
Abstract
Key message
Host resistances in PI 197088 cucumber to downy and powdery mildew pathogens are conferred by 11 (3 with major effect) and 4 (1 major effect) QTL, respectively, and three of which are co-localized.
Abstract
The downy mildew (DM) and powdery mildew (PM) are the two most important foliar diseases of cucurbit crops worldwide. The cucumber accession PI 197088 exhibits high-level resistances to both pathogens. Here, we reported QTL mapping results for DM and PM resistances with 148 recombinant inbred lines from a cross between PI 197088 and the susceptible line ‘Coolgreen’. Phenotypic data on responses to natural DM and PM infection were collected in multi-year and multi-location replicated field trials. A high-density genetic map with 2780 single nucleotide polymorphisms (SNPs) from genotyping-by-sequencing and 55 microsatellite markers was developed, which revealed genomic regions with segregation distortion and mis-assemblies in the ‘9930’ cucumber draft genome. QTL analysis identified 11 and 4 QTL for DM and PM resistances accounting for more than 73.5 and 63.0% total phenotypic variance, respectively. Among the 11 DM resistance QTL, dm5.1, dm5.2, and dm5.3 were major-effect contributing QTL, whereas dm1.1, dm2.1, and dm6.2 conferred susceptibility. Of the 4 QTL for PM resistance, pm5.1 was the major-effect QTL explaining 32.4% phenotypic variance and the minor-effect QTL pm6.1 contributed to disease susceptibility. Three PM QTL, pm2.1, pm5.1, and pm6.1, were co-localized with DM QTL dm2.1, dm5.2, and dm6.1, respectively, which was consistent with the observed linkage of PM and DM resistances in PI 197088. The genetic architecture of DM resistance in PI 197088 and another resistant line WI7120 (PI 330628) was compared, and the potential of using PI 197088 in cucumber breeding for downy and powdery mildew resistances is discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cucurbit downy mildew (DM) is caused by the obligate oomycete Pseudoperonospora cubensis (Berk. & Curt.) Rostovzev, while powdery mildew (PM) is caused mainly by the fungus Podosphaera xanthii (Fr.) Braun and Shishkoff (syn. P. fusca). They are the two most economically important foliar diseases of all cucurbits worldwide. The symptoms of DM on susceptible cucumbers are characterized by water-soaked lesions limited by leaf veins that turn chlorotic and necrotic with brown/black sporulation on the abaxial leaf surface, eventually causing the death of entire plants (Oerke et al. 2006). The PM fungus does not usually kill the plants. PM appears as white or grayish-white patches on the plant leaves, petioles, stems, and fruits, sometimes causing chlorosis due to decreased photosynthesis (Sitterly 1972; Pérez-García et al. 2009).
Early breeding efforts identified many cucumber lines with both DM and PM resistances, such as ‘Puerto Rico 37’ and the Indian accession PI 197087 (Roque and Adsuar 1939; Smith 1948; Epps and Barnes 1952; Kooistra 1968). Van Vliet and Meysing (1974, 1977) found that the DM and PM resistance in the cultivar ‘Poinsett’ (derived from PI 197087) was often closely linked, but Fanourakis and Simon (1987) failed to draw the association between DM and PM resistances in other cucumber lines that derive their resistance from PI 197087, such as WI2757 and Gy2. PI 197088 is an accession collected from a region of India close to where PI 197087 originated. It has consistently exhibited a high level of resistance to PM (Clark 1975; Zijlstra and Groot 1992; Block and Reitsma 2005; Nie et al. 2015a). El Jack and Munger (1983) observed partial dominance of PM resistance in PI 197088 at the early growing stage. Morishita et al. (2003) suggested that the PM resistance in PI 197088-5, a selection of PI 197088, is due to two genes, one recessive and another incompletely dominant. In PI 197088-1, Sakata et al. (2006) identified four temperature-dependent quantitative trait loci (QTL) for PM resistance on chromosomes 1, 5, 6, and 7. With a detached-leaf assay, Fukino et al. (2013) detected seven QTL on chromosomes 1–6 in CS-PMR1 (also derived from PI 197088). More recently, Berg et al. (2015) and Nie et al. (2015a) identified a candidate gene for the major-effect PM resistance QTL pm5.1 (syn. pm-h), and found that the resistance is due to loss of function of the susceptibility gene CsMLO1 (or CsMLO8) on chromosome 5. Nie et al. (2015b) further found that a 1-bp insertion in this candidate gene in PI 197088 resulted in a premature stop codon and PM resistance.
As compared with PM resistance, a complete picture of the genetic basis of DM resistance in PI 197088 is still lacking. Large-scale screening tests identified PI 197088 with high-level resistance to prevailing field DM strains in the US (Call et al. 2012a, b). Caldwell et al. (2011) and Shetty et al. (2014) reported three DM resistance QTL in PI 197088 that were located on chromosomes 2, 4, and 5, respectively. Yoshioka et al. (2014) identified 10 QTL on chromosomes 1, 3, 5, 6, and 7 from the cross between CS-PMR1 (derived from PI 197088) and Santou. Several studies also identified DM resistance QTL in other cucumber lines. Szczechura et al. (2015) identified three DM resistance QTL on chromosome 5 from PI 197085. Wang et al. (2016) reported five QTL in WI7120 (PI 330628) on chromosomes 2, 4, 5, and 6, and revealed the dynamics of DM resistance through disease progress. In field screening tests, VandenLangenberg (2015) observed the segregation of DM resistance in F2 populations developed from crosses among PI 197088, PI 330628, and PI 605996 that may suggest different genetic basis of DM resistance among these accessions. VandenLangenberg and Wehner (2016) also observed different responses to DM natural infection among several accessions over plant development stages indicating the dynamics of active QTL during the process of DM resistance.
In the PI 197088-derived breeding line CS-PMR1, seven QTL for DM and PM resistances were found to be co-localized (dm1.2/pm1.1, dm1.3/pm1.2, dm3.1/pm3.1, dm5.1/pm5.1, dm5.2/pm5.2, and dm5.3/pm5.3, dm6.1/pm6.1) (Fukino et al. 2013; Yoshioka et al. 2014). DM/PM resistances in these studies were evaluated with single DM/PM strain by leaf-disc assays. It is not known if the host plants will perform the same way when infected with mixed DM/PM strains under field conditions. Moreover, due to the low resolution of genetic maps developed in the previous studies, the precise locations of mapped DM and PM QTL are not known, which prevents fine mapping and cloning of the major-effect QTL.
A robust and high-density genetic map is useful for many marker-based applications such as fine mapping of genes/QTL, as well as improvement of the genome assembly. With the availability of cucumber draft genomes (e.g., Huang et al. 2009; Yang et al. 2012) and use of next-generation sequencing technologies, several high-density linkage maps in cucumber have been developed (e.g., Yang et al. 2012; Wei et al. 2014; Xu et al. 2015; Zhou et al. 2015; Rubinstein et al. 2015; Zhu et al. 2016). These maps can significantly improve the detection power of minor-effect QTL for target traits and reduce the confidence LOD interval of the QTL. For example, using a high-density map consisting of 2084 bins with unique recombination events, Zhou et al. (2015) were able to pinpoint the candidate genes for cucumber scab resistance locus in a 180-kb region.
The main objective of this study is to conduct QTL mapping and reveal the genetic architecture of DM and PM resistances in PI 197088. An F7:8 RIL population from the cross between PI 197088 and Coolgreen was phenotyped for responses to natural infection of DM and PM pathogens under field conditions in multiple years. A high-resolution linkage map with saturated SNP and SSR markers was developed from the same cross and used for QTL analysis in this study.
Materials and methods
Plant materials
An RIL population derived from the cross between PI 197088 and Coolgreen was used to identify QTL for DM and PM resistances in the present study. PI 197088, a cucumber accession collected from India, is highly resistant to DM and PM diseases. Coolgreen, a cucumber variety developed by Asgrow Seed Co. (part of Seminis Vegetable Seeds Inc.), is highly susceptible to both pathogens. The PI 197088 × Coolgreen population with 169 F6:7 RILs was developed at the Department of Horticultural Science at North Carolina State University (VandenLangenberg 2015). The whole population was evaluated for DM resistance in field trials. A subset of 148 RILs was self-pollinated to advance to the F7:8 generation in the Walnut Street Research Greenhouse at the University of Wisconsin-Madison, and used for PM resistance studies and SSR/SNP genotyping.
Phenotypic data collection
Field evaluation of responses to natural infection of DM pathogen
For DM phenotypic data collection, in the summer field seasons of 2013, 2014, and 2015, 169 RILs and the two parental lines were directly seeded in test plots at the Horticultural Crops Research Stations of North Carolina State University in Clinton and Castle Hayne, North Carolina, which were designated as CL2013 (31/22 °C day/night temperature; 86% RH), CL2014 (31/21 °C day/night temperature; 81% RH), CL2015 (32/22 °C day/night temperature; 78% RH), CH2013 (31/23 °C day/night temperature; 79% RH), and CH2014 (31/22 °C day/night temperature; 76% RH), respectively. All entries were planted on 27 June 2013, 30 June 2014, and 30 June 2015 at Clinton, and 20 June 2013 and 26 June 2014 at Castle Hayne, respectively; the plants were grown with recommended horticultural practices (VandenLangenberg 2015).
All experiments used a randomized complete block design (RCBD) with two replications and 15 plants per plot. Plots were exposed to natural DM pathogen infection that was promoted by overhead irrigation during the growing season as needed, about three times per week (VandenLangenberg 2015). In each trial, disease severity was assessed three times with 1 week apart starting at 41 days after planting (dap) (first rating). The rating scale was based on percentage of symptomatic leaf area (per plot) with a scale from 0 to 9, where 0 = no damage, 1 = 1–10%, 2 = 11–20%, 3 = 21–30%, 4 = 31–40%, 5 = 41–50%, 6 = 51–60%, 7 = 61–70%, 8 = 71–80%, and 9 = 81–100% (or dead) (Jenkins and Wehner 1983).
Field evaluation for response to natural infection of powdery mildew
Phenotypic data of PM resistance among 148 F7:8 RILs of PI 197088 × Coolgreen were collected from field trials in 2014, 2015, and 2016 at the University of Wisconsin Hancock Agricultural Research Station (HARS) at Hancock, Wisconsin, which were designated as WI2014 (28/13 °C day/night temperature, 72% RH), WI2015 (27/14 °C day/night temperature, 80% RH), and WI2016 (27/14 °C day/night temperature, 78% RH), respectively. Each trial was an RCBD consisting of two replications with six plants per replication per line. Plots were exposed to natural epidemics of PM. Disease severity of each plot was evaluated ~ 80 days after planting when two parental lines showed clearly distinctive symptoms of PM infection. Disease evaluation was based on sporulation level on leaves using disease rating scales 1–9, where 1 = 0–10% surface area covered with PM spores, 2 = 11–20%, 3 = 21–30%, 4 = 31–40%, 5 = 41–50%, 6 = 51–60%, 7 = 61–70%, 8 = 71–80%, and 9 = 81–100% (He et al. 2013).
Statistical analysis of phenotypic data
Analysis of variance (ANOVA) was performed with the R/lme4 package to estimate the genetic and environmental effects (Bates et al. 2014). DM resistance trait was fitted in a linear mixed-effects model (LMM) with location and year effects for each rating time, while PM disease score was fitted in an LMM with year effects only. The best linear unbiased predictors (BLUPs) were also extracted from the models and used for further QTL analysis.
Heritability estimates were calculated from variance components from the mixed-effects model. Since the dominance variance and covariance of additive and dominant effects are equal to zero in the F6:7 and F7:8 RIL populations, heritability was considered to be narrow-sense (Cockerham 1983).
Genotyping
Bulked segregant analysis for DM resistance with microsatellite markers
Before the whole-genome marker data were available, we performed bulked segregant analysis (BSA) to identify markers associated with DM resistance. Two bulks, highly resistant and highly susceptible, were constructed based on the mean of three DM rating scores in CL2013 experiment by pooling equal quantities of DNA from eight resistant and eight susceptible RIL plants, respectively. SSR markers described in Ren et al. (2009), Cavagnaro et al. (2010), and Yang et al. (2012) were used to screen PI 197088 and Coolgreen; polymorphic ones were then applied to the two bulks. Polymorphic markers between bulks were used for further single marker analysis. Additional adjacent markers were also explored to genotype the 148 RILs and the genotypic data were integrated with SNP markers for genetic map construction.
Specific length amplified fragment (SLAF) sequencing (SLAF-Seq)
The 148 F7:8 RILs were genotyped with next-generation sequencing-based SLAF-Seq, which is an improved genotyping-by-sequencing (GBS) technology with distinguishing characteristics in deeper sequencing, reduced sequencing costs, and dual barcode system for large populations (Sun et al. 2013). The SLAF-Seq library was constructed following Wei et al. (2014) and sequenced with an Illumina HiSeq 2500 sequencer (Illumina, San Diego, CA, USA), which was performed at the Beijing Biomarker Technologies Corporation (http://www.biomarker.com.cn).
Raw SLAF-Seq reads were demultiplexed based on barcodes, and low-quality reads (quality score < 20) were filtered out. Reads of 100 base pair from the same samples were mapped onto the 9930 cucumber draft genome sequence (http://www.icugi.org, V2.0) using the SOAPdenovo2 software (Luo et al. 2012). All sequences aligned to the same position were defined as an SLAF locus. SLAF loci that contained fewer than three SNPs (single nucleotide polymorphism) with an average of 3 × depths of coverage were kept for genetic map construction.
Linkage map construction with SNP and SSR markers
The physical order of all SNP markers on the 9930 draft genome assembly (V2.0, http://www.icugi.org/) was examined with paired recombination fraction (RF) by LOD score for a test of RF = 0.5 using the function est.rf in R/qtl (Broman et al. 2003). On the RF-LOD plot, off-position and unlinked markers on each linkage group could be easily inspected manually. Next, the function mstmap in R/ASMap was used to re-order and adjust the marker positions, which was based on the minimum-spanning-tree (MST) algorithm and is efficient and fast for handling dense maps (Wu et al. 2008; Taylor and Butler 2015). After correcting errors in marker order, the RF between each marker pair was re-examined. On the RF plot, the correctness of the order of markers was judged with the following criteria: nearby markers are clearly associated, and distant markers showed no association. To integrate the SSR loci onto the SNP-based map, all SSR markers were assigned to unknown chromosomes first, and the function tryallpositions in R/qtl was then used to try all possible positions for each marker by keeping all other markers fixed. Finally, the genetic distances were re-estimated using the function est.map in R/qtl which was based on the likelihood through the hidden Markov chains algorithm (LHMC or Lander–Green algorithm. Lander et al. 1987). The resulting map with both SNP and SSR markers was then used for further analysis. The Kosambi mapping function was applied during the calculation of genetic map distance.
For genetic and physical map integration, the SLAF reads of mapped SNPs were used to search for homologous sequences in the Gy14 V1.0 draft genome assembly using BLAST + (Camacho et al. 2009) with a cut-off E value < 1.0E−10.
QTL analysis
Single marker analysis (SMA)
SMA was conducted for polymorphic SSR markers between the two bulks by performing ANOVA to test if variation among genotypic classes was significant and to estimate additive effects for each marker (P < 0.05).
QTL analysis with SNP-based high-density genetic map
QTL analysis for PM and DM resistances was performed in the R/qtl package (Arends et al. 2010, 2014). Preliminary analysis with both simple interval mapping (SIM) and composite interval mapping (CIM) methods indicated multiple QTL for DM resistance on one chromosome. Often, a non-existing (‘ghost’) QTL may appear between two-linked QTL. To reduce the chance in detecting ghost QTL, the multiple-QTL mapping (MQM) method was employed which also strengthened the identification of QTL in coupling and repulsion phases. The function mqmaugmentdata was first used for filling in missing genotypes, and the cofactors were determined by function scanone and initial mqmscan with a forward stepwise approach. Then, the function mqmscan was employed again for unsupervised backward elimination to identify significant QTL. The significance of each QTL was tested by the function mqmpermutation for each trait using 1000 permutations at a significance threshold of 0.05. All QTL identified by different environments and their interactions were further investigated in R/lme4 by fitting linear models to test their statistical significance. All significant QTL and QTL interactions were kept for fitting a full model to evaluate the additive effects and percentage of phenotypic variance (R 2) explained (Jamann et al. 2015). The support intervals for these QTL were calculated using a 1.5 LOD drop interval.
Results
Performance of mildew resistances in the RIL population
Phenotypic variations of responses to downy mildew infection in RIL population
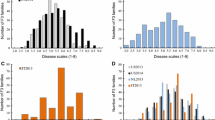
Phenotypic data on DM inoculation responses of the 169 PI 197088 × Coolgreen RILs were collected in 3 years (2013, 2014, and 2015) at the Horticultural Crops Research Station at Clinton (CL) and Castle Hayne (CH) of North Carolina State University. In general, with the progress of the disease development in the field, plants in the population became increasingly diseased over time at all five environments (CL2013, CL2014, CL2015, CH2013, and CH2014). The observed ratings followed a normal distribution that covered a large range of ratings at each environment and date (Fig. 1a; supplemental Table S1).

Violin and box plots depicting phenotypic distribution of mean DM (a) and PM (b) disease scores among PI 197088 × Coolgreen RILs at three rating times across five DM) or three (PM) environments
From the first rating to the third rating, the average correlations (Spearman’s r s) of disease ratings among environments increased from 0.50 to 0.69 (P < 0.01), implying the accrued genotypic effects in late stages of DM development (supplemental Fig. S1). Thus, the increased (narrow-sense) heritability estimates were observed from 0.83 to 0.88 across the three rating times in all environments (Table 1). Although the heritability was as high as 0.88, ANOVA indicated that the year effects on the phenotype variance were significant. The interaction effects of genotype-by-location and genotype-by-year were also significant at all ratings, although the three-way genotype-by-location-by-year interaction became less significant from the first to the third ratings (Table 1). Therefore, QTL analysis was performed with data from each environment and BLUPs to minimize the environmental effect. QTL mapping was also conducted for the three ratings to study the dynamic resistance to DM disease in PI 197088.
Phenotypic variations of PM resistance and its correlation with DM resistance
Data for responses to PM natural infection of the 148 RIL population were collected for 3 years (2014–2016) at HARS of the University of Wisconsin. Violin and box plots depicting phenotypic distribution of PM disease scores in the 3 years are shown in Fig. 1b. As shown in Fig. S1, disease scores for the PM among the 3 years were highly and significantly correlated (r s = 0.62–0.80) with non-normal distribution. The disease score of F1 plants was similar to that of the susceptible parent Coolgreen indicating the recessive nature of PM resistance in PI 197088 (Table S1). The interaction effects between genotype and year were detected, and the heritability was as high as 0.90 (Table 2).
The Spearman’s rank correlation coefficients (r s) between BLUPs of DM and PM disease resistance scores of three rating times ranged from 0.61 to 0.66 (P < 0.01) (Fig. 2) suggesting possible common genetic basis of resistances against the two pathogens in PI 197088.

Phenotypic analysis of BLUPS of DM and PM disease scores by rating times showing good correlation of DM and PM resistances in PI 197088. Bar graphs across the diagonal are phenotypic distribution of DM (three ratings) and PM (BLUPs from three ratings) disease scores. The values and plots above and below the diagonal are pairwise Spearman’s rank correlation coefficients and scatter plot between pairs of data sets, respectively. *P < 0.05; **P < 0.01; ***P < 0.001
Genotyping and linkage map construction
Bulked segregant analysis and SMA identified significant marker–DM resistance associations
Among 1536 SSR markers screened between PI 197088 and Coolgreen, 422 were polymorphic (27.5%), of which, 312 evenly distributed across the 7 chromosomes were applied to the resistant and susceptible bulks, and 19 were polymorphic between bulks. The 19 markers were distributed in five chromosomes (Chr1, 3, 4, 5, and 6). Single marker analysis revealed 16 out of the 19 markers that were significantly associated with DM resistance (Table S2) suggesting at least six QTL were underlying DM resistance in PI 197088.
Analysis of SLAF markers
SLAF-seq of 148 RIL plants generated 12.83 Gb data containing 110,421,590 pair-end reads, of which 87.7 and 93.0% bases had quality scores higher than 30 (Q30) or Q20, respectively. Among 58,886 high-quality SLAFs identified, 11,620 (19.7%) were polymorphic between the two parental lines (Table 3). The following criteria were used to filter the SLAF markers: (1) sequence depth in both parents > 10 × coverage; (2) marker missing data rate < 10% in RILs; (3) no duplicated (co-segregating) markers among 148 RIL plants (bin mapping); and (4) a stringent threshold was used for distinguishing genotypic errors and segregation distortion at 0.5% level cutoff with a Bonferroni correction [− log10(P) > 6.2]. Finally, 2780 SNP markers were kept for genetic map construction (Table 3).
Segregation distortion (SD) analysis
Each codominant SNP marker was tested for significant deviation from the expected Mendelian segregation ratio with χ 2 tests and the P value for each marker was extracted (calculated in R). Since cucumber is a diploid with seven pairs of chromosomes, at least 14 independent genomic regions are expected. A threshold of at least 0.05/14 ~ 0.004 (− log10(P) ≈ 2.40) with Bonferroni correction would be needed to obtain a genome-wide error rate of α = 0.05. Thus, a region with the SNP genotype frequencies [− log10(P)] larger than 2.40 and at least five tightly linked distorted SNP bins were considered in distorted region. The SD patterns in the RIL population were visualized by plotting the − log10(P) value of each marker along the seven chromosomes (Fig. 3; marker order was based on their physical positions in 9930 assembly V2.0). Among the 2780 SNPs, 346 (12.4%) showed SD, which were located on chromosomes 1, 2, 3, 5, and 6 with all in favor of the Coolgreen alleles (Table 3). Notably, 95% (330 out of 346) of loci in SD were located in chromosomes 1 (204), 2 (42), and 5 (84), which physically occupied 20, 5.7, and 10.6 Mbp of each chromosome in the 9930 V2.0 assembly (Fig. 3; Table 3).

Chromosomal distribution of SNPs with segregation distortion. Seven chromosomes were separated by alternate blue and orange colors. Black horizontal line is the threshold for determining segregation distortion at P < 0.05 with Bonferroni correction for genome-wide error (− log10(P) = 2.40). On the X-axis, SNPs on each chromosome are ordered according to their physical positions in 9930 V2.0. The Y-axis corresponds to the − log10(P) value. The SNPs with − log10(P) > 6.2 were trimmed off as genotyping errors
Genetic map construction and evaluation
Marker ordering with a large number of markers is challenging for linkage map construction, which is critical for accurate QTL mapping. To obtain a reliable marker order, first, the 2780 SNP obtained from SLAF-Seq were ordered by their physical positions based on the 9930 draft genome assembly V2.0. The recombination fraction (RF) of each pair of the SNP markers was then examined. As shown in the RF plot (supplemental Fig. S2A), the majority of the markers were in the right order for linkage map construction, but several from chromosomes 3, 5, 6, and 7 were misplaced.
To optimize the off-position markers, SNPs were re-order in R/ASMap and generated a new RF plot which displayed as a good order of markers with nearby markers clearly associated, and no distant markers showed any association (Fig. S2B). The genetic distances were estimated, and the linkage map was constructed according to the new marker order (Table S3).
To examine the quality and explore the utility of the genetic map, 19 SSR markers from the SMA and 36 neighboring SSR markers were chosen to genotype the 148 RILs, and the genotypic data were integrated with 2780 SNP markers to construct a new genetic map. The new RF plot also displayed a good order (Fig. S2C). Importantly, all 55 SSR markers that embedded in the nearly 3000 SNP markers seemed to be located at their correct physical positions indicating a good marker ordering of the genetic map. Moreover, some SSR markers were able to fill large gaps left on the SNP-only map thus improving the quality of genetic map (Table S3). Overall, the final genetic map contained 2835 SNP and SSR marker loci that spanned 832.4 cM with an average marker interval of 0.3 cM (Table 3).
High-density map allowed detection of mis-assemblies in the 9930 V2.0 cucumber draft genome
During linkage map construction, few off-position markers were identified from the RF plot by aligning with 9930 draft genome assembly V2.0 (Fig. S2A). Since bin map was used in this study, the bin size of these out-of-order markers was investigated. The corresponding sequences were extracted from the 9930 V2.0 assembly and aligned with Gy14 assembly V1.0 (Yang et al. 2012). The map positions of these marker-associated sequences were consistent, respectively, to their physical locations in the Gy14 V1.0 assembly, but inconsistent with those in the 9930 V2.0 assembly (supplemental Table S4). This suggested that these four bins in the 9930 draft genome assembly were mis-assembled.
A whole-genome alignment was further conducted among the high-density linkage map, the 9930 draft genome assembly V2.0, and the Gy14 assembly V1.0. As shown in supplemental Fig. S3, in addition to the four mis-assembled intra-chromosomal regions, the alignment also revealed two inter-chromosomal regions on chromosomes 5 and 7 in 9930 draft genome assembly V2.0. A group of markers showed cross over at the top of chromosomes 5 and 7. The collinearity between genetic and their physical positions, and the detailed mis-assembled regions could be found in Table S3.
QTL analysis of downy and powdery mildew resistances in PI 197088
The RIL means and BLUPs of DM and PM disease scores from each rating time of all experiments were used for QTL analysis with the MQM approach. A global view of all QTL detected with individual data sets across the seven chromosomes is provided in Fig. S4. Details of each identified QTL including peak location, LOD value, 1.5-LOD-support interval, additive effect, and percentages of total phenotypic variances explained (R 2) are provided in Table 4 (based on BLUPs across three ratings) and supplemental Table S5 (based on mean disease scores of individual rating). From Table S5, slight shifts of the peak position were observed across rating times and environments for DM and PM resistances, but the 1.5 support LOD intervals of these QTL were highly consistent. Therefore, the following analysis and discussion used the results of QTL analysis from BLUPs, which were more accurate due to its incorporation of the environmental effects.
QTL for downy mildew resistance
Eleven QTL were detected across three rating times which were located on six chromosomes with each QTL explaining 1.98–30.97% of the phenotypic variance (Table 4). A careful examination of QTL and their effects over three rating times reveal the dynamic effects of the QTL on disease development. The QTL dm5.1, dm5.2, and dm5.3 were all major-effect QTL (R 2 = 19.68–30.97%) with negative additive effects suggesting that these QTL contribute to DM resistance (reduced disease scores). The three QTL were detected at all three rating times of all environments indicating their important roles in conferring DM defense at entire plant growth stage (Fig. 4). Epistatic interaction between dm5.2 and dm5.3 was observed at first and second rating times, which explained 2.56% of the phenotypic variance (Table 4).

LOD profiles (a) and genotypic effects at peak marker locations (b) of three DM resistance QTL in PI 197088 on chromosome 5 detected with BLUPs of mean disease scores and MQM at three rating times. Horizontal dashed line in a represents LOD threshold at alpha = 0.05 after 1000 permutations. In b, red, green, and blue lines represent the first, second, and third ratings, respectively. ‘a’ and ‘b’ are alleles from PI 197088 and Coolgreen, respectively. Error bars are ± 1 SE
The QTL dm4.1 at chromosome 4 had moderate effect on conferring DM resistance at all rating times (R 2 = 13.7%). From the 1.5-LOD interval, dm4.1 was likely the same as the major-effect QTL dm4.1 detected in WI7120 (PI 330628) for DM resistance (Wang et al. 2016) (see “Discussion”). The phenotypic variance explaining by dm3.2 decreased from 13.4, 10.0, to 5.3% at three rating times (early to late), respectively, whereas dm2.1 and dm4.1 showed increased effects (larger phenotypic variance explained) in later rating. Interestingly, the alleles from PI 197088 at dm1.1, dm2.2, and dm6.2 loci all exhibited positive additive effects suggesting that they all confer susceptibility to DM infection. These three susceptible QTL explained relatively small proportion of the phenotypic variance and were detected only at the second and third rating times.
The magnitudes of additive effects of the 11 QTL that were estimated by the peak markers across five environments are plotted in Fig. S5. The effects of all alleles at location CH dropped dramatically from 2013 to 2014, but were stable at location CL with only a slight difference from 2013 to 2015. This could be reflected by population distribution of disease scores, as shown in Fig. 1: CH2013 had the highest and CH2014 had the lowest population mean, while CL2013, CL2014, and CL2015 had similar population means across five environments. The possible reason may be due to the variation of DM epidemics at CH, which was close to the ocean with strong sea breeze which may promote dispersal of DM spores through wind currents.
QTL analysis of powdery mildew resistance in PI 197088
Using BLUPs from three environments (WI2014, WI2015, and WI2016), four QTL, pm1.1, pm2.1, pm5.1, and pm6.1, were identified, which explained 1.82–32.38% of the phenotypic variance (Table 4; Fig. S6). The major-effect QTL, pm5.1, peaked at 115.0 cM that flanked by the SNP marker Marker5_24647715 (114.30 cM, 24.65 Mb of chromosome 5) and SSR15196 (115.10 cM, 25.03 Mb of chromosome 5). Within this region, a candidate gene for PM resistance, CsMLO1 (CsMLO8), was previously identified (at 24.83 Mb) (Berg et al. 2015; Nie et al. 2015a, b). Nie et al. (2015b) found that in PI 197088, CsMLO1 has a 1-bp insertion resulting in a premature stop codon and PM resistance. Very likely, pm5.1 detected in the present study was the same as CsMLO1 identified in the previous study. This result exemplified the importance of a high-quality and high-density genetic map (of course, high-quality phenotypic data) in improving the power of QTL detection.
The other three QTL, pm1.1, pm2.1, and pm6.1, had relatively small effects on contributing to PM resistance with each explaining 1.82–5.56% of the phenotypic variance. Among them, pm1.1 and pm2.1 were also identified in other PM resistant cucumber lines, such as S06 and WI2757 (Liu et al. 2008; He et al. 2013). The QTL pm6.1 is novel, but it had positive additive effects conferring PM susceptibility in PI 197088.
Co-localization of DM and PM resistance QTL
By a cursory examination at the map locations of the DM and PM QTL detected in the present study, it was clear that three DM and PM QTL pairs had overlapped 1.5-LOD intervals, which included dm2.1/pm2.1, dm5.3/pm5.1, and dm6.2/pm6.1 (Table 4; Fig. S7). However, the peak locations of dm2.1 and dm5.3 were obviously different from that of pm2.1 and pm5.1, respectively, suggesting that they may be different but linked loci conferring resistances to the two pathogens. On the other hand, dm6.2 and pm6.1 shared the same peak location, and both contributed to disease development (positive additive effects) which may imply both QTL share the same genetic base. The co-localization of DM and PM QTL also explained the observed high correlation between DM and PM phenotypes in PI 197088.
Discussion
SD regions, high-density map, and improvement of genome assembly
Segregation distortion (SD) is a widespread phenomenon in plants and animals, in which the frequencies of segregating alleles skew from the expected Mendelian ratios (Lyttle 1991). In cucumber, SD has been observed in a number of studies. For example, in the RIL populations derived from a cross between Gy14 and the wild cucumber (C. sativus var. hardwickii) line PI 183967, Ren et al. (2009) found preferential transmission of ‘wild’ alleles in a number of chromosomal regions. In the Gy14 × 9930 RIL population, the transmission favored the alleles from the male parent (Rubinstein et al. 2015). In the present study, five SD regions were identified in the PI 197088 × Coolgreen RIL population with all alleles in favor of Coolgreen (Fig. 3; Table 3).
In many studies, markers with SD were often removed during linkage map construction for the reason that SD may introduce errors in map distance estimation and marker ordering, and thus affect QTL mapping results (e.g., Lorieux et al. 1995). More recent studies indicated that SD has little or no effect on mapping accuracy and, in some cases, even improves QTL mapping power (e.g., Hackett and Broadfoot 2003; Xu 2008; Zhang et al. 2010; Bartholome et al. 2015). In the present study, 12.5% mapped SNP loci (346 of 2780) were in SD, 95% of which were distributed in chromosomes 1, 2, and 5 (Fig. 3; Table 3). Notably, 60.1, 11.2, and 16.7% markers in the three chromosomes were in SD regions (Table 3). If these markers in SD were excluded in linkage analysis, the map lengths of the two chromosomes would be much shorter, and a significant portion of Chr1 (69.1%) and Chr 5 (37.9%) will be missing from the resulting genetic map. More importantly, three QTL (dm1.1, dm2.1, and dm3.1) were inside these SD regions; their map locations would otherwise be inaccurate without these markers. This piece of information is also important from a marker-assisted breeding perspective. If the gene for a target trait is located within an SD region, the trait will also show SD (e.g., Pan et al. 2017). If the favorable allele in the SD region contributes to the desirable trait, the selection for this target trait would be easier. On the other hand, if the contributing allele is unfavorable in SD, marker-assisted selection (MAS) would be an efficient way to increase the frequency of the favorable allele. Therefore, among the three QTL, using MAS to select alleles of dm2.1 and dm3.1 that conferred DM resistance from PI 197088 may accelerate the breeding process.
The high-density genetic map developed herein helped identify mis-assemblies in the 9930 draft genome (V2.0). In 9930 V2.0, the two inter-chromosomal mis-assembled regions on chromosomes 5 and 7 have been reported in several studies (Sun et al. 2013; Wei et al. 2014; Zhou et al. 2015; Rubinstein et al. 2015), which were confirmed in the present work (Fig. S3). In addition, using this high-density genetic map, four small mix-assembled sequences ranged from 7.8 to 281 kb were also identified in 9930 V2.0, which provided additional evidence to improve the 9930 draft genome assembly in the future (Fig. S3; Table S3).
QTL identification through bulk segregant analysis and whole-genome scan
Bulk segregant analysis (BSA) and whole-genome scan are two widely used strategies for QTL mapping in plants. While BSA counts the recombination events in individuals with extreme phenotypes, genome-wide scan examines all of them. In this study, we employed both methods, which allowed us to compare their efficiency in detecting DM resistance QTL. Using whole-genome scan with saturated SNP linkage map, we identified 11 QTL with each explaining 2.0–31.0% of the phenotypic variance (Tables 4 and S5). In BSA, of the 19 marker polymorphic between the highly resistant and highly susceptible bulks, single marker analysis revealed 16 that were significantly associated with DM resistance (Table S2). The 16 markers in four chromosomes (3, 4, 5, and 6) were located in the 1.5 LOD intervals of six QTL, dm3.2, dm4.1, dm5.1, dm5.2, dm5.3, and dm6.1 that were identified by the whole-genome scan. While the association was non-significant or likely a false positive for SSR15108 or SSR14596 (Chr1) and SSR14934 (Chr6), BSA failed to detect any marker associated with dm2.1. Interestingly, the three DM resistance QTL (dm1.1, dm2.1, and dm6.2) that were detected with whole-genome scan but not BSA (Table S2) were all contributing to DM susceptibility (Table 4).
This comparison suggested that BSA is an efficient option in detecting QTL with large-to-moderate effects on the observed phenotypic variance (Zou et al. 2016). Multiple reasons can explain the lower QTL detection power or identification of false positive markers using BSA in the present study. Only eight phenotypically extreme plants were selected for each bulk based on data from a single environment (CL2013). Thus, the bulks may not necessary cover enough informative recombinants, and environmental effect may shift the order of resistant level of each individual. Plants carrying susceptibility-contributing QTL were likely excluded in the highly resistant bulk. The limited number of SSR marker polymorphic between the two bulks may reduce the chance of detecting possible marker–trait associations. To increase the polymorphic markers between bulks, high-throughput bulk-sequencing (BSA-seq or QTL-seq) would be an effective alternative. For example, Win et al. (2017) identified three additional QTL in DM resistance by BSA-seq compared to genome-wide scan with non-saturated SSR linkage map. In this study, we used SMA to test the association of polymorphic markers and phenotypes, which required genotyping of the entire population. For BSA-seq with high-density markers, other statistics methods are available to test the association between detected markers and phenotypes without the genotyping of entire population (Magwene et al. 2011; Takagi et al. 2013). In rice, chickpea and other crops, BSA-seq has been proven to delimit QTL into ~ 1 Mb region using 20–50 extreme individuals in each bulk (Takagi et al. 2013; Singh et al. 2016). Therefore, BSA-Seq would be cost-effective and efficient for QTL mapping, although in most cases, it does not and cannot replace fine mapping and cloning of QTL in bi-parental segregating population.
Co-localization of DM and PM resistances
PI 197088 has been known to possess resistances to multiple diseases including DM and PM (Staub et al. 2002). In this study, QTL mapping identified three pairs of DM and PM QTL with overlapping LOD-support intervals (dm2.1/pm2.1, dm5.3/pm5.1, and dm6.2/pm6.1) (Table 4), suggesting that they may be closely linked or actually have the same genetic basis. This can explain the significant correlation between DM and PM phenotypes. In particular, the increased correlation from 0.61 to 0.66 (P < 0.01) between PM and DM from first to third rating times could account for the increased genetic effects of dm2.1 and dm6.2 at a later stage.
Linkage of DM and PM resistance has been reported in cucumber and several other crops such as melon, tomato, and Arabidopsis (Van Vliet and Meysing 1977; Perchepied et al. 2005; Van Damme et al. 2009; Fukino et al. 2013; Huibers et al. 2013; Yoshioka et al. 2014; Gao et al. 2015). A notable example is the Arabidopsis dmr1 (downy mildew resistant 1) gene, which encodes a homoserine kinase that mediates resistance to both DM (Hyaloperonospora arabidopsidis) and PM (Oidium neolycopersici) diseases (Van Damme et al. 2009; Huibers et al. 2013). In cucumber, CsMLO1 has been shown to be the underlying the pm5.1 major-effect QTL, which was co-localized with dm5.3 but with different peak locations. Therefore, very likely, dm5.3 and pm5.1 are two different but closely linked disease resistance loci, although further investigation is needed to confirm this.
DM resistance QTL in PI 197088
Several previous studies have identified DM resistance QTL in PI 197088. Caldwell et al. (2011) and Shetty et al. (2014) detected three QTL on chromosomes 2, 4, and 5 from PI 197088, but the confidence intervals of these QTL spanned nearly two-thirds of the respective chromosomes. Shetty et al. (2014) proposed a haplotype of R-S-R for DM resistance in the distal region of the long arm of chromosome 5; the two R (DM resistance) blocks likely correspond to dm5.2 and dm5.3, respectively, from the present study. Using a single isolate of the DM pathogen and a detached-leaf assay or greenhouse artificial inoculation in the CS-PMR1 × Santou RIL population, Yoshioka et al. (2014) conducted QTL mapping of DM resistance in CS-PMR1 (derived from PI 197088) and detected 10 QTL, of which 7 (dm1.2, dm1.3, dm3.1, dm3.2, dm5.1, dm5.2, and dm5.3) were from CS-PMR1 and 3 (dm1.1, dm6.1, and dm7.2) from Santou that has moderate resistance to DM. However, among them, only four (dm3.1, dm5.1, dm5.2, and dm5.3) showed largely consistent chromosome locations with the QTL detected by the present study (based on 1.5 LOD interval of each QTL). In addition, the major-effect QTL for DM resistance identified by Yoshioka et al. (2014) was dm1.1 conferred by Santou. In contrast, dm5.1, dm5.2, and dm5.3 detected herein were all major-effect QTL contributed by PI 197088 with each contributing 19.7-31.0% to the total observed phenotypic variance (Table 4). In addition, we detected dm4.1 with moderate effect, which was not identified in the study by Yoshioka et al. (2014).
A number of reasons may contribute to the discrepancies of results among these studies. Yoshioka et al. (2014) used a single isolate of the DM pathogen to inoculate the plants in the detached-leaf assay or greenhouse screening, whereas the present study used natural inoculation with field strains. The population of the DM pathogen in the field is a mixture with many isolates varying in the degree of pathogenicity or virulence (Lebeda and Urban 2007; Quesada-Ocampo et al. 2012; Lebeda et al. 2013; reviewed by Cohen et al. 2015). The four common QTL (dm3.1, dm5.1, dm5.2, and dm5.3) detected by both studies may confer resistance to DM pathogen strains at both locations. It is not known if the different inoculation methods and different targets for inoculation (cotyledons or the first true leaf vs. whole plants) used in the two studies may affect the outcome of QTL mapping. In addition, many other factors such as the methods used in QTL detection, the marker density of genetic map for QTL analysis, or the development stages for phenotypic data collection may all affect the power of QTL detection, especially minor-effect QTL (He et al. 2013; Wang et al. 2016).
The potential of PI 197088 and other resistance sources in breeding for DM resistance
Among over 1200 cucumber lines examined, PI 197088 was the top performer for DM resistance in multi-year and multi-location screening tests (Call et al. 2012a, b). PI 197088 is being used widely in commercial breeding programs (e.g., Caldwell et al. 2011; Shetty et al. 2014). The present study offers some new perspectives on the use of PI 197088 as a resistance source in cucumber breeding with both pros and cons. First, two pairs of PM and DM resistance QTL (dm2.1/pm2.1 and dm5.3/pm5.1) were co-localized; especially, both dm5.3 (R 2 = 31%) and pm5.1 (R 2 = 32%) were major-effect QTL with large positive effects on disease resistance (Table 4), which makes it more convenient to develop cucumber lines with resistance to both pathogens. In the present study, phenotypic data of the RIL population to DM inoculation responses were scored at 41, 48, and 55 days after planting (dap). QTL analysis identified 3 QTL, dm1.1, dm2.2, and dm6.2 that had positive additive effects. That is, these QTL conferred susceptibility to DM infection in PI 197088. The two QTL, dm2.2 and dm6.2, were mapped at later rating times (48 and 55 dap) and explained increasing phenotypic variance. In field experiments, VandenLangenberg and Wehner (2016) observed that PI 197088 developed disease symptoms faster at later growth stages as compared to PI 330628 (WI7120) and PI 605996 (supplemental Fig. S8). Our finding of DM susceptibility QTL in PI 197088 in this study may explain why PI 197088 did not hold its resistance after 42 dap and showed similar resistance level as Ashley and Poinsett 76 (Fig. S8). These susceptible QTL may be responsible for the reduced resistance in PI 197088 at later plant growth stages. Thus, to enhance the utility of PI 197088, the use of molecular markers in tracking of these alleles would be necessary to minimize their negative effects on DM resistance from PI 197088.
Of the 11 DM resistance QTL identified in PI 197088, two (dm2.1 and dm3.1) were mapped in SD regions, which are all in favor of alleles from the susceptible parental line Coolgreen. Using a traditional breeding scheme, these DM resistance alleles would have a lower-than-expected frequency in the progeny, which may even get lost under stringent selection. Thus, a large breeding population would be needed to ensure the selection of PI 197088 resistance alleles. However, the use of molecular markers to track these alleles should be efficient to increase their frequency during selection.
PI 197088 and PI 330628 (WI7120) are two PI lines with the highest level of resistance against the post-2004 strains(s) in US cucumber fields (Call et al. 2012b). QTL mapping in WI7120 revealed four QTL for DM resistance including dm4.1 and dm5.1 as the major-effect (R 2 = 15–30%), as well as dm2.1 and dm6.1 as moderate or minor-effect QTL; all contributing alleles to DM resistance were from WI7120 (no susceptible QTL) (Wang et al. 2016). The result may explain the fact that WI7120 has fewer DM resistant QTL than PI 197088 but exhibited similar level of resistance at the early growing stage and can maintain a higher level of resistance than PI 197088 for weeks past flowering stage (VandenLangenberg and Wehner 2016). In addition, none of the four QTL was located in SD region. Therefore, WI7120 is probably a preferable source of downy mildew resistance in cucumber breeding.
Author contribution statement
YQW and TCW designed and supervised the experiments. YHW and YQW collected powdery mildew phenotypic data. KV and TCW developed the F6:7 RIL population and collected the field data for downy mildew phenotypes. CW performed SLAF library construction for SLAF-seq. YHW performed the research and analyzed the data. YHW and YQW wrote the manuscript with inputs from other co-authors. All authors reviewed and approved this submission.
References
Arends D, Prins P, Jansen RC, Broman KW (2010) R/qtl: high-throughput multiple QTL mapping. Bioinformatics 26:2990–2992
Arends D, Prins P, Broman KW, Jansen RC (2014) Tutorial-multiple-QTL mapping (MQM) analysis for R/qtl. http://www.rqtl.org/tutorials/MQM-tour.pdf. Accessed Nov 2017
Bartholome J, Mandrou E, Mabiala A et al (2015) High-resolution genetic maps of Eucalyptus improve Eucalyptus grandis genome assembly. New Phytol 206:1283–1296
Bates D, Mächler M, Bolker B, Walker S (2014) Fitting linear mixed-effects models using lme4. J Stat Softw. https://doi.org/10.18637/jss.v067.i01
Berg JA, Appiano M, Martínez MS et al (2015) A transposable element insertion in the susceptibility gene CsaMLO8 results in hypocotyl resistance to powdery mildew in cucumber. BMC Plant Biol 15:243
Block CC, Reitsma KR (2005) Powdery mildew resistance in the US National plant germplasm system cucumber collection. Hortscience 40:416–420
Broman KW, Wu H, Sen S, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19:889–890
Caldwell D, Chan E, de Vries J et al (2011) Methods and compositions for identifying downy mildew resistant cucumber plants. United States patent US 2011/0126309 A1
Call AD, Criswell AD, Wehner TC et al (2012a) Resistance of cucumber cultivars to a new strain of cucurbit downy mildew. Hortscience 47:171–178
Call AD, Criswell AD, Wehner TC et al (2012b) Screening cucumber for resistance to downy mildew caused by Pseudoperonospora cubensis (Berk. and Curt.) Rostov. Crop Sci 52:577–592
Camacho C, Coulouris G, Avagyan V et al (2009) BLAST plus: architecture and applications. BMC Bioinform 10:421
Cavagnaro PF, Senalik DA, Yang L et al (2010) Genome-wide characterization of simple sequence repeats in cucumber (Cucumis sativus L.). BMC Genom 11:569
Clark LR (1975) Powdery mildew resistance in plant introductions of cucumber in Iowa. Plant Dis Report 59:1024–1028
Cockerham CC (1983) Covariances of relatives from self-fertilization. Crop Sci 23:1177–1180
Cohen Y, VandenLangenberg KM, Wehner TC et al (2015) Resurgence of Pseudoperonospora cubensis: the causal agent of cucurbit downy mildew. Phytopathology 105:998–1012
El Jack A, Munger H (1983) Two sources conferring partial dominant resistance to powdery mildew (Sphaerotheca fuliginea Poll.) in cucumber. Cucurbit Genet Coop Rep 6:7–8
Epps W, Barnes W (1952) The increased susceptibility of the Palmetto cucumber to downy mildew in South Carolina. Plant Dis Report 36:14–15
Fanourakis NE, Simon PW (1987) Analysis of genetic linkage in the cucumber. J Hered 78:238–242
Fukino N, Yoshioka Y, Sugiyama M et al (2013) Identification and validation of powdery mildew (Podosphaera xanthii)-resistant loci in recombinant inbred lines of cucumber (Cucumis sativus L.). Mol Breed 32:267–277
Gao D, Appiano M, Huibers RP et al (2015) Natural loss-of-function mutation of EDR1 conferring resistance to tomato powdery mildew in Arabidopsis thaliana accession C24. Mol Plant Pathol 16:71–82
Hackett C, Broadfoot L (2003) Effects of genotyping errors, missing values and segregation distortion in molecular marker data on the construction of linkage maps. Heredity 90:33–38
He X, Li Y, Pandey S et al (2013) QTL mapping of powdery mildew resistance in WI 2757 cucumber (Cucumis sativus L.). Theor Appl Genet 126:2149–2161
Huang S, Li R, Zhang Z et al (2009) The genome of the cucumber, Cucumis sativus L. Nat Genet 41:1275–1281
Huibers RP, Loonen AEHM, Gao D et al (2013) Powdery mildew resistance in tomato by impairment of SlPMR4 and SlDMR1. PLoS One 8:e67467
Jamann T, Balint-Kurti P, Holland JB (2015) QTL mapping using high-throughput sequencing. Springer Science + Business Media, New York
Jenkins SF, Wehner TC (1983) A system for the measurement of foliar diseases of cucumber. Cucurbit Genet Coop Rep 6:10–12
Kooistra E (1968) Powdery mildew resistance in cucumber. Euphytica 17:236–244
Lander ES, Green P, Abrahamson J et al (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Lebeda A, Urban J (2007) Temporal changes in pathogenicity and fungicide resistance in Pseudoperonospora cubensis populations. Acta Hortic 731:327–336
Lebeda A, Pavelková J, Sedláková B, Urban J (2013) Structure and temporal shifts in virulence of Pseudoperonospora cubensis populations in the Czech Republic. Plant Pathol 62:336–345
Liu L, Yuan X, Cai R et al (2008) Quantitative trait loci for resistance to powdery mildew in cucumber under seedling spray inoculation and leaf disc infection. J Phytopathol 156:691–697
Lorieux M, Goffinet B, Perrier X et al (1995) Maximum-likelihood models for mapping genetic markers showing segregation distortion. 1. Backcross populations. Theor Appl Genet 90:73–80
Luo R, Liu B, Xie Y et al (2012) SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1:18
Lyttle T (1991) Segregation distorters. Annu Rev Genet 25:511–557
Magwene PM, Willis JH, Kelly JK (2011) The statistics of bulk segregant analysis using next generation sequencing. PLoS Comput Biol 7:e1002255
Morishita M, Sugiyama K, Saito T, Sakata Y (2003) Review: powdery mildew resistance in cucumber. Jpn Agric Res Q 37:7–14
Nie J, He H, Peng J et al (2015a) Identification and fine mapping of pm5.1: a recessive gene for powdery mildew resistance in cucumber (Cucumis sativus L.). Mol Breed 35:7
Nie J, Wang Y, He H et al (2015b) Loss-of-Function mutations in CsMLO1 confer durable powdery mildew resistance in cucumber (Cucumis sativus L.). Front Plant Sci 6:1–14
Oerke E, Steiner U, Dehne H, Lindenthal M (2006) Thermal imaging of cucumber leaves affected by downy mildew and environmental conditions. J Exp Bot 57:2121–2132
Pan Y, Liang X, Gao M et al (2017) Round fruit shape in WI7239 cucumber is controlled by two interacting quantitative trait loci with one putatively encoding a tomato SUN homolog. Theor Appl Genet 130:573–586
Perchepied L, Bardin M, Dogimont C, Pitrat M (2005) Relationship between loci conferring downy mildew and powdery mildew resistance in melon assessed by quantitative trait loci mapping. Phytopathology 95:556–565
Pérez-García A, Romero D, FernÁndez-OrtuÑo D et al (2009) The powdery mildew fungus Podosphaera fusca (synonym Podosphaera xanthii), a constant threat to cucurbits. Mol Plant Pathol 10:153–160
Quesada-Ocampo LM, Granke L, Olsen J et al (2012) The genetic structure of Pseudoperonospora cubensis populations. Plant Dis 96:1459–1470
Ren Y, Zhang Z, Liu J et al (2009) An integrated genetic and cytogenetic map of the cucumber genome. PLoS One 4:e5795
Roque A, Adsuar J (1939) New cucumber varieties resistant to the downy mildew. In: Annual Report Agri Expt Station of Puerto Rico fiscal year 1937–1938, pp 45–46
Rubinstein M, Katzenellenbogen M, Eshed R et al (2015) Ultrahigh-density linkage map for cultivated cucumber (Cucumis sativus L.) using a single-nucleotide polymorphism genotyping array. PLoS One 10:e0124101
Sakata Y, Kubo N, Morishita M et al (2006) QTL analysis of powdery mildew resistance in cucumber (Cucumis sativus L.). Theor Appl Genet 112:243–250
Shetty N, van Kooten H, Sipeyre B et al (2014) Downy Mildew Resistant Cucumber Plants. United States patent US 8859859 B2
Singh VK, Khan AW, Jaganathan D et al (2016) QTL-seq for rapid identification of candidate genes for 100-seed weight and root/total plant dry weight ratio under rainfed conditions in chickpea. Plant Biotech J 14:2110–2119
Sitterly WR (1972) Breeding for disease resistance in cucurbits. Annu Rev Phytopathol 10:471–490
Smith P (1948) Powdery mildew resistance in cucumber. Phytopathology 38:1027–1028
Staub JE, Dane F, Reitsma K et al (2002) The formation of test arrays and a core collection in cucumber using phenotypic and molecular marker data. J Am Soc Hortic Sci 127:558–567
Sun X, Liu D, Zhang X et al (2013) SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS One 8:e58700
Szczechura W, Staniaszek M, Klosinska U, Kozik E (2015) Molecular analysis of new sources of resistance to Pseudoperonospora cubensis (Berk. et Curt.) Rostovzev in cucumber. Russ J Genet 51:974–979
Takagi H, Abe A, Yoshida K et al (2013) QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J 74:174–183
Taylor J, Butler D (2015) ASMap: linkage map construction using the MSTmap algorithm. R package version 0.4-5. http://CRAN.R-project.org/package=ASMap. Accessed Nov 2017
Van Damme M, Zeilmaker T, Elberse J et al (2009) Downy mildew resistance in Arabidopsis by mutation of HOMOSERINE KINASE. Plant Cell 21:2179–2189
Van Vliet GJA, Meysing WD (1974) Inheritance of resistance to Pseudoperonospora cubensis Rost. in cucumber (Cucumis sativus L.). Euphytica 23:251–255
Van Vliet GJA, Meysing WD (1977) Relation in the inheritance of resistance to Pseudoperonospora cubensis Rost and Sphaerotheca fuliginea Poll. in cucumber (Cucumis sativus L.). Euphytica 26:793–796
VandenLangenberg KM (2015) Studies on downy mildew resistance in cucumber (Cucumis sativus L.). Dissertation, North Carolina State University
VandenLangenberg KM, Wehner TC (2016) Downy mildew disease progress in resistant and susceptible cucumbers tested in the field at different growth stages. Hortscience 51:984–988
Wang Y, VandenLangenberg KM, Wehner TC et al (2016) QTL mapping for downy mildew resistance in cucumber inbred line WI7120 (PI 330628). Theor Appl Genet 129:1493–1505
Wei QZ, Wang YZ, Qin XD et al (2014) An SNP-based saturated genetic map and QTL analysis of fruit-related traits in cucumber using specific-length amplified fragment (SLAF) sequencing. BMC Genom 15:10
Win KT, Vegas J, Zhang C et al (2017) QTL mapping for downy mildew resistance in cucumber via bulked segregant analysis using next-generation sequencing and conventional methods. Theor Appl Genet 130:199–211
Wu Y, Bhat PR, Close TJ, Lonardi S (2008) Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph. PLoS Genet 4:e1000212
Xu S (2008) Quantitative trait locus mapping can benefit from segregation distortion. Genetics 180:2201–2208
Xu X, Xu R, Zhu B et al (2015) A high-density genetic map of cucumber derived from specific length amplified fragment sequencing (SLAF-seq). Front Plant Sci 5:1–8
Yang L, Koo D-H, Li Y et al (2012) Chromosome rearrangements during domestication of cucumber as revealed by high-density genetic mapping and draft genome assembly. Plant J 71:895–906
Yoshioka Y, Sakata Y, Sugiyama M, Fukino N (2014) Identification of quantitative trait loci for downy mildew resistance in cucumber (Cucumis sativus L.). Euphytica 198:265–276
Zhang L, Wang S, Li H et al (2010) Effects of missing marker and segregation distortion on QTL mapping in F2 populations. Theor Appl Genet 121:1071–1082
Zhou Q, Miao H, Li S et al (2015) A sequencing-based linkage map of cucumber. Mol Plant 8:961–963
Zhu W, Huang L, Chen L et al (2016) A high-density genetic linkage map for cucumber (Cucumis sativus L.): based on specific length amplified fragment (SLAF) sequencing and QTL analysis of fruit traits in cucumber. Front Plant Sci 7:11
Zijlstra S, Groot SPC (1992) Search for novel genes for resistance to powdery mildew (Sphaerotheca fuliginea) in cucumber (Cucumis sativus). Euphytica 64:31–37
Zou C, Wang P, Xu Y (2016) Bulked sample analysis in genetics, genomics and crop improvement. Plant Biotechnol J 14:1941–1955
Acknowledgements
The authors thank Kristin Haider for technical help. This research is supported by a U.S. Department of Agriculture (USDA)-Specialty Crop Research Initiative grant (SCRI, project# 2011-51181-30661) and the National Institute of Food and Agriculture, U.S. Department of Agriculture, under award number 2015-51181-24285. Names are necessary to report factually on available data; however, the USDA neither guarantees nor warrants the standard of the product, and the use of the name by USDA implies no approval of the product to the exclusion of others that may also be suitable. USDA is an equal opportunity provider and employer.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Communicated by Sanwen Huang.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, Y., VandenLangenberg, K., Wen, C. et al. QTL mapping of downy and powdery mildew resistances in PI 197088 cucumber with genotyping-by-sequencing in RIL population. Theor Appl Genet 131, 597–611 (2018). https://doi.org/10.1007/s00122-017-3022-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-017-3022-1