
Abstract
Key message
A key candidate gene, GRMZM2G110141, which could be used in marker-assisted selection in maize breeding programs, was detected among the 16 genetic loci associated with waterlogging tolerance identified through genome-wide association study.
Abstract
Waterlogging stress seriously affects the growth and development of upland crops such as maize (Zea mays L.). However, the genetic basis of waterlogging tolerance in crop plants is largely unknown. Here, we identified genetic loci for waterlogging tolerance-related traits by conducting a genome-wide association study using maize phenotypes evaluated in the greenhouse under waterlogging stress and normal conditions. A total of 110 trait-single nucleotide polymorphism associations spanning 16 genomic regions were identified; single associations explained 2.88–10.67% of the phenotypic variance. Among the genomic regions identified, 14 co-localized with previously detected waterlogging tolerance-related quantitative trail loci. Furthermore, 33 candidate genes involved in a wide range of stress-response pathways were predicted. We resequenced a key candidate gene (GRMZM2G110141) in 138 randomly selected inbred lines and found that variations in the 5ʹ-UTR and in the mRNA abundance of this gene under waterlogging conditions were significantly associated with leaf injury. Furthermore, we detected favorable alleles of this gene and validated the favorable alleles in two different recombinant inbred line populations. These alleles enhanced waterlogging tolerance in segregating populations, strongly suggesting that GRMZM2G110141 is a key waterlogging tolerance gene. The set of waterlogging tolerance-related genomic regions and associated markers identified here could be valuable for isolating waterlogging tolerance genes and improving this trait in maize.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Waterlogging is an important abiotic stress leading to losses in crop production (Bailey-Serres et al. 2012). During its lifecycle, maize (Zea mays L.) frequently encounters waterlogging stress due to poor drainage and/or long periods of rainfall (Visser et al. 2003). Waterlogging of soil directly results in reduced levels of oxygen in tissues and gas diffusion between cells (Voesenek and Bailey-Serres 2013), which restrict aerobic respiration and result in a decrease in soil pH (Fukao and Bailey-Serres 2004; Setter et al. 2009). To survive under waterlogging conditions, numerous morphological, transcriptional, and metabolic changes are induced in plants (Bailey-Serres and Colmer 2014; Bailey-Serres and Voesenek 2008; Lee et al. 2011; Nanjo et al. 2011; Narsai et al. 2009; Zou et al. 2010), which ultimately influence growth and vigor and alter growth parameters such as morphological and biomass-related traits.
Identifying waterlogging tolerance-related quantitative trait loci (QTL) is challenging. However, these QTL have direct applications in maize breeding. Using markers tightly linked to waterlogging tolerance, it is possible to transfer tolerance genes to less tolerant lines to develop hybrids with high tolerance to waterlogging. To date, more than 100 QTL conferring waterlogging tolerance have been mapped on all 10 maize chromosomes. Since the availability of oxygen in roots under waterlogging conditions is vital for plant survival, plants have evolved suites of adaptive traits that ensure gas exchange while avoiding oxygen loss (Colmer and Voesenek 2009). Approximately 14 QTL for root aerenchyma formation were mapped onto all 10 maize chromosomes except chromosome 4 (chr4) and chr6 (Mano and Omori 2008; Mano et al. 2007, 2008, 2012). In addition, two QTL for adventitious root formation were mapped to chr4 and chr8 (Mano et al. 2005), and one locus for barrier formation against radial oxygen loss was mapped to chr3 (Watanabe et al. 2017). The differential responses of plants to waterlogging directly affect root and shoot growth, as well as leaf senescence. Eighteen QTL for root length (Osman et al. 2013; Qiu et al. 2007; Zhang et al. 2012), 24 QTL for plant height (Osman et al. 2013; Qiu et al. 2007; Zhang et al. 2012), and two QTL for leaf injury (LI) (Mano et al. 2006, 2012) were mapped to different genomic loci on the 10 maize chromosomes. These changes ultimately affect biomass-related traits. Many QTL for biomass-related traits have also been identified and mapped in maize, including six QTL for root fresh weight, 14 QTL for root dry weight, three QTL for shoot fresh weight, 15 QTL for shoot dry weight, and 11 QTL for total dry weight (root dry weight and shoot dry weight) (Mano et al. 2006; Osman et al. 2013; Qiu et al. 2007; Zhang et al. 2012). Moreover, more than 14 QTL for yield and yield-related traits, such as root lodging, brace root, and stem lodging under waterlogging conditions in the field have been mapped (Zaidi et al. 2015).
These studies have produced large amounts of data that pave the way to understanding the genetic basis of waterlogging tolerance. However, in practice, breeders prefer to select traits with high heritability and easily identified characteristics or indicators. Therefore, morphological characteristics and yield-related traits are usually used as indicators of waterlogging tolerance, although examining physiological and developmental traits could well reveal the intrinsic biological characteristics of waterlogging tolerance. It is challenging to map/clone candidate genes that are directly associated with these traits. Comparative transcriptome/proteome analysis of plants under waterlogging conditions is usually performed to identify candidate genes primarily involved in signaling, transcription, and efficient anaerobic ATP production. Such analyses have demonstrated that many candidate genes encoding proteins of unknown function are involved in plant survival under low-oxygen conditions in Arabidopsis thaliana and maize (Lee et al. 2011; Mustroph et al. 2010; Narsai et al. 2011; Yu et al. 2015; Zou et al. 2010).
Maize germplasm collections consisting of inbred lines have abundant genetic diversity and rapid linkage disequilibrium (LD) decay, making maize an ideal crop for genome-wide association study (GWAS) (Yan et al. 2011). An association panel consisting of a global germplasm collection of 368 maize inbred lines was recently assembled (Yang et al. 2010; Li et al. 2013) and was successfully used to dissect the genetic basis of complex agronomic traits, such as oil biosynthesis and metabolic diversity in maize kernels (Li et al. 2013; Wen et al. 2014), as well as drought tolerance at the seedling stage (Liu et al. 2013; Mao et al. 2015; Wang et al. 2016). In the present study, we used this association panel to (i) estimate the phenotypic variability of morphological and biomass-related traits of seedlings under waterlogging conditions, (ii) identify waterlogging tolerance-associated variants and potential candidate genes, and (iii) develop molecular markers for improving waterlogging tolerance. The results of this study improve our understanding of the genetic basis of waterlogging tolerance and should facilitate the identification of desirable alleles for the genetic improvement of waterlogging tolerance in maize.
Materials and methods
Plant materials and growth conditions
Seed of the 368 inbred lines comprising the association panel (Fu et al. 2013; Li et al. 2013) were planted in a greenhouse with a controlled temperature (~ 28/22 °C day/night cycle), a 14-h/10-h light/dark cycle, and 70% average humidity. The growth substrate and waterlogging treatment were similar to those described previously (Qiu et al. 2007; Zhang et al. 2012). Briefly, 12 uniform seedlings of each line were planted in a plastic pot (20 cm in diameter and 30 cm deep) containing 3.5 kg quartz sand and 630 mL sterile deionized water. Two independent planting experiments were conducted using a randomized complete-block design with three replicates. The waterlogging treatment was carried out at the second-leaf stage by maintaining a 2–3-cm water layer above the quartz sand surface for 6 days (d). A parallel experiment that ensured normal growth was conducted as a control.
Phenotypic evaluation and statistical analysis
To evaluate the phenotypic responses of the inbred lines to waterlogging stress, seven seedling traits including plant height (PHT, cm), root length (RL, cm), shoot fresh weight (SFW, g), root fresh weight (RFW, g), shoot dry weight (SDW, g), root dry weight (RDW, g), and total dry weight (TDW, g, TDW = SDW + RDW) under control and waterlogging conditions were measured after 6 d of waterlogging stress. Leaf injury (LI) was scored only in plants under waterlogging conditions, as it was not observed under control conditions. The LI index was scored as described by Mano and Omori (2013) to determine the degree of leaf chlorosis. If a given seedling did not show leaf chlorosis, it was given an LI score of 0. If ~ 50% of the first leaf of a given seedling was chlorotic, it was given an LI of 0.5. A plant with a completely chlorotic first leaf was scored as 1.0, and a plant with a completely chlorotic first leaf and a 50% chlorotic second leaf was given a score of 1.5. Sampling, drying, and measurements were performed using previously described methods (Qiu et al. 2007). Briefly, the seedlings were carefully removed from the pot, and the roots were gently washed under running water. RL was measured from the coleoptilar node to the tip of the longest root, and PHT was measured from the coleoptilar node to the tip of the longest leaf. The roots were then separated from the plant, and RFW and SFW of each replicate per genotype for control and waterlogging treatment were measured, after which the plant tissues were rapidly transferred into an oven and dried until a constant weight was achieved. The average value of 12 seedlings was calculated to represent the trait phenotype of a line for one replicate, and the average value of three replicates per genotype was calculated to represent the trait phenotype under control or treatment conditions. Additionally, the seven secondary traits of each genotype were calculated based on the phenotypic value of a given trait under waterlogging condition to that under control treatment (e.g., RPHT = PHTtreatment/PHTcontrol), which revealed substantial genotypic responses to waterlogging stress. These secondary traits were defined as relative phenotypes, such as RPHT for relative phenotype of PHT, RRL for RL, RSFW for SFW, RRFW for RFW, RSDW for SDW, RRDW for RDW, and RTDW for TDW.
To evaluate the effect of waterlogging stress on plant phenotypes in the association panel, the mixed model was performed as follow: phenotype ~ T + (1|G) + (1|E) + (1|G*T) + (1|G*E) (T, treatment; G, genotype; E, environment; G*T, interaction of G and T; G*E, interaction of G and E), in which statistical significance of factor T was evaluated using Wald test, and significance of the other factors was evaluated using the likelihood ratio test (LRT). The average phenotype of seven seedling traits under control and treatment conditions, seven secondary traits, and LI phenotype in each environment were used to estimate best linear unbiased prediction (BLUP) of these traits via mixed models, in which G and E were set as a random effect, and the heritability (H2) of these trait was separately calculated as H2 = δ 2 g /(δ 2 G + δ 2 E /n + δ 2 e /nr) (δ 2 G genetic variance; δ 2 E environment variance; δ 2 e error; n number of experiments, and r number of replicates). These statistical analyses were conducted using the lme4 package in R software (R Development Core Team 2013; version 3.4.4; http://www.r-project.org/).
Genome-wide association study (GWAS)
GWAS was performed using BLUP data for seven secondary traits and LI phenotype via a compressed mixed linear model (cMLM) using TASSEL software (v3.0), which accounts for population structure (Q matrix) and familial kinship (K matrix) (Yu et al. 2005; Zhang et al. 2010). The 56,110 genomic SNPs (single nucleotide polymorphisms) from the MaizeSNP50 BeadChip (Li et al. 2012a, b) and 525,105 SNPs from transcriptomic sequencing (Fu et al. 2013) with a minor allele frequency (MAF) of ≥ 5% were merged. The merged data sets included 558529 SNPs that were used to perform the GWAS analyses with the inbred lines in the association panel. The standard cMLM was applied using default settings (P3D for variance component analysis; compression level set to optimum level), in which Q and K matrix were estimated as previously described (Fu et al. 2013). Briefly, a total of 16,338 SNPs with < 20% missing data and MAF > 5% were used to estimate population structure and kinship coefficient (Fu et al. 2013). The population structure was inferred by STRUCTURE (Pritchard et al. 2000), and the kinship matrix was calculated using the method of Loiselle et al. (Loiselle et al. 1995). The equation of standard MLM for GWAS could be expressed as: y = Wv + Xβ + Zu + e, where y is a vector of a phenotype; v and β are unknown fixed effects representing marker effects and non-marker effects, respectively; and u is a vector of size n (number of individuals) for unknown random polygenic effects having a distribution with mean of zero and covariance matrix of G = 2 Kδ 2 a , where K is the kinship (co-ancestry) matrix with element kij (i, j = 1, 2,…, n) calculated from either a set of genetic markers or pedigrees and δ 2 a is an unknown genetic variance. W, X and Z are the incidence matrices for v, β, and u, respectively, and e is a vector of random residual effects that are normally distributed with zero mean and covariance R = Iδ 2 e , where I is the identity matrix and δ 2 e is the unknown residual variance (Zhang et al. 2010). The Me (effective number of independent markers) method, implemented in Genetic type 1 error calculator (GEC) software, was used to estimate the Me for the 558,529 SNPs (Li et al. 2012a, b), and a uniform threshold was set to declare the significance of trait–SNP associations using a Bonferroni correction for multiple tests (p ≤ 1/Me). The proportion of genotypic variance explained (PVE) by a single SNP was estimated via cMLM using TASSEL3.0. The extent of local linkage disequilibrium (LD) for each significant SNP and all other SNPs on the same chromosome was also evaluated through TASSEL3.0, in which each significant SNP was set as a covariate factor. A genomic locus where the LD between the significant SNP and nearby SNPs decayed to r2 = 0.2 was defined as a local LD-based QTL interval. All significant SNPs with overlapping genomic regions were classified into a QTL interval. The genes associated with these SNPs were summarized and annotated using the MaizeGDB (www.MaizeGDB.org).
Association analysis of candidate gene GRMZM2G110141
To evaluate the genetic diversity of a candidate gene GRMZM2G110141 associated with LI phenotype, four pairs of primers (Table S1) were designed to amplify the promoter (1 kb), 5′- and 3′-UTR (untranslated region) and all introns and exons of this gene in 138 randomly selected inbred lines from the association panel using the B73 genome sequence as a reference (http://www.maizegdb.org/, version 5b.60). All amplified sequences were aligned using MEGA version 5 (http://www.megasoftware.net/). Nucleotide polymorphisms including SNPs and InDels were identified, and variants with MAF ≥ 5% were used for association analysis. Variants significantly associated with the LI index were identified using the cMLM model as mentioned before.
Detection of desirable alleles in two recombinant inbred line populations
Four maize inbred lines (B73, BY804, BY815, and K22) were applied to construct recombinant inbred lines (RILs), and a total of 181 RILs derived from B73 × BY804 (population1, pop1) and 197 RILs derived from BY815 × K22 (population2, pop2) were generated (Liu et al. 2017; Pan et al. 2017), which were genotyped at GRMZM2G110141 using gene-specific primers (Table S1). Polymorphisms were visualized using a Fragment Analyzer™ (AATI, North Brunswick, NJ USA). Waterlogging treatment of maize seedlings was conducted as described before. The LI indices of the parental lines and RILs were scored after 6 d of waterlogging stress. The alleles associated with low LI were designated as desirable alleles, and statistical analysis was performed via ANOVA.
The mRNA abundance analysis of GRMZM2G110141
The mRNA abundance of the candidate gene GRMZM2G110141 under normal and waterlogging conditions was assayed in 43 inbred lines that were randomly selected from the 138 resequenced samples. Root samples were separately collected from seedlings grown under normal (0 h), 4 h stress (4 h), and 3 d stress (3 d) treatment. Roots from five seedlings were pooled for RNA extraction using TRIZOL reagent (Invitrogen, Gaithersburg, MD, USA). Total RNA was treated with RNase-free DNase (Invitrogen). The purified RNA was used to synthesize single-stranded cDNA using recombinant M-MLV reverse transcriptase (Invitrogen). Quantitative reverse-transcription PCR (qRT-PCR) was performed using gene-specific primers (Table S1) with 2 × iTaq™ Universal SYBR Green Supermix (BioRad, Hercules, CA, USA). ZmActin1 (GRMZM2G126010) was employed as the internal control to normalize the expression data. Relative mRNA abundance were calculated according to the 2−ΔΔCT (cycle threshold) method (Livak and Schmittgen 2001).
Results
Waterlogging stress leads to extensive phenotypic variations in the association panel
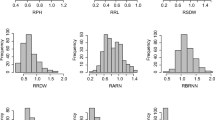
To estimate the effect of genotype in maize plants under waterlogging stress, we measured the phenotypes of eight traits under control and waterlogging conditions. The phenotypic values of each trait fit a normal distribution. Relative phenotypic values for root-related traits such as RRL, RRFW, and RRDW were seriously affected by waterlogging (Fig. 1a), and RL, RFW, and RDW decreased by more than 20% under waterlogging compared to normal condition, whereas shoot-related traits such as PHT, SFW, and SDW decreased by less than 8% in response to waterlogging (Table 1). The LI varied from 0 to 2.2, showing abundant variation (Fig. 1b, Supplementary Fig. 1). Statistical analysis (Table 1) demonstrated that all traits studied significantly differed among genotypes, indicating that the variance of these traits is genetically controlled. The relative phenotypic values of morphological- and biomass-related traits were significantly positively correlated between any two traits, including RPHT, RRL, RSFW, RRFW, RSDW, RRDW, and RTDW, whereas LI was significantly negatively correlated with seven other traits (Table S2). Heritability values from 0.691 to 0.967 were obtained for the measured traits and relative phenotypic values under control and waterlogging conditions (Table S3).

Phenotypic variation in waterlogging tolerance-related traits in the maize diversity panel. a Boxplot of the relative phenotypic value for the seven morphological and biomass traits analyzed. RPHT, relative phenotypic value for PHT; RRL for RL; RSFW for SFW; RRFW for RFW; RSDW for SDW; RRDW for RDW and RTDW for TDW. b Distribution of leaf injury across the 368 diverse maize accessions under waterlogging stress. The relative phenotypic value of each trait was calculated based on the phenotypic value of a given trait under waterlogging conditions compared to control treatment (e.g., RPHT = PHTtreatment/PHTcontrol). RL root length, PHT plant height, RFW root fresh weight, SFW shoot fresh weight, RDW root dry weight, SDW shoot dry weight, TDW total dry weight
Identification of significant loci for waterlogged-related traits through GWAS
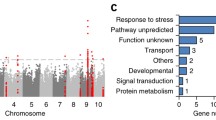
To identify waterlogging tolerance-associated loci, we performed a GWAS using the cMLM model that simultaneously control Q and kinship matrix, and 110 trait-SNP associations across the eight traits with a cutoff of p ≤ 3.99 × 10−6 (1/Me) were identified (Figs. S2, S3, Table S4), in which Me value was computed using the methodology described by Li et al. (2012a, b). These SNPs were mainly distributed on chr1, 2, 4, 5, 6, and 7. The PVE by single SNP ranged from 2.88 to 10.67%, and 69 SNPs had a PVE of > 5%. A total of 75 SNPs were significantly associated with the relative phenotypic values of biomass-related traits (RRFW, RSFW, RRDW, RSDW, and RTDW), and 12 SNPs were significantly associated with the relative phenotypic values of morphological-related traits (RPHT and RRL). We identified 23 SNPs on chr6 associated with LI, which had the highest PVE. Moreover, six SNPs were associated with two different traits: chr4.S_185037528 on chr4 was associated with RSDW and RTDW, and the five other SNPs on chr5 were associated with RSFW and RTDW. Under more stringent criteria (p ≤ 0.01/Me = 3.99 × 10−8), we identified five trait–SNP associations on chr4, 6, 7, and 10. Of these, chr4.S_185037528, chr4.S_183906006, chr6.S_150592850, chr7.S_109329219, and chr10.S_121414571 are located within GRMZM2G151516 (encoding the chloroplastic protein kinase APK1B), GRMZM5G829928 (encoding PTAC6; plastid transcriptionally active 6), GRMZM2G110141 (encoding DUF1682 family protein, a protein of unknown function), GRMZM2G092525 (encoding a protein whose expression is responsive to nematode infection), and GRMZM2G087824 (encoding an expressed protein of unknown function), respectively.
We evaluated the extent of the local LD of each significant SNP identified by GWAS (Fig. S4). The average value was 10.9 Mb. The most extensive LD decay was found on chr4, which ranged from 20 to 55.05 Mb, with an average of 51.01 Mb. However, the average LD decay was only 4.58 Mb, except on chr4. We merged the significant leading SNPs detected in the association panel for all traits and found 16 loci where multiple significant GWAS signals clustered in local LD-based QTL regions (r2 > 0.2), which represented 16 waterlogging-associated genomic loci: we named these loci QTL1 to QTL16 (Table 2). The average locus interval was 16 Mb. Five loci were covered by more than 10 significant SNPs, and 14 loci were supported by more than one significant SNP. These QTL are distributed on all chromosomes except chr3 and 8. Moreover, 14 loci co-localized with at least one previously identified QTL and seven loci co-localized with at least three QTL (Table 2, Table S5) (Mano et al. 2005, 2006, 2007, 2008, 2012; Osman et al. 2013; Qiu et al. 2007; Zaidi et al. 2015; Zhang et al. 2012).
Identifying a favorable allele of GRMZM2G110141 for waterlogging tolerance
Because QTL12, covering 23 LI-associated SNPs and the most significant LI-associated SNP (p = 1.79E−08) (Table S4), closely co-localized with a previously reported QTL (Osman et al. 2013), we further analyzed this QTL. We analyzed the haplotype block in the 100-kb genomic region flanking the most significant LI-associated SNP. We identified four blocks in this region, including block#2 and block#4, which covered a 67-kb and 6-kb genomic region, respectively, each encoding two genes (Fig. 2). The most significant SNP (chr6.S_150592850) is involved in block#4 and is located in gene GRMZM2G110141 (Fig. 2). Expression profiling of four candidate genes revealed that GRMZM2G110141 was highly expressed in seedling leaves, seedling roots, and the leaf meristem (Fig. S5), indicating its importance for seedling development. Furthermore, 12 SNPs in GRMZM2G110141 were found to be associated with LI (Table S4). Therefore, we resequenced a 3.2-kb genomic fragment containing GRMZM2G110141 in 138 randomly selected inbred lines. A total of 128 SNPs and 20 insertion/deletions (InDels) were detected. We again analyzed the association of each polymorphism with LI using the cMLM model and calculated the pairwise LD of these polymorphisms (Fig. 3a). The variants within the 5′-UTR (21 SNPs and 3 InDels) were significantly associated with LI (Bonferroni threshold p < 6.76 × 10−5), and these 24 LI-associated variants were mapped within a LD block (Fig. 3b). The 138 maize genotypes were classified into two haplotype (Hap) groups based on the significant variants (p < 6.76 × 10−5) (Fig. 3c). Hap1 had a significantly lower LI than Hap2 (p = 1.25E−10), and Hap1 was therefore designated as the favorable/tolerant allele (Fig. 3d), which could explain 26.2% of the phenotypic variation. Notably, some variants in the LD, such as SNP-329, SNP-275, SNP-271, and SNP-269, occurred in three motifs (CAAT-box, CAAAT-box, and TATCCAT/C-motif), which are important cis-acting elements in regulating gene expression. Moreover, three InDels (InDel-131, InDel-106, and InDel-75) resulting in a 13-bp deletion were also found in the tolerant lines. These results suggest that 5′-UTR variants of GRMZM2G110141 might lead to changes in gene expression, in turn altering the tolerance of the inbred lines to waterlogging stress.

Genomic location of the SNP locus associated with LI, and haplotype block analysis of chromosome 6. Genes within these regions are indicated in the diagram. LI leaf injury

Natural variations in GRMZM2G110141 are significantly associated with waterlogging tolerance in maize. a Association analysis of genetic variation in GRMZM2G110141 with waterlogging tolerance in maize. A schematic diagram of the 3.2-kb genomic region of GRMZM2G110141, including the promoter, 5′-UTR, three exons, two introns, and 3′-UTR. The locations of the start codon and stop codon are labeled as ATG and TAA, respectively. The region of the 5′-UTR is highlighted by a red rectangle. The p value is shown on a –log10 scale. b The pattern of pairwise LD of DNA polymorphisms in the promoter region and 5′-UTR. c Haplotypes of GRMZM2G110141 in maize genotypes BY815, BY804, JIAO51, K22, TY11, and B73 (reference genome). The site of the start codon (ATG) is designated as “+1.” A total of 24 DNA polymorphisms are significantly associated with maize waterlogging tolerance and are located in the 5′-UTR of GRMZM2G110141. These polymorphisms are shaded red. The CAAT-box, CAAAT-box, and TATCCAT/C-motif are different cis elements in the 5′-UTR. The location of the primer (indicated by a blue arrow) was used for genotyping the InDel polymorphism of GRMZM2G110141. d Comparison of LI for Hap1 and Hap2 in the 138 resequenced lines. e Comparison of GRMZM2G110141 expression between the waterlogging-tolerant and sensitive alleles. The gene expression levels were determined in the roots of 43 randomly selected lines under control, 4 h stress, and 3 d stress conditions. Hap1 represents tolerant alleles and Hap2 represents sensitive alleles. N is the genotype numbers of the two alleles. Statistical significance was determined by analysis of variance (ANOVA). LI leaf injury, Hap haplotype
To test the hypothesis, we measured the relative mRNA abundance of GRMZM2G110141 using qRT-PCR in plants under well-watered (control, before waterlogging treatment), short-term (4 h), and long-term (3 d) waterlogging stress in 43 randomly selected inbred lines. The mRNA abundance of GRMZM2G110141 was significantly higher in Hap1 than in Hap2 under stress conditions (Fig. 3e). Moreover, under short-term (4 h) and long-term (3 d) waterlogging conditions, the mRNA abundance of GRMZM2G110141 was significantly negatively correlated with LI (4 h, r = − 0.577, p = 0.030; 3 d, r = − 0.562, p = 0.037) but was not significantly correlated with LI under normal conditions (r = − 0.402, p = 0.314) (Fig. S6), indicating that the differences in GRMZM2G110141 expression contribute to the variance in LI in these inbred lines under waterlogging stress. Therefore, the variants in the 5′-UTR of GRMZM2G110141 might be the important causal variation conferring waterlogging stress tolerance in maize seedlings.
GRMZM2G110141 alleles can be used to identify waterlogging-tolerant genotypes
We designed a pair of PCR primers flanking the three InDels (13 bp) within the 5′-UTR to distinguish the alleles of GRMZM2G110141 (Fig. 3c). We analyzed two RIL populations (pop1 and pop2) and their parents to estimate the effect of the haplotype. Both BY804 and BY815 harbor GRMZM2G110141 alleles of the Hap1 genotype, whereas those of B73 and K22 are of the Hap2 genotype. The amplicons were 148-bp and 161-bp long for Hap1 and Hap2, respectively (Fig. S7). We compared the waterlogging tolerance of the four parental lines and RILs derived from these lines (Fig. 4). After 6 d of waterlogging stress, the LI of lines harboring Hap1 was significantly lower (0.31 for BY804 and 0.34 for BY815) than that of lines harboring Hap2 (1.19 for B73 and 1.34 for K22) (Fig. 4). Each line in pop1 and pop2 was genotyped by PCR and also subjected waterlogging stress treatment for 6 d. We determined the LI of each line and identified 88 and 102 lines with the Hap1 genotype and 93 and 95 lines with the Hap2 genotype in pop1 and pop2, respectively. The average LI index of RILs with Hap1 was 0.46 in pop1 and 0.52 in Pop2 (Fig. 4), which were significantly lower than those of lines with Hap2 in pop1 (1.15) and pop2 (1.21), respectively (p < 0.01) (Fig. 4). These results suggest that Hap1 of GRMZM2G110141 might be an allele that confers waterlogging tolerance, which could therefore be used to improve this trait in maize.

Favorable alleles of GRMZM2G110141 improve waterlogging tolerance in maize. a Phenotypic responses of B73 and BY804 to 6 d of waterlogging stress. b Phenotypic responses of K22 and BY815 to 6 d of waterlogging stress. c Leaf injury (LI) indices of B73, BY804, Hap1, and Hap2 in pop1. d LI of K22, BY815, Hap1, and Hap2 in pop2. N indicates the genotype numbers of the two alleles. Statistical significance was determined by analysis of variance (ANOVA). pop1, population 1 derived from a cross between B73 and BY804; pop2, population 2 derived from a cross between K22 and BY815; Hap haplotype
Discussion
Genetic architecture of waterlogging tolerance-related traits in maize
Waterlogging stress severely impairs the growth and development of land plants. Tolerance to waterlogging is a complex process that is not only determined by induced adaptive traits (Colmer and Voesenek 2009) but is also influenced by environmental factors, such as temperature and the duration of flooding. Here, we analyzed the genetic architecture of waterlogging tolerance-related traits in maize seedlings via GWAS. We identified 110 trait–SNP associations located within 16 genomic loci defined by an LD-based QTL interval (p < 1/Me = 3.66 × 10−6) (Table 2, Table S4), and a total of 76 trait–SNP associations were also identified under more stringency level (p < 1/N = 1.79 × 10−6). This number is markedly higher than the number of loci previously discovered in a GWAS using 144 inbred lines from Chinese germplasm (Table S6) (Zhang et al. 2012), mainly due to the diversity of the germplasms used, marker density, and population size (Table S6), which directly affect the detection power of association mapping (Yan et al. 2011). Moreover, we used the Me method to declare the significance threshold for trait–SNP associations in the present study, which considered the LD among SNPs that were not independent among tests (Li et al. 2012a, b). However, the Bonferroni method used in a previous study (Zhang et al. 2012) failed to consider the LD among SNPs, leading to an overly conservative p value correction (Pahl and Schafer. 2010).
The cross-validation of the mapping results from GWAS and linkage mapping provides important clues to screen vital target loci for waterlogging tolerance. Approximately 87.5% of loci identified in our GWAS were co-localized with at least one previously identified QTL (Mano et al. 2005, 2006, 2007; Mano et al. 2008, 2012; Osman et al. 2013; Qiu et al. 2007; Zaidi et al. 2015; Zhang et al. 2012) and seven loci (QTL5, 8, 11, 13, 14, 15, and 16) co-localized with at least three QTL (Table 2, Table S5), indicating that these seven loci are the most important QTL hotspots associated with waterlogging tolerance, which would become the key target loci to be applied to screen candidate genes and genetic improvement in future. On the other hand, two loci (QTL4 on chr2 and QTL10 on chr5) did not co-localize with previously identified QTL, suggesting that these loci might be new genetic loci that function in waterlogging tolerance. These data demonstrate that our GWAS platform is sufficiently robust to detect a number of genomic loci associated with waterlogging tolerance.
The stress signaling was sensed and transmitted to whole plants although the waterlogging stress was imposed on the root system. Here we used eight root- and leaf-related traits to characterize waterlogging tolerance variation, which may reflect different aspects of seedlings response after waterlogging stress. Our GWAS analysis indicated that different loci may control different response. For example, QTL8 on chr5 is associated with RSFW, RSDW, and RTDW, and two QTL associated with PHT (Osman et al. 2013) were also detected in this locus, suggesting that QTL8 might control plant height; QTL5 on chr4 is associated with RSDW and RTDW, and previously identified QTL in this locus are associated with adventitious root formation, root/shoot dry weight, plant height, and LI (Mano et al. 2005, 2012; Osman et al. 2013; Qiu et al. 2007), suggesting that QTL5 affects the development of the whole plant; QTL11 on chr6 is associated with RTDW, and five QTL associated with plant height, root/shoot dry weight, root/shoot fresh weight, and total dry weight were found at this locus, demonstrating that QTL11 controls biomass-related genetic loci; QTL14 on chr7 is associated with RPHT, while previously identified QTL are primarily associated with root-related traits such as root angle and root lodging (Zaidi et al. 2015). These results also provide evidence that the root system directly senses waterlogging and determines the level of plant tolerance to stress, which is genetically controlled in maize. Moreover, these QTL associated with specific trait, such as QTL8 controlling PHT, would help in our understanding of the genetic basis and provide valuable information for breeding.
Markers developed from GRMZM2G110141 could be used to improve waterlogging tolerance in maize
The polymorphic SNPs of the association panel were mainly detected based on RNA sequencing dataset from maize kernels (Fu et al. 2013). These SNPs are directly located within functionally important genes. In the current study, we identified 33 candidate genes associated with significant SNPs (Table S4), most of which are involved in a wide range of molecular functions, such as sucrose metabolism (GRMZM2G477236), stress responses (GRMZM2G335618), and signaling (GRMZM5G822947) and are responsive to waterlogging stress (Bailey-Serres et al. 2012; Yu et al. 2015; Zou et al. 2010). Molecular markers linked to functional loci could be developed to screen hybrid and inbred lines for the presence of favorable alleles. The favorable alleles could then be transferred from one inbred line to another via backcross breeding using marker-assisted selection to improve waterlogging tolerance in maize. Thus, we focused on the most significant peak associated with LI on chr6. Four candidate genes located within the peak SNP span two LD blocks, and only GRMZM2G110141 was highly expressed in seedling leaves and roots (Fig. 2). Although this gene was annotated as encoding a protein of unknown function, previous transcriptome and proteome studies have revealed that numerous proteins with no known biological function are involved in low-oxygen survival in Arabidopsis and maize (Lee et al. 2011; Mustroph et al. 2010; Yu et al. 2015; Zou et al. 2010). Resequencing and analysis of favorable alleles of GRMZM2G110141 associated with the most significant SNP further indicated that the sequence variation in the 5′-UTR affects waterlogging tolerance in the natural population (Fig. 3). The data from two RILs helped confirm the notion that Hap1 is a favorable allele for waterlogging tolerance (Fig. 4). The functional markers derived from GRMZM2G110141 could be used as direct targets to genetically improve waterlogging tolerance in maize seedlings on a large scale via high-throughput genotyping.
Conclusion
In the present study, a complex genetic architecture underlying natural variation of waterlogging tolerance in maize seedlings was unraveled through GWAS. A large panel coupled with a high density of SNPs enabled us to detect dozens of significant association loci. Previously identified QTL were summarized and compared with our GWAS signals to check their validity. As a result, we found that most of the LD-based QTL interval derived from GWAS signal could co-localize with at least one previously identified QTL. A total of 33 candidate genes that involved in wide range of stress-response pathway were associated with significant SNPs, and the key candidate GRMZM2G110141 was further validated via resequencing, analysis of mRNA abundance and confirmation of favorable alleles in two RILs. Our study provide some clues to the strategic concept of genetic improvement of waterlogging tolerance, and the hotspot association regions and candidate genes identified might open the door to help create waterlogging-tolerant varieties of maize.
Author contribution statement
FQ and FY designed the study. FQ and ZZ supervised the study. KL and DD performed the experiments. FY, XZ and HZ analyzed the data. FY, ZZ, BH, and FQ prepared the manuscript, and all authors read and approved the manuscript.
References
Bailey-Serres J, Colmer TD (2014) Plant tolerance of flooding stress–recent advances. Plant, Cell Environ 37:2211–2215
Bailey-Serres J, Voesenek LA (2008) Flooding stress: acclimations and genetic diversity. Annu Rev Plant Biol 59:313–339
Bailey-Serres J, Lee SC, Brinton E (2012) Waterproofing crops: effective flooding survival strategies. Plant Physiol 160:1698–1709
Colmer T, Voesenek L (2009) Flooding tolerance: suites of plant traits in variable environments. Funct Plant Biol 36:665–681
Fu J, Cheng Y, Linghu J, Yang X, Kang L, Zhang Z, Zhang J, He C, Du X, Peng Z, Wang B, Zhai L, Dai C, Xu J, Wang W, Li X, Zheng J, Chen L, Luo L, Liu J, Qian X, Yan J, Wang J, Wang G (2013) RNA sequencing reveals the complex regulatory network in the maize kernel. Nat Commun 4:2832
Fukao T, Bailey-Serres J (2004) Plant responses to hypoxia–is survival a balancing act? Trends Plant Sci 9:449–456
Lee SC, Mustroph A, Sasidharan R, Vashisht D, Pedersen O, Oosumi T, Voesenek LA, Bailey-Serres J (2011) Molecular characterization of the submergence response of the Arabidopsis thaliana ecotype Columbia. New Phytol 190:457–471
Li MX, Yeung JM, Cherny SS, Sham PC (2012a) Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum Genet 131:747–756
Li Q, Yang X, Xu S, Cai Y, Zhang D, Han Y, Li L, Zhang Z, Gao S, Li J, Yan J (2012b) Genome-wide association studies identified three independent polymorphisms associated with a-tocopherol content in maize kernels. PLoS ONE 7:e36807
Li H, Peng Z, Yang X, Wang W, Fu J, Wang J, Han Y, Chai Y, Guo T, Yang N, Liu J, Warburton ML, Cheng Y, Hao X, Zhang P, Zhao J, Liu Y, Wang G, Li J, Yan J (2013) Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat Genet 45:43–50
Liu S, Wang X, Wang H, Xin H, Yang X, Yan J, Li J, Tran LS, Shinozaki K, Yamaguchi-Shinozaki K, Qin F (2013) Genome-wide analysis of ZmDREB genes and their association with natural variation in drought tolerance at seedling stage of Zea mays L. PLoS Genet 9:e1003790
Liu J, Huang J, Guo H, Lan L, Wang H, Xu Y, Yang X, Li W, Tong H, Xiao Y, Pan Q, Qiao F, Raihan MS, Liu H, Zhang X, Yang N, Wang X, Deng M, Jin M, Zhao L, Luo X, Zhou Y, Li X, Zhan W, Liu N, Wang H, Chen G, Li Q, Yan J (2017) The conserved and unique genetic architecture of kernel size and weight in maize and rice. Plant Physiol 175:774–785
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408
Loiselle BA, Sork VL, Nason J, Graham C (1995) Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82:1420–1425
Mano Y, Omori F (2008) Verification of QTL controlling root aerenchyma formation in a maize × teosinte” Zea nicaraguensis” advanced backcross population. Breed Sci 58:217–223
Mano Y, Omori F (2013) Flooding tolerance in interspecific introgression lines containing chromosome segments from teosinte (Zea nicaraguensis) in maize (Zea mays subsp. mays). Ann Bot 112:1125–1139
Mano Y, Omori F, Muraki M, Takamizo T (2005) QTL mapping of adventitious root formation under flooding conditions in tropical maize (Zea mays L.) seedlings. Breed Sci 55:343–347
Mano Y, Masanori M, Tadashi T (2006) Identification of QTL controlling flooding tolerance in reducing soil conditions in maize (Zea mays L.) seedlings. Plant prod sci 9:176–181
Mano Y, Omori F, Takamizo T, Kindiger B, Bird RM, Loaisiga CH, Takahashi H (2007) QTL mapping of root aerenchyma formation in seedlings of a maize × rare teosinte “Zea nicaraguensis” cross. Plant Soil 295:103–113
Mano Y, Omori F, Kindiger B, Takahashi H (2008) A linkage map of maize × teosinte Zea luxurians and identification of QTLs controlling root aerenchyma formation. Mol Breed 21:327–337
Mano Y, Omori F, Takeda K (2012) Construction of intraspecific linkage maps, detection of a chromosome inversion, and mapping of QTL for constitutive root aerenchyma formation in the teosinte Zea nicaraguensis. Mol Breeding 29:137–146
Mao H, Wang H, Liu S, Li Z, Yang X, Yan J, Li J, Tran LS, Qin F (2015) A transposable element in a NAC gene is associated with drought tolerance in maize seedlings. Nat Commun 6:8326
Mustroph A, Lee SC, Oosumi T, Zanetti ME, Yang H, Ma K, Yaghoubi-Masihi A, Fukao T, Bailey-Serres J (2010) Cross-kingdom comparison of transcriptomic adjustments to low-oxygen stress highlights conserved and plant- specific responses. Plant Physiol 152:1484–1500
Nanjo Y, Maruyama K, Yasue H, Yamaguchi-Shinozaki K, Shinozaki K, Komatsu S (2011) Transcriptional responses to flooding stress in roots including hypocotyl of soybean seedlings. Plant Mol Biol 77:129–144
Narsai R, Howell KA, Carroll A, Ivanova A, Millar AH, Whelan J (2009) Defining core metabolic and transcriptomic responses to oxygen availability in rice embryos and young seedlings. Plant Physiol 151:306–322
Narsai R, Rocha M, Geigenberger P, Whelan J, van Dongen JT (2011) Comparative analysis between plant species of transcriptional and metabolic responses to hypoxia. New Phytol 190:472–487
Osman KA, Tang B, Wang Y, Chen J, Yu F, Li L, Han X, Zhang Z, Yan J, Zheng Y, Yue B, Qiu F (2013) Dynamic QTL analysis and candidate gene mapping for waterlogging tolerance at maize seedling stage. PLoS ONE 8:e79305
Pahl R, Schafer H (2010) PERMORY: an LD-exploiting permutation test algorithm for powerful genome-wide association testing. Bioinformatics 26:2093–2100
Pan Q, Xu Y, Li K, Peng Y, Zhan W, Li W, Li L, Yan J (2017) The genetic basis of plant architecture in 10 maize recombinant inbred line populations. Plant Physiol 175:858–873
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Qiu F, Zheng Y, Zhang Z, Xu S (2007) Mapping of QTL associated with waterlogging tolerance during the seedling stage in maize. Ann Bot 99:1067–1081
R Development Core Team (2013) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/
Setter TL, Waters I, Sharma SK, Singh KN, Kulshreshtha N, Yaduvanshi NP, Ram PC, Singh BN, Rane J, McDonald G, Khabaz-Saberi H, Biddulph TB, Wilson R, Barclay I, McLean R, Cakir M (2009) Review of wheat improvement for waterlogging tolerance in Australia and India: the importance of anaerobiosis and element toxicities associated with different soils. Ann Bot 103:221–235
Visser E, Voesenek L, Vartapetian B, Jackson M (2003) Flooding and plant growth. Ann Bot 91:107–109
Voesenek LA, Bailey-Serres J (2013) Flooding tolerance: O2 sensing and survival strategies. Curr Opin Plant Biol 16:647–653
Wang X, Wang H, Liu S, Ferjani A, Li J, Yan J, Yang X, Qin F (2016) Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nat Genet 48:1233–1241
Watanabe K, Takahashi H, Sato S, Nishiuchi S, Omori F, Malik AI, Colmer TD, Mano Y, Nakazono M (2017) A major locus involved in the formation of the radial oxygen loss barrier in adventitious roots of teosinte Zea nicaraguensis is located on the short-arm of chromosome 3. Plant, Cell Environ 40:304–316
Wen W, Li D, Li X, Gao Y, Li W, Li H, Liu J, Liu H, Chen W, Luo J (2014) Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights. Nat Commun 5:3438
Yan J, Warburton M, Crouch J (2011) Association mapping for enhancing maize (Zea mays L.) genetic improvement. Crop Sci 51:433–449
Yang X, Gao S, Xu S, Zhang Z, Prasanna BM, Li L, Li J, Yan J (2010) Characterization of a global germplasm collection and its potential utilization for analysis of complex quantitative traits in maize. Mol Breed 28:511–526
Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2005) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Yu F, Han X, Geng C, Zhao Y, Zhang Z, Qiu F (2015) Comparative proteomic analysis revealing the complex network associated with waterlogging stress in maize (Zea mays L.) seedling root cells. Proteomics 15:135–147
Zaidi PH, Rashid Z, Vinayan MT, Almeida GD, Phagna RK, Babu R (2015) QTL mapping of agronomic waterlogging tolerance using recombinant inbred lines derived from tropical maize (Zea mays L.) germplasm. PLoS ONE 10:e0124350
Zhang Z, Ersoz E, Lai CQ, Todhunter RJ, Tiwari HK, Gore MA, Bradbury PJ, Yu J, Arnett DK, Ordovas JM (2010) Mixed linear model approach adapted for genome-wide association studies. Nat Genet 42:355–360
Zhang X, Tang B, Yu F, Li L, Wang M, Xue Y, Zhang Z, Yan J, Yue B, Zheng Y, Qiu F (2012) Identification of major QTL for waterlogging tolerance using genome-wide association and linkage mapping of maize seedlings. Plant Mol Biol Re 31:594–606
Zou X, Jiang Y, Liu L, Zhang Z, Zheng Y (2010) Identification of transcriptome induced in roots of maize seedlings at the late stage of waterlogging. BMC Plant Biol 10:189
Acknowledgements
We greatly thank Dr. Yingjie Xiao for providing help in statistic analysis. This research was supported by the Fundamental Research Funds for the Central Universities (Program No. 2662015PY102) and supported by National Natural Science Foundation of China (Project 31571675).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Antonio Augusto Franco Garcia.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yu, F., Liang, K., Zhang, Z. et al. Dissecting the genetic architecture of waterlogging stress-related traits uncovers a key waterlogging tolerance gene in maize. Theor Appl Genet 131, 2299–2310 (2018). https://doi.org/10.1007/s00122-018-3152-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-018-3152-0