A, Di Stasio E et al (2006) The effects of calcitriol and nicotinamide on residual pancreatic beta-cell function in patients with recent-onset Type 1 diabetes (IMDIAB XI). Diabet Med 23:920–923
Borissova AM, Tankova T, Kirilov G et al (2003) The effect of vitamin D3 on insulin secretion and peripheral insulin sensitivity in type 2 diabetic patients. Int J Clin Pract 57:258–261
Norman AW, Frankel JB, Heldt AM (1980) Vitamin D deficiency inhibits pancreatic secretion of insulin. Science 209:823–825
Fadda GZ, Akmal M, Lipson LG et al (1990) Direct effect of parathyroid hormone on insulin secretion from pancreatic islets. Am J Physiol 258:E975–E984
Kolb H, Mandrup-Poulsen T (2005) An immune origin of type 2 diabetes? Diabetologia 48:1038–1050
Schleithoff SS, Zittermann A, Tenderich G et al (2006) Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: a double-blind, randomized, placebo-controlled trial. Am J Clin Nutr 83:754–759
Equils O, Naiki Y, Shapiro AM et al (2006) 1,25-Dihydroxyvitamin D inhibits lipopolysaccharide-induced immune activation in human endothelial cells. Clin Exp Immunol 143:58–64
Jablonski KL, Chonchol M, Pierce GL (2011) 25-Hydroxyvitamin D deficiency is associated with inflammation-linked vascular endothelial dysfunction in middle-aged and older adults. Hypertension 57:63–69
Reid D, Toole BJ, Knox S et al (2011) The relation between acute changes in the systemic inflammatory response and plasma 25-hydroxyvitamin D concentrations after elective knee arthroplasty. Am J Clin Nutr 93:1006–1011
Scragg R, Sowers M, Bell C (2007) Serum 25-hydroxyvitamin D, ethnicity, and blood pressure in the Third National Health and Nutrition Examination Survey. Am J Hypertens 20:713–719
Fraser A, Williams D, Lawlor DA (2010) Associations of serum 25-hydroxyvitamin D, parathyroid hormone and calcium with cardiovascular risk factors: analysis of 3 NHANES cycles (2001–2006). PLoS One 5:e13882
Martins D, Wolf M, Pan D et al (2007) Prevalence of cardiovascular risk factors and the serum levels of 25-hydroxyvitamin D in the United States: data from the Third National Health and Nutrition Examination Survey. Arch Intern Med 167:1159–1165
Hypponen E, Boucher BJ, Berry DJ et al (2008) 25-Hydroxyvitamin D, IGF-1, and metabolic syndrome at 45 years of age: a cross-sectional study in the 1958 British Birth Cohort. Diabetes 57:298–305
Ganji V, Zhang X, Shaikh N et al (2011) Serum 25-hydroxyvitamin D concentrations are associated with prevalence of metabolic syndrome and various cardiometabolic risk factors in US children and adolescents based on assay-adjusted serum 25-hydroxyvitamin D data from NHANES 2001–2006. Am J Clin Nutr 94:225–233
Al Mheid I, Patel R, Murrow J et al (2011) Vitamin D status is associated with arterial stiffness and vascular dysfunction in healthy humans. J Am Coll Cardiol 58:186–192
Yiu YF, Chan YH, Yiu KH et al (2011) Vitamin D deficiency is associated with depletion of circulating endothelial progenitor cells and endothelial dysfunction in patients with type 2 diabetes. J Clin Endocrinol Metab 96:E830–E835
Poole KE, Loveridge N, Barker PJ et al (2006) Reduced vitamin D in acute stroke. Stroke 37:243–245
Zittermann A, Schleithoff SS, Tenderich G (2003) Low vitamin D status: a contributing factor in the pathogenesis of congestive heart failure? J Am Coll Cardiol 41:105–112
Scragg R, Jackson R, Holdaway IM et al (1990) Myocardial infarction is inversely associated with plasma 25-hydroxyvitamin D3 levels: a community-based study. Int J Epidemiol 19:559–563
Cigolini M, Iagulli MP, Miconi V et al (2006) Serum 25-hydroxyvitamin D3 concentrations and prevalence of cardiovascular disease among type 2 diabetic patients. Diabetes Care 29:722–724
Forman JP, Giovannucci E, Holmes MD et al (2007) Plasma 25-hydroxyvitamin D levels and risk of incident hypertension. Hypertension 49:1063–1069
Forman JP, Curhan GC, Taylor EN (2008) Plasma 25-hydroxyvitamin D levels and risk of incident hypertension among young women. Hypertension 52:828–832
Pittas AG, Chung M, Trikalinos T et al (2010) Systematic review: Vitamin D and cardiometabolic outcomes. Ann Intern Med 152:307–314
Burgaz A, Orsini N, Larsson SC (2011) Blood 25-hydroxyvitamin D concentration and hypertension: a meta-analysis. J Hypertens 29:636–645
Margolis KL, Martin LW, Ray RM et al (2012) A prospective study of serum 25-hydroxyvitamin D levels, blood pressure, and incident hypertension in postmenopausal women. Am J Epidemiol 175:22–32
Pittas AG, Sun Q, Manson JE et al (2010) Plasma 25-hydroxyvitamin D concentration and risk of incident type 2 diabetes in women. Diabetes Care 33:2021–2023
Pittas AG, Nelson J, Mitri J et al (2012) Plasma 25-hydroxyvitamin D and progression to diabetes in patients at risk for diabetes: an ancillary analysis in the Diabetes Prevention Program. Diabetes Care 35:565–573
Mitri J, Muraru MD, Pittas AG (2011) Vitamin D and type 2 diabetes: a systematic review. Eur J Clin Nutr 65:1005–1015
Forouhi NG, Ye Z, Rickard AP et al (2012) Circulating 25-hydroxyvitamin D concentration and the risk of type 2 diabetes: results from the European Prospective Investigation into Cancer (EPIC)-Norfolk cohort and updated meta-analysis of prospective studies. Diabetologia 55:2173–2182
Parker J, Hashmi O, Dutton D et al (2010) Levels of vitamin D and cardiometabolic disorders: systematic review and meta-analysis. Maturitas 65:225–236
Wang TJ, Pencina MJ, Booth SL et al (2008) Vitamin D deficiency and risk of cardiovascular disease. Circulation 117:503–511
Thomas GN, Hartaigh B, Bosch JA et al (2012) Vitamin D levels predict all-cause and cardiovascular disease mortality in subjects with the metabolic syndrome: the Ludwigshafen Risk and Cardiovascular Health (LURIC) Study. Diabetes Care 35:1158–1164
Sun Q, Pan A, Hu FB (2012) 25-Hydroxyvitamin D levels and the risk of stroke: a prospective study and meta-analysis. Stroke 43:1470–1477
Brondum-Jacobsen P, Nordestgaard BG, Schnohr P et al (2012) 25-Hydroxyvitamin D and symptomatic ischemic stroke: An Original Study and Meta-Analysis. Ann Neurol. doi:10.1002/ana.23738
Chowdhury R, Stevens S, Ward H et al (2012) Circulating vitamin D, calcium and risk of cerebrovascular disease: a systematic review and meta-analysis. Eur J Epidemiol 27:581–591
Brondum-Jacobsen P, Benn M, Jensen GB et al (2012) 25-Hydroxyvitamin d levels and risk of ischemic heart disease, myocardial infarction, and early death: population-based study and meta-analyses of 18 and 17 studies. Arterioscler Thromb Vasc Biol 32:2794–2802
Scragg R (2011) Vitamin D and public health: an overview of recent research on common diseases and mortality in adulthood. Public Health Nutr 14:1515–1532
Grandi NC, Breitling LP, Brenner H (2010) Vitamin D and cardiovascular disease: systematic review and meta-analysis of prospective studies. Prev Med 51:228–233
Pilz S, Iodice S, Zittermann A (2011) Vitamin D status and mortality risk in CKD: a meta-analysis of prospective studies. Am J Kidney Dis 58:374–382
Witham MD, Crighton LJ, Gillespie ND (2010) The effects of vitamin D supplementation on physical function and quality of life in older patients with heart failure: a randomized controlled trial. Circ Heart Fail 3:195–201
Shane E, Mancini D, Aaronson K et al (1997) Bone mass, vitamin D deficiency, and hyperparathyroidism in congestive heart failure. Am J Med 103:197–207
Pilz S, Marz W, Wellnitz B et al (2008) Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J Clin Endocrinol Metab 93:3927–3935
Boxer RS, Kenny AM, Cheruvu VK et al (2010) Serum 25-hydroxyvitamin D concentration is associated with functional capacity in older adults with heart failure. Am Heart J 160:893–899
Gotsman I, Shauer A, Zwas DR et al (2012) Vitamin D deficiency is a predictor of reduced survival in patients with heart failure; vitamin D supplementation improves outcome. Eur J Heart Fail 14:357–366
Anderson JL, May HT, Horne BD et al (2010) Relation of vitamin D deficiency to cardiovascular risk factors, disease status, and incident events in a general healthcare population. Am J Cardiol 106:963–968
Rienstra M, Cheng S, Larson MG et al (2011) Vitamin D status is not related to development of atrial fibrillation in the community. Am Heart J 162:538–541
Smith MB, May HT, Blair TL et al (2011) Vitamin D excess is significantly associated with risk of atrial fibrillation. Circulation 124:A14699
Isaia G, Giorgino R, Rini GB et al (2003) Prevalence of hypovitaminosis D in elderly women in Italy: clinical consequences and risk factors. Osteoporos Int 14:577–582
Oplander C, Volkmar CM, Paunel-Gorgulu A et al (2009) Whole body UVA irradiation lowers systemic blood pressure by release of nitric oxide from intracutaneous photolabile nitric oxide derivates. Circ Res 105:1031–1040
Wood AD, Secombes KR, Thies F et al (2012) Vitamin D3 supplementation Has no effect on conventional cardiovascular risk factors. a parallel-group, double-blind, placebo-controlled RCT. J Clin Endocrinol Metab 97:3557–3568
Carter GD (2012) 25-hydroxyvitamin D: a difficult analyte. Clin Chem 58:486–488
Witham MD, Nadir MA, Struthers AD (2009) Effect of vitamin D on blood pressure: a systematic review and meta-analysis. J Hypertens 27:1948–1954
Wu SH, Ho SC, Zhong L et al (2010) Effects of vitamin D supplementation on blood pressure. South Med J 103:729–737
Larsen T, Mose FH, Bech JN (2012) Effect of cholecalciferol supplementation during winter months in patients with hypertension: a randomized, placebo-controlled trial. Am J Hypertens 25:1215–1222
Thadhani R, Appelbaum E, Pritchett Y et al (2012) Vitamin D therapy and cardiac structure and function in patients with chronic kidney disease: the PRIMO randomized controlled trial. JAMA 307:674–684
Wang H, Xia N, Yang Y (2012) Influence of vitamin D supplementation on plasma lipid profiles: a meta-analysis of randomized controlled trials. Lipids Health Dis 11:42
Perez-Castrillon JL, Vega G, Abad L et al (2007) Effects of Atorvastatin on vitamin D levels in patients with acute ischemic heart disease. Am J Cardiol 99:903–905
Rejnmark L, Vestergaard P, Heickendorff L (2010) Simvastatin does not affect vitamin D status, but low vitamin D levels are associated with dyslipidemia: results from a randomised, controlled trial. Int J Endocrinol 2010:957174
Yavuz B, Ertugrul DT, Cil H et al (2009) Increased levels of 25 hydroxyvitamin D and 1,25-dihydroxyvitamin D after rosuvastatin treatment: a novel pleiotropic effect of statins? Cardiovasc Drugs Ther 23:295–299
Schwartz JB (2009) Effects of vitamin D supplementation in atorvastatin-treated patients: a new drug interaction with an unexpected consequence. Clin Pharmacol Ther 85:198–203
Witham MD, Dove FJ, Khan F et al (2012) Effects of vitamin D supplementation on markers of vascular function after myocardial infarction—a randomised controlled trial. Int J Cardiol. doi:10.1016/j.ijcard.2012.03.054
Witham MD, Dove FJ, Dryburgh M et al (2010) The effect of different doses of vitamin D(3) on markers of vascular health in patients with type 2 diabetes: a randomised controlled trial. Diabetologia 53:2112–2119
Witham MD, Dove FJ, Sugden JA et al (2012) The effect of vitamin D replacement on markers of vascular health in stroke patients—a randomised controlled trial. Nutr Metab Cardiovasc Dis 22:864–870
Tarcin O, Yavuz DG, Ozben B et al (2009) Effect of vitamin D deficiency and replacement on endothelial function in asymptomatic subjects. J Clin Endocrinol Metab 94:4023–4030
Sugden JA, Davies JI, Witham MD et al (2008) Vitamin D improves endothelial function in patients with type 2 diabetes mellitus and low vitamin D levels. Diab Med 25:320–325
Harris RA, Pedersen-White J, Guo DH et al (2011) Vitamin D3 supplementation for 16 weeks improves flow-mediated dilation in overweight African-American adults. Am J Hypertens 24:557–562
Hsia J, Heiss G, Ren H et al (2007) Calcium/vitamin D supplementation and cardiovascular events. Circulation 115:846–854
de Boer IH, Tinker LF, Connelly S et al (2008) Calcium plus vitamin D supplementation and the risk of incident diabetes in the Women’s Health Initiative. Diabetes Care 31:701–707
Trivedi DP, Doll R, Khaw KT (2003) Effect of four monthly oral vitamin D3 (cholecalciferol) supplementation on fractures and mortality in men and women living in the community: randomised double blind controlled trial. BMJ 326:469
Elamin MB, Abu Elnour NO, Elamin KB et al (2011) Vitamin D and cardiovascular outcomes: a systematic review and meta-analysis. J Clin Endocrinol Metab 96:1931–1942
Wang L, Manson JE, Song Y et al (2010) Systematic review: vitamin D and calcium supplementation in prevention of cardiovascular events. Ann Intern Med 152:315–323
Chlebowski RT, Johnson KC, Kooperberg C et al (2008) Calcium plus vitamin D supplementation and the risk of breast cancer. J Natl Cancer Inst 100:1581–1591
Marckmann P, Agerskov H, Thineshkumar S et al (2012) Randomized controlled trial of cholecalciferol supplementation in chronic kidney disease patients with hypovitaminosis D. Nephrol Dial Transplant 27:3523–3531
Kandula P, Dobre M, Schold JD et al (2011) Vitamin D supplementation in chronic kidney disease: a systematic review and meta-analysis of observational studies and randomized controlled trials. Clin J Am Soc Nephrol 6:50–62
Palmer SC, McGregor DO, Macaskill P (2007) Meta-analysis: vitamin D compounds in chronic kidney disease. Ann Intern Med 147:840–853
de Zeeuw D, Agarwal R, Amdahl M et al (2010) Selective vitamin D receptor activation with paricalcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL study): a randomised controlled trial. Lancet 376:1543–1551
Cheng J, Zhang W, Zhang X et al (2012) Efficacy and safety of paricalcitol therapy for chronic kidney disease: a meta-analysis. Clin J Am Soc Nephrol 7:391–400
George PS, Pearson ER, Witham MD (2012) Effect of vitamin D supplementation on glycaemic control and insulin resistance: a systematic review and meta-analysis. Diabet Med 29:e142–e150
Avenell A, Cook JA, Maclennan GS et al (2009) Vitamin D supplementation and type 2 diabetes: a substudy of a randomised placebo-controlled trial in older people (RECORD trial, ISRCTN 51647438). Age Ageing 38:606–609
Rajasree S, Rajpal K, Kartha CC et al (2001) Serum 25-hydroxyvitamin D3 levels are elevated in South Indian patients with ischemic heart disease. Eur J Epidemiol 17:567–571
Durup D, Jorgensen HL, Christensen J (2012) A reverse J-shaped association of all-cause mortality with serum 25-hydroxyvitamin D in general practice: the CopD study. J Clin Endocrinol Metab 97:2644–2652
de Boer IH, Levin G, Robinson-Cohen C et al (2012) Serum 25-hydroxyvitamin D concentration and risk for major clinical disease events in a community-based population of older adults: a cohort study. Ann Intern Med 156:627–634
Bolland MJ, Avenell A, Baron JA et al (2010) Effect of calcium supplements on risk of myocardial infarction and cardiovascular events: meta-analysis. BMJ 341:c3691
Avenell A, Maclennan GS, Jenkinson DJ et al (2012) Long-term follow-up for mortality and cancer in a randomized placebo-controlled trial of vitamin D(3) and/or calcium (RECORD trial). J Clin Endocrinol Metab 97:614–622
Bolland MJ, Grey A, Avenell A (2011) Calcium supplements with or without vitamin D and risk of cardiovascular events: reanalysis of the Women’s Health Initiative limited access dataset and meta-analysis. BMJ 342:d2040
Rejnmark L, Avenell A, Masud T et al (2012) Vitamin D with calcium reduces mortality: patient level pooled analysis of 70,528 patients from eight major vitamin D trials. J Clin Endocrinol Metab 97:2670–2681
Conflicts of interest
Dr Witham has received grant funding for vitamin D research from Chief Scientist Office, Scottish Government, Diabetes UK, Heart Research UK, Chest Heart and Stroke Scotland, Tenovus Tayside, ME Research UK.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Beveridge, L.A., Witham, M.D. Vitamin D and the cardiovascular system. Osteoporos Int 24, 2167–2180 (2013). https://doi.org/10.1007/s00198-013-2281-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-013-2281-1