
Abstract
Key message
Thirteen potentially new leaf rust resistance loci were identified in a Vavilov wheat diversity panel. We demonstrated the potential of allele stacking to strengthen resistance against this important pathogen.
Abstract
Leaf rust (LR) caused by Puccinia triticina is an important disease of wheat (Triticum aestivum L.), and the deployment of genetically resistant cultivars is the most viable strategy to minimise yield losses. In this study, we evaluated a diversity panel of 295 bread wheat accessions from the N. I. Vavilov Institute of Plant Genetic Resources (St Petersburg, Russia) for LR resistance and performed genome-wide association studies (GWAS) using 10,748 polymorphic DArT-seq markers. The diversity panel was evaluated at seedling and adult plant growth stages using three P. triticina pathotypes prevalent in Australia. GWAS was applied to 11 phenotypic data sets which identified a total of 52 significant marker–trait associations representing 31 quantitative trait loci (QTL). Among them, 29 QTL were associated with adult plant resistance (APR). Of the 31 QTL, 13 were considered potentially new loci, whereas 4 co-located with previously catalogued Lr genes and 14 aligned to regions reported in other GWAS and genomic prediction studies. One seedling LR resistance QTL located on chromosome 3A showed pronounced levels of linkage disequilibrium among markers (r 2 = 0.7), suggested a high allelic fixation. Subsequent haplotype analysis for this region found seven haplotype variants, of which two were strongly associated with LR resistance at seedling stage. Similarly, analysis of an APR QTL on chromosome 7B revealed 22 variants, of which 4 were associated with resistance at the adult plant stage. Furthermore, most of the tested lines in the diversity panel carried 10 or more combined resistance-associated marker alleles, highlighting the potential of allele stacking for long-lasting resistance.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Wheat (Triticum aestivum L.) is a major source of calories and protein in the human diet (Shewry and Hey 2015). However, the current global production is insufficient to meet the demand of a rapidly growing world population (Grassini et al. 2013). At the same time, wheat yields are consistently threatened by increasing climatic variations (Asseng et al. 2015) and rapidly evolving pests and pathogens (Chaves et al. 2013). Leaf rust (LR) caused by Puccinia triticina Eriks., is one of the most common and geographically widespread wheat diseases worldwide. LR causes more annual yield losses globally compared to losses attributed to stem and stripe rust (Bolton et al. 2008; Huerta-Espino et al. 2011). Among various disease management strategies, the cultivation of resistant wheat cultivars is the most effective and environment-friendly strategy (Kolmer et al. 2013).
Genetic resistance against LR is broadly categorised into seedling or all-stage resistance and adult plant resistance (APR). To date, 77 genes confering resistance to LR (Lr) have been successfully characterised, of which the majority confer seedling resistance (McIntosh et al. 2017). Typically, seedling resistance is controlled by a single gene with major effect that interacts with the pathogen in a ‘gene-for-gene’ relationship (Flor 1971). Usually, the seedling genes are pathogen race-specific and confer a hypersensitive response (HR)—a cell death phenomenon preventing the pathogen spread (Ellis et al. 2014; Mondal et al. 2016). This exerts intense selective pressure on the pathogen population, thus quickly rendering the deployed resistance gene ineffective (Burdon et al. 2014; Li et al. 2014; Niks et al. 2015). In contrast, APR is usually effective at the post-seedling growth stages, is either controlled by multiple genes each with minor effect or a single gene with major effect. Some APR genes provide partial resistance that is effective against all races of a given pathogen species (i.e. race-nonspecific) (Lagudah 2011; McCallum et al. 2012; Burdon et al. 2014). Mostly APR genes interact additively and enhance resistance to a level of immunity (Singh et al. 2014). Some APR genes confer pleiotropic resistance against multiple diseases, for instance Lr34, Lr46, and Lr67 provide partial resistance to LR, stripe rust, stem rust, and powdery mildew diseases of wheat (Lagudah 2011; Risk et al. 2012; Ellis et al. 2014).
To date, six Lr genes (including seedling and APR) have been cloned: Lr1 (Cloutier et al. 2007), Lr10 (Feuillet et al. 2003), Lr21 (Huang et al. 2003), Lr22a (Thind et al. 2017), Lr34 (Krattinger et al. 2009), and Lr67 (Moore et al. 2015). This has enabled the development of gene-specific molecular markers for rapid gene identification via marker-assisted selection (MAS). Markers further assist in pyramiding of 4–5 APR or seedling resistance genes or in combinations to generate durable rust resistant wheat cultivars (Ellis et al. 2014; Singh et al. 2014). To maintain and/or broaden the genetic diversity of durable rust resistance, the identification of novel genetic sources of resistance is required. One approach for the genetic enrichment of elite breeding pools is to exploit landraces by introducing genetic diversity from germplasm collections (Lopes et al. 2015; Sehgal et al. 2015; Kumar et al. 2016). More than 850,000 wheat accessions are stored in gene banks, representing a rich genetic resource to reinstate the variation of genetic bottlenecks (e.g. from domestication or selective breeding). Many of these accessions are already adapted to very specific target environments, possessing exclusive advantageous characteristics, such as resistances towards specific biotic and abiotic stresses (Mitrofanova 2012; Huang and Han 2014; Lopes et al. 2015), including resistance to rust diseases (Cavanagh et al. 2013; Lopes et al. 2015; Rinaldo et al. 2016; Vikram et al. 2016).
For instance, the Lr genes Lr52 and Lr67 (Hiebert et al. 2010; Bansal et al. 2013), and the stripe rust gene Yr47 (Bansal et al. 2011) were identified in wheat landraces from the Watkins collection. Another historical yet relatively unexploited diverse wheat collection is the “N. I. Vavilov Institute of Plant Genetic Resources” (VIR) in St Petersburg, Russia, collected by the Russian botanist and geneticist N. I. Vavilov and his colleagues in the early 1900s. Recent studies have reported a large variety of novel alleles in the VIR wheat collection, revealing the promising basis for the genetic improvement of resistances to various biotic and abiotic stresses (Mitrofanova 2012; Sadovaya et al. 2015; Riaz et al. 2016a; Riaz et al. 2017). However, determining the genomic regions underpinning these resistances is challenging.
Traditionally, quantitative trait loci (QTL) mapping is used to identify underlying genetic variations that co-segregate with a trait of interest using a bi-parental mapping population (Koornneef et al. 2004; Zhu et al. 2008). Although traditional QTL mapping is successful, it is fundamentally limited to the comparative low allelic diversity of the two crossing parents and low recombination events which impair the mapping resolution (Zhu et al. 2008). Alternatively, genome-wide association studies (GWAS) represent a powerful tool to dissect the genetic architecture of complex traits in natural populations, such as germplasm collections (Zhu et al. 2008; Hall et al. 2010), by detecting genomic regions that are in linkage disequilibrium (LD) with genes affecting the trait of interest. Due to a greater number of historical chromosomal recombinations accumulated over a large number of generations in natural populations, GWAS can position QTL at a much higher resolution (Yu and Buckler 2006; Semagn et al. 2010).
Here, we present a large-scale association study for seedling and APR to LR under controlled and field conditions in a highly diverse panel of 295 bread wheat lines from the VIR. Using high-density Diversity Arrays Technology (DArT-seq) markers and multi-year phenotypic data sets, we were able to map previously undescribed QTL for resistance against three major P. triticina pathotypes that are prevalent in Australia. We anticipate that this study provides breeders with a rich basis for the improvement of durable LR resistances in future wheat cultivars. Ongoing work based on these findings will help to functionally validate the significance of candidate genes in the identified novel genomic regions.
Materials and methods
Plant materials and genotyping
A diversity panel of 295 homozygous single seed descent (SSD) bread wheat lines from VIR, representing species-wide genetic diversity (Riaz et al. 2017), was selected for the assessment of LR response. DNA of each wheat line was extracted following the protocol recommended by Diversity Arrays Technology (DArT) (http://www.diversityarray.com) and the whole panel was genotyped with the DArT genotyping-by-sequencing (GBS) platform using the DArT-seq wheat PstI complexity reduction method, as described by Li et al. (2015), which returned a total of 56,306 raw DArT-seq markers. The DArT-seq markers are presence–absence dominant markers extracted in silico from sequences obtained from genomic representations. The raw marker data was filtered to retain only markers with ≤ 10% missing values, a minor allele frequency ≥ 3% and lines with ≤ 20% missing values, resulting in a selection of 10,748 high-quality, polymorphic DArT-seq markers for the subsequent genetic analyses. All used DArT-seq markers were ordered according to their genetic positions in a high-resolution DArT-seq consensus map (version 4.0), provided by Dr. Andrzej Kilian (Diversity Arrays Technology Pty Ltd, Canberra, Australia).
As described by Riaz et al. (2016a), the diversity panel was also screened for the polymerase chain reaction (PCR)-based markers cssfr5 (Lagudah et al. 2009), csLV46 (Lagudah, unpublished data) and SNP1-TM4 (Moore et al. 2015), which facilitated identification of the known LR APR genes Lr34, Lr46, and Lr67, respectively.
Evaluation of leaf rust resistance
For the resistance screening, we used the three P. triticina pathotypes (pt), namely pt 104-1,2,3,(6),(7),11,13, pt 76-1,3,5,7,9,10,12,13 + Lr37 and pt 104-1,3,4,6,7,8,10,12 + Lr37 (Table 1), which are prevalent in the eastern and western wheat growing regions of Australia (Park 2016). A summary of the experiments performed in this study at the seedling and adult plant stage for scoring LR response across years and pathotypes is presented in Table 2.
Integrated seedling and adult plant phenotyping
The 295 SSD lines in the diversity panel were evaluated using the integrated seedling and adult plant phenotyping method under controlled conditions, as described by Riaz et al. (2016a). For seedling infection, the P. triticina pathotype pt 104-1,2,3,(6),(7),11,13 was used (Table 2). Briefly, the diversity panel was sown in a standard glasshouse with diurnal temperatures (i.e. 22/17 °C day/night) and 12 h photoperiod. At the two-leaf stage, plants were inoculated using a P. triticina urediniospore suspension (Riaz et al. 2016a, b). Twelve days post-inoculation, seedlings were scored using the 0–4 Stakman scale (Stakman et al. 1962). Afterwards, plants were transferred to a temperature-controlled growth facility where the plants were subjected to “speed breeding” or “accelerated growth conditions” (AGC) by adopting a 12-h cycling temperature (22/17 °C) and 24 h photoperiod, which helps the plants to attain the adult plant stage rapidly (Riaz et al. 2016b; Watson et al. 2017). After 2 weeks, plants were re-inoculated using the same P. triticina pathotype 104–1,2,3,(6),(7),11,13 (Table 2). Twelve days post-inoculation, LR response was recorded for the flag-2 leaf using the 0–4 Stakman scale (Stakman et al. 1962). This provides representative measures of the adult plant response to LR, which are similar to field-based measures (Riaz et al. 2016b). The LR response in this experiment was converted from the 0–4 to 0–9 scale to standardise data sets across all experiments (Ziems et al. 2014; Riaz et al. 2016b) and for subsequent GWAS analyses. Lines that depicted a LR response < 7 on the 0–9 scale were considered resistant.
Field trials
The SSD lines in the diversity panel were subjected to LR screening in the field over a 3-year period (2014, 2015, and 2016) at the Redlands Research Facility (27°31′40.8″S 153°15′05.7″E), Queensland, Australia, as detailed by Riaz et al. (2016b). Six seeds of each SSD line were sown as un-replicated hill plots, whereas four standards with known disease responses (i.e. Thatcher, Avocet, Avocet + Lr34, and Avocet + Lr46) were replicated five times throughout the test material to detect spatial variation in the nursery. About 5 weeks after sowing, the LR epidemic was initiated by transplanting rust-infected wheat (Morocco) seedlings into the field among the spreader rows. When the disease was sufficiently established on susceptible standards [i.e. Thatcher was scored 20 moderately susceptible to susceptible (MSS) in the field in 2014, 9 in the field in 2015, and 8 in the field in 2016], all SSD lines were assessed for LR response.
In 2014, the diversity panel was assessed for disease response in the LR nursery inoculated with P. triticina pathotype 104–1,2,3,(6),(7),11,13 (Table 2). The disease response for each line was assessed on a whole plot basis using the modified Cobb scale (Peterson et al. 1948). The disease severity data and IT were used to calculate the coefficient of infection (CI), as reported by Loegering (1959). Disease scoring was conducted at 70, 77, 86, and 96 days after sowing (DAS). Therefore, these multiple phenotypic data sets represent different time-points during the epidemic development in the nursery. The CI values of each disease score were then divided by 10 to convert to 0–9 scale. The converted scores were used to visualise the density distribution of LR response across phenotypic data sets.
In 2015, the LR nursery was inoculated with a mixture of two P. triticina pathotypes, namely pt 104-1,2,3,(6),(7),11,13 and pt 76-1,3,5,7,9,10,12,13 + Lr37 (Table 2). Plants were assessed on a whole plot basis for disease response three times during the season (i.e. 78, 85, and 101 DAS) using the 1–9 scale where 1 = very resistant and 9 = very susceptible, as reported by Bariana et al. (2007). In 2016, the LR nursery was inoculated with a mixture of three P. triticina pathotypes, namely pt 104-1,2,3,(6),(7),11,13, pt 76-1,3,5,7,9,10,12,13 + Lr37 and pt 104-1,3,4,6,7,8,10,12 + Lr37 (Table 2). Therefore, the 2016 nursery comprised the most virulent composition of P. triticina pathotypes compared to nurseries conducted in 2014 and 2015. Plants were evaluated for disease response twice (i.e. 71 and 84 DAS) using the 1–9 scale, as detailed above.
In all the field trials, a threshold for ‘resistance’ to LR was determined as any line depicting a disease response ≤ 5 based on the 1–9 scale, where resistance was deemed as “moderately resistant to moderately susceptible” (MRMS) or better. Each disease reading within a field environment was regarded as a unique phenotypic data set and subsequently used for GWAS. The field phenotypic data sets were referred as Field_2014_1, Field_2014_2, Field_2014_3, Field_2014_4, Field_2015_1, Field_2015_2, Field_2015_3, Field_2016_1, and Field_2016_2.
Population structure, genetic diversity, and linkage disequilibrium
The population structure and genetic diversity for the diversity panel were previously described by Riaz et al. (2017). Briefly, population structure was estimated using the partitioning around medoids clustering algorithm and ‘Jaccard distance’ in R (Team 2014). The optimal number of clusters (i.e. k = 2) was determined using the ‘fpc’ package (Hennig 2014). Pairwise LD between markers were measured as r 2 (Hao et al. 2007). LD decay, the relationship between LD and genetic map distance between marker pairs in cM, was estimated as a locally estimated scatterplot smoothing (LOESS) curve and the LD cut-off threshold was set at r 2 = 0.1. The LD decay for the A, B, and D genomes was estimated for the whole population and the previously described clusters.
Genome-wide association analysis, allele stacking, and haplotype construction
Genome-wide marker-trait associations were calculated for data from a total of 11 phenotypic data sets (seedling, AGC and the field trials), using the R package GenABEL (Aulchenko et al. 2007). The applied mixed linear model was adjusted for population stratification by including identity-by-state estimates (kinship matrix) for genotype pairs and a principal component adjustment that uses the first four principal components as covariates. The significant cut-off value was arbitrarily set at − log10(P) = 3.5. Overlapping significant markers for different environments that were located at the same chromosomal position within a 5 cM window were considered the same QTL. Based on the predicted direction of the allele effect on the resistance score (e.g. negative effect \(\overset{\wedge}{=}\) resistance allele) (Table 3), we assigned resistance alleles for each significant marker.
To investigate an effect of accumulated alleles for LR resistance at the independent loci on the disease score, we assigned the lines to groups, based on the absolute number of resistance-associated alleles possessed and compared their relative disease indices that were calculated as
where the disease index LRi is the accumulated relative value of a line’s disease score in experiment k in relation to the population mean in this experiment k over all n field experiments. Lines with high indices (above 0) are relatively more susceptible to LR infection than lines with indices below 0.
Two QTL were selected for haplotype analysis and subsequent network analysis: (1) a seedling QTL on chromosome 3A (qNV.Lr-3A.3) because it was deemed a new QTL with large effect, and (2) a QTL conferring APR (qNV.Lr-7B.2) on the long arm of chromosome 7B which was detected across many phenotypic data sets and reported by numerous previous studies. Haplotypes for seedling resistance and APR were constructed on the basis of LD around the respective identified QTL on chromosomes 3A (qNV.Lr-3A.3) and 7B (qNV.Lr-7B.2). All surrounding markers with pairwise r 2 values > 0.8 were included in the haplotype analysis, resulting in 7 and 22 haplotype variants, respectively. Haplotype networks, showing TCS genealogies between haplotype variants (Clement et al. 2000), were calculated using PopART (http://popart.otago.ac.nz.) (Leigh and Bryant 2015). The network nodes were coloured according to the average disease rating in the respective haplotype groups. A Tukey’s test was performed to test for significant phenotypic differences between the haplotype groups. The origin information for lines within each haplo-group was used to visualise the geographic distribution of these haplotypes in the diversity panel.
Alignment of QTL identified in this study with previously reported Lr genes and QTL
For comparison, QTL identified in this study and already catalogued Lr genes (McIntosh et al. 2017) were projected onto the common integrated map developed by Maccaferri et al. (2015) using MapChart software version 2.3 (Voorrips 2002). A QTL was considered potentially new if the genetic distance was ≥ 5 cM of the reported Lr gene or QTL. Eight recent GWAS studies (Kertho et al. 2015; Jordan et al. 2015; Gao et al. 2016; Li et al. 2016; Aoun et al. 2016; Pasam et al. 2017; Turner et al. 2017; Kankwatsa et al. 2017) and two genomic prediction studies (Daetwyler et al. 2014; Juliana et al. 2017) using high-throughput marker platforms were considered for QTL comparison.
In silico annotation of significant markers
The genomic regions identified in this study were subjected to homology search for syntenic regions in Brachypodium distachyon and rice (Oryza sativa L.) genome. The marker sequences were annotated against the protein sequences to determine putative molecular functions, which could lead to the possible identification of candidates for disease resistance across species. The homology search was performed using EnsemblPlants; http://plants.ensembl.org/index.html (Kersey et al. 2016).
Results
Disease response
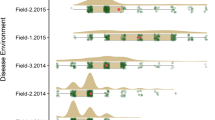
Of the 288 lines in the diversity panel tested for LR response, 76.4% lines were seedling susceptible, and 23.6% lines were resistant (Fig. 1a, b). At the adult stage under AGC, 46.5% lines were resistant, and 53.5% showed a susceptible response (Fig. 1a; Supplementary material 1). In the 2014 field trial, 63.0, 75.4, and 63.0% of the tested 284 lines were resistant at the first three disease assessments (70, 77, and 86 DAS). However, at the fourth disease assessment (96 DAS) when the lines were at the flag leaf stage, and inoculum pressure in the nursery was highest, 71.2% lines displayed susceptibility, while only 28.8% lines displayed resistance (Fig. 1a). In 2015, 29.8% of the evaluated 288 lines showed resistance and 70.2% demonstrated susceptibility at the first disease assessment (78 DAS), while only 9% of the lines showed a resistant disease response at the third reading (101 DAS) (Fig. 1a). In 2016, of the 261 tested lines, 56.7 and 27.6% were resistant for the disease assessments performed at 71 and 85 DAS, respectively. The full description of disease responses observed for all lines in the diversity panel is provided in Supplementary material 1.

a Violin plots illustrating the density distribution of leaf rust response for lines in the diversity panel based on 11 phenotypic data sets. The disease data for environments AGC and field (2014, 2015 and 2016) were converted to the 0–9 scale (9 = very susceptible) to allow comparison across all data sets. The red line displays the median, the top and bottom of the thick vertical bars represent first and third quartiles, respectively, and the green fill shows disease density estimates (n = 248). b A sample of the seedling leaf rust responses observed for the diversity panel
Marker properties, population structure, and linkage disequilibrium
After filtering, a total of 10,748 polymorphic mapped markers along with three PCR-based markers for known APR genes (Lr34, Lr46, and Lr67) were used for LD analysis and GWAS. Lower marker density and marker coverage were evident for the D genome compared to A and B genomes. Analysis of population structure in the diversity panel was previously described by Riaz et al. (2017), where distinct clustering was observed on the basis of cultivation status and geographical origin. The diversity panel was divided into two clusters (k = 2), containing 171 and 124 lines, respectively. Analysis of LD decay revealed strong differences between the three subgenomes. Overall, LD between marker pairs decayed quickly in the A and B genomes, especially in the latter, where the r 2 LOESS curve never exceeded the threshold line. In contrast, LD in the D genome was very pronounced, and LOESS curves did not drop below the threshold line until 19 cM for cluster 1 and 21 cM for cluster 2 (Fig. 2).

Linkage disequilibrium (LD) decay as a function of genetic distance (cM) in A, B and D genomes for the diversity panel. LD was estimated for the whole population (black dotted line), and cluster 1 (red line) and cluster 2 (blue dotted line) as defined by Riaz et al. (2017). The LD decay was the point where the locally estimated scatterplot smoothing (LOESS) curves intersect the LD, whereas the threshold for LD decay was at r 2 = 0.1 (black line)
Marker–trait associations
A total of 52 significant markers (p < 0.001) were associated with LR resistance (Table 3; Supplementary material 2). Six markers were detected at the seedling stage and 46 markers at the adult stage (Table 3; Supplementary material 2). Most of the significant markers (n = 32) were detected in 2015 field environments. Manhattan plots depicting association between significant markers and LR response in different environments were displayed in Supplementary material 3. By considering chromosome position and LD between adjacent markers, a total of 31 QTL regions were assigned. These QTL were located on chromosomes 1A, 1B, 2A, 2B, 3A, 3B, 4A, 5A, 5B, 6A, 6B, 7A, 7B, and 7D (Table 3). Of the 31 QTL, 29 were associated with resistance at the adult stage and one QTL each was found to be associated with seedling (i.e. detected only at the seedling stage) and all-stage resistance (i.e. detected both at the seedling and adult stage) (Table 3). The QTL qNV.Lr-2B.3 (all-stage resistance) and qNV.Lr-7B.2 (adult plant stage) were detected in many of the environments. The gene-specific marker cssfr5 for known APR gene Lr34 on chromosome 7D (Lagudah et al. 2009) was the only loci among the three PCR markers used that could be detected in GWAS with –log10 (p value) between 3.9 and 6.19 for the different field trials, and was designated QTL qNV.Lr-7D (Table 3).
Of the 31 QTL, 13 were identified as being new LR resistance loci (Table 3). Among the other 18 QTL, 4 were co-located with the catalogued Lr genes, namely Lr3 on chromosome 6B, Lr64 on 6A, Lr14 (a and b alleles), Lr68, LrBi16 and LrFun on 7B, and Lr34 on 7D (McIntosh et al. 2017; Table 3 and Supplementary material 3). The remaining 14 QTL identified in our study were in alignment with the candidate regions reported in other GWAS studies (Table 3, Supplementary material 4). An in silico annotation of the identified significant markers showed that most sequences were uncharacterised regarding their molecular function (Table 3). However, 12 markers corresponded to the putative proteins carrying domains involved in disease resistance mechanisms, such as leucine rich repeat (LRR), NB-ARC, P-loop_NTPase, zinc finger, CCHC-type, RNA-dependent DNA polymerase, protein kinase-like domain, cyclin-like F-box, galectin, carbohydrate recognition domain, glycosyl transferase family 29, glycosyl transferase family 31, Ran GTPase, small GTP-binding protein, ABC transporter and domain of unknown function-DUF1618 (Table 3).
Haplotype analysis and allele stacking
A new QTL (qNV.Lr-3A.3) on the long arm of chromosome 3A (116.7–117.0 cM) represented by two highly significant markers for seedling LR resistance [− log10(p value) = 6.26/4.1] which were in high LD (r 2 = 0.7), was selected for subsequent haplotype analysis (Table 3, Fig. 3a). This large effect QTL was considered a new genomic region conferring seedling resistance because it did not align with any previously reported Lr genes or QTL (Fig. 3b). Screening of allelic variation in our diversity panel resulted in seven different haplotype variants (qNV.Lr-3A.3—hap1–hap7), where hap1 was the most frequent variant in our diversity panel (frequency = 92.5%) (Fig. 3b). Hap2 was present in 4.7% of the lines while all other variants only occurred in 1% of the lines each. Inter-group comparisons of the disease responses for the first three haplotype groups showed that hap1 was associated with a significantly higher susceptibility to LR (8 on a 0–9 scale) than hap2 and hap3, where the median disease response ranged between 3.6 and 5.5, respectively (Fig. 3c). The lines carrying hap1 are geographically widespread and originate from 28 countries, including Russia (n = 48), India (n = 37), and Pakistan (n = 30). The lines carrying hap2 were from Armenia (n = 3), Azerbaijan (n = 3), Russia (n = 2), Pakistan (n = 1), Ethiopia (n = 1), and five were of unknown origin while hap3 was from Ukraine (n = 1) and two were of unknown origin (Fig. 3d). Interestingly, of the 14 lines carrying the resistant haplotype (hap2), only one line was deemed to also carry the known APR genes Lr34 and Lr46 (Supplementary material 5).

Haplotype analysis of QTL qNV.Lr-3A.3 on chromosome 3A associated with resistance to leaf rust at the seedling stage. a Chromosomal position of QTL qNV.Lr-3A.3 (116.7–117.0 cM based on the DArT-seq consensus map version 4.0 provided by Dr. Andrzej Kilian) and linkage disequilibrium for associated markers. b Haplotype network displaying seven haplotype variants, where the size of the node is proportional to the number of lines carrying that haplotype variant while colour indicates the mean disease response for those lines (0–9 scale, where 9 = very susceptible). c Box plots displaying the disease response for the lines carrying the three most common haplotypes. d The geographic distribution of the three most common haplotypes in the diversity panel
We also constructed a haplotype on the basis of the identified APR QTL qNV.Lr-7B.2 on the long arm of chromosome 7B (126.0–130.6 cM) represented by 11 highly significant markers associated with LR resistance at the adult stage. Interestingly, several previously reported Lr genes and QTL have been reported in the region, including Lr14 (a and b alleles) (Dyck and Samborski 1970; Terracciano et al. 2013), Lr68 (Herrera-Foessel et al. 2012), LrBi16 (Zhang et al. 2011), and LrFun (Xing et al. 2014) (Fig. 4a). Around the identified QTL, the five DArT-seq markers (i.e. 1207290, 1117456, 1214960, 1134022, and 2304335) in very high LD (r 2 > 0.75) were used for the haplotype analysis (Table 3; Fig. 4a). In total, 22 haplotype variants were identified in our panel, of which hap1 and hap2 were the most frequent (78.3 and 7.8%, respectively). To construct the TCS haplotype network, only the variants which occurred at least twice in the panel (i.e. hap1-hap9) were used (Fig. 4b). Tukey’s test and a comparison of median values for seven haplotypes showed that genotypes in hap1 were significantly more susceptible to LR (8 on a 1–9 scale) in all screenings of 2015 (Fig. 4c). Four haplotypes (hap2-hap5) displayed less susceptibility across three phenotypic data sets in 2015, where the median value of each haplotype across phenotypic data sets was variable i.e. hap2 (5–8 on a 1–9 scale), hap3 (4–7 on a 1–9 scale), hap4 (3–8 on a 1–9 scale) and hap5 (4–5.5 on a 1–9 scale) (Fig. 4c). The lines carrying hap2 originated from Russia (n = 4), India (n = 2), Armenia (n = 1), and 16 were of unknown origin. The lines carrying hap3 were from Russia (n = 2), unknown origin (n = 2) and one each from Iraq, Spain, and India. The hap4 originated from China (n = 2), and one each from Russia, India, and Ukraine. The hap5 was present in a line from Pakistan (n = 1) (Fig. 4d).

Haplotype analysis of QTL qNV.Lr-7B on chromosome 7B associated with resistance to leaf rust at the adult plant stage. a Chromosomal position of the QTL qNV.Lr-7B.3 (128.6–130.6 cM based on the DArT-seq consensus map version 4.0 provided by Dr. Andrzej Kilian) and comparison with catalogued Lr genes. The linkage disequilibrium block highlighted for the five associated markers. b Haplotype network displaying the nine most common haplotype variants, where the size of the node is proportional to the number of lines carrying that haplotype variant while colours indicate mean disease response for those lines (1–9 scale, where 9 = very susceptible). c Box plots displaying the disease response by lines carrying seven most common haplotypes in three phenotypic data sets in 2015. d The geographic distribution of the five most common haplotypes in the diversity panel
To test the effect of an accumulation of alleles for LR resistance at the independent loci, we assigned the lines from the diversity panel to groups, based on the absolute number of resistance-associated alleles possessed. This resulted in 13 different groups, ranging from two lines that carried ≤ 5 resistance-associated alleles, up to three lines that carried 29 or more (Fig. 5; Supplementary material 6). A comparison of their indices which represent the average LR response of a line in relation to the overall population evaluated in field trials from 2014 to 2016 revealed a very clear linear trend. While lines that combined relatively few of the identified resistance-associated alleles showed a comparatively high disease index, resistance to LR continuously increased with additional resistance-associated alleles. In total, 51 lines were detected that carry 19 or more resistance-associated alleles and showed index levels largely below zero (Fig. 5; Supplementary material 6).

The effect of resistance-associated favourable alleles at quantitative trait loci for resistance to leaf rust response in the diversity panel. The field relative disease index is calculated using phenotypic data sets from field trials only. The frequency of lines carrying favourable alleles is also presented
Discussion
Novel sources of LR resistance
Deployment of resistant cultivars is the most economical and effective method to control rust diseases in the field (Ellis et al. 2014). However, deployed resistance genes can easily be overcome due to the rapid evolution of the pathogen and limited genetic diversity for resistance factors in modern wheat germplasm. Crop domestication and later selective breeding in modern breeding programs have led to a dramatic loss of genetic diversity in many important crop species, such as rice (Oryza sativa var. japonica) (Huang et al. 2012), maize (Zea mays subsp. mays L.) (Wright et al. 2005) and hexaploid bread wheat (Reif et al. 2005). The exploitation of genetic resources from the primary gene pool of wheat is considered a promising approach to identify new and durable resistance factors that can be utilised for the improvement of modern high-yielding varieties (Mujeeb-Kazi et al. 2013). The primary gene pool includes wild and early domesticated relatives of wheat, landraces, old cultivars and breeding lines. The use of landraces compared to wild relatives is advantageous as they carry homologous chromosomes that can easily recombine with hexaploid wheat (Wulff and Moscou 2014). As advances in genotyping technologies provide high-throughput genome information at an unprecedented resolution and low costs, vast germplasm collections stored in many gene banks worldwide represent a rich and now accessible genetic treasure chest for modern wheat improvement (Voss-Fels and Snowdon 2016). We have identified potentially new genomic regions that are highly associated with LR resistance at seedling and adult stage in the Vavilov wheat diversity panel. Analysis of LD for the three genomes revealed that LD decayed rapidly in the A and B genomes in both population clusters, reflecting the high level of allelic diversity in the diversity panel. In our study, the highest LD was estimated for the D genome, which was also reported in numerous previous studies (Nielsen et al. 2014; Wang et al. 2014a; Zegeye et al. 2014; Voss-Fels et al. 2015). Across experiments, the lines identified as resistant include landraces, cultivars and breeding lines originating from different countries of the world (i.e. Russia, Kazakhstan, India, Pakistan, Ukraine, and China), thus providing diverse sources to achieve durable resistance in various eco-geographic contexts (Gurung et al. 2014; Maccaferri et al. 2015). These resources harbour promising novel resistances against a rapidly evolving pathogen. For instance, in Australia, a recent exotic introduction of P. triticina pathotype 104–1,3,4,6,7,8,10,12 + Lr37 carried virulence on five Lr genes (Lr12, Lr13, Lr20, Lr27 + 31, and Lr37) which were widely deployed in cultivars (Cuddy et al. 2016; Park 2016). Thus, the identification of resistant lines in this study not only provides new sources of resistance, but likely different alleles for already known genes, which can help to enhance genetic diversity in modern wheat breeding programs.
Alignment of putative QTL to previously reported Lr genes and QTL
A large number of QTL were detected in our study (n = 31). Almost half (n = 13) of the identified QTL were considered novel, while the remainder (n = 18) aligned with previously reported QTL and/or catalogued Lr genes (discussed in Supplementary material 7). Interestingly, a locus corresponding to the seedling resistance gene Lr3 on chromosome 6B was detected in our study, despite the use of P. triticina pathotypes that were virulent for Lr3. This suggests the probable presence of alternate alleles of the already ‘extinct’ resistance loci or a tightly linked gene with distinct resistance functions. The lack of availability of tightly linked or gene-specific markers for the known Lr genes hinders the ability to precisely position these genes on the respective chromosome. Further, allelism testing can also be performed to determine the association between the detected loci and previously reported genes and/or the QTL. A large number of QTL (n = 30) were identified in GWAS using more than one adult phenotype dataset. Of these, only six QTL were detected across different adult phenotypic data sets. This might be due to the fact the genomic regions underpinning APR often interact with the plant growth stage, inoculum pressure and the temperature conditions, thus affecting the resistance phenotype. An interesting region identified across both seedling, AGC and field data sets was QTL qNV.Lr-2B.3 on chromosome 2B, which contained seven associated markers. Within this genomic region, we identified several candidate genes (i.e. NB-ARC, P-loop_NTPase, zinc finger, CCHC-type, and RNA-dependent DNA polymerase) that are known to encode proteins involved in pathogen recognition and subsequent activation of innate immune responses that lead to programmed cell death. It is well known that R genes tend to occur in clusters in plant genomes and give rise to many haplotypes via recombination (Friedman and Baker 2007; van Ooijen et al. 2008). Such ‘hotspots’ for resistance QTL could involve various combinations of classical R genes and other race-nonspecific genes (Burdon et al. 2014). A good example is the QTL region qNV.Lr-7B.2 which contains seedling resistance gene Lr14b (Dyck and Sambroski 1970) and APR gene Lr68 (Herrera-Foessel et al. 2012). It should be noted that QTL detected at the adult plant stage could also harbour genes regulating physiological characteristics, rather than classical R genes. For instance, in sorghum (Sorghum bicolor (L.) Moench), several QTL for resistance to rust (Puccinia purpurea) were found to co-locate with QTL for maturity and plant height (Wang et al. 2014b). These genetic factors could offer durable resistance to rust diseases.
Haplotype analysis
In GWAS, single marker scans are performed to understand the underlying genetic architecture of disease resistance in natural populations. In addition, a more powerful approach is to perform a haplotype analysis based on closely linked markers which are more likely to be inherited together as a block (Hayes et al. 2007). Haplotype analyses which typically depict marker-trait associations at a higher resolution due to an increased information content compared to bi-allelic molecular markers, such as SNPs or DArT markers, have been successfully applied in identifying genomic regions involved in effective Fusarium head blight resistance on wheat chromosome arm 3BS (Hao et al. 2012). In the present study, we performed haplotype analyses for two QTL, the seedling QTL qNV.Lr-3A.3 and the APR QTL qNV.Lr-7B.2. Therefore, we followed previous studies and jointly defined markers in strong LD with the two identified QTL as a haplotype block (Hao et al. 2012; Diaz et al. 2011). Analysis of LD around the seedling QTL (qNV.Lr-3A.3) displayed a high level of LD between two associated markers, suggesting a high level of allelic fixation. One of the lines carrying the resistant haplotype (hap2) of QTL qNV.Lr-3A.3 was also found positive to carry known APR genes Lr34 and Lr46, thus providing a combination of R and APR genes/QTL. Such gene/QTL combinations are promising to achieve longer lasting resistance in elite cultivars. Similarly, of the markers located in the APR QTL qNV.Lr-7B.2 on chromosome 7B, five markers with very high LD were considered a haplotype block. The results revealed a broad allelic variation for this chromosomal fragment and showed that four haplotype groups (hap2-hap5) were associated with a reduction in susceptibility across three phenotypic data sets in 2015. This might be explained by the fact that this chromosomal region is known to carry Lr genes such as Lr14 (a and b alleles), Lr68, LrBi16 and LrFun (McIntosh et al. 2017).
Pyramiding of resistance-associated alleles for durable rust resistance
It has been well described that durable rust resistance in wheat can be achieved by pyramiding multiple QTL (Ellis et al. 2014; Mundt 2014). In our study, a comparatively high number of loci with variable LR resistance in the field were detected with a high fraction of lines that carried more than 20 resistance-associated alleles. Identification of lines with a large number of favourable alleles has been reported by previous studies (Kollers et al. 2014; Naruoka et al. 2015; Muleta et al. 2017). The landraces are the traditional varieties which were selected by the farmers in the field preferably for agronomic traits, but at the same time they were also indirectly selected for disease resistance (Zeven 2002). Since the rust pathogen has co-evolved with wheat landraces for thousands of years in the same environment, diverse resistance alleles and their combinations exist in the host population keeping epidemic development in check (Thrall and Burdon 2000; Ordonez and Kolmer 2007). Since the landraces in the seed bank were removed from their environmental context, it is hypothesised that they might hold new allelic variations against the modern P. triticina pathotypes. Detection of a large number of resistance alleles showed that these resistance-associated alleles have accumulated in landraces over time and occurred at variable frequencies (i.e. high, low, and rare) in the population. In particular, rare alleles are known to provide resistance to diseases and environmental stresses (Vikram et al. 2016). Therefore, the utilization of landraces for trait introgression may greatly increase the genetic diversity and frequency of rare alleles into modern wheat breeding programs. In our study, we were able to show that there is a close relationship between the level of LR resistance and number of resistance alleles from independent loci, highlighting the high potential of allele stacking for rust improvement in future cultivars. Combining seedling resistance and APR alleles is most effective and promising to provide sustainable resistance levels and also reduce the fitness cost associated with APR (Nelson 1978; Ellis et al. 2014; Consortium 2016). For example, durable resistance using combinations of resistances (seedling and APR) against stripe rust was achieved in Western Europe, while combining multiple minor genes provided durable resistance to stem rust and powdery mildew at the International Maize and Wheat Improvement Center (CIMMYT), Mexico (Singh et al. 2011; Basnet et al. 2014; Ellis et al. 2014; Singh et al. 2014; Brown 2015).
The detection of a large number of favourable alleles is promising. However, simultaneous consideration of all alleles in a breeding program is often challenging. To overcome this challenge, a small subset of alleles can be targeted by designing specific crosses in a breeding program, thus supporting recombination of favourable alleles at many loci. Furthermore, implementing trait introgression via MAS allows selection for various traits in early generations and can easily eliminate undesirable allele combinations. Recent advancements in genomic approaches such as marker-assisted backcrossing, whole-genome scans, genomic prediction and genomic selection enable rapid combination of multiple alleles in a single variety (Liu et al. 2014). Together with the latest advancements in plant phenotyping approaches and rapid generation advance systems, such as “Speed Breeding” (Watson et al. 2017), the development of rust resistant wheat cultivars can be accelerated (Hickey et al. 2012; Riaz et al. 2016b). Segregating populations can be screened and individuals selected by “phenotyping-on-the-go” during line development. The identified resistance can be rapidly introgressed using the rapid generation advance system in breeding programs. This could help fast-track the introgression of new LR resistance from the Vavilov wheat diversity panel into elite genetic backgrounds for future cultivars. The strategy is not limited to LR, as phenotyping methods adapted to speed breeding have been designed for other important traits in wheat, including stripe rust (Hickey et al. 2012), stem rust (Riaz and Hickey 2017), yellow spot (Dinglasan et al. 2016), seed dormancy (Hickey et al. 2009), and root architecture (Richard et al. 2015).
Author contribution statement
AR, SP, EA, LH, and KVF conceived and designed the study; AR collected the data; AR, NK and SP performed PCR marker screening, EL contributed ideas and provided PCR markers for screening; AR and KVF analysed the data; AR wrote the manuscript; OA and OM provided geographic information about the Vavilov wheat collection; GP guided in designing the field nurseries and provided LR inoculum; RS contributed ideas for the haplotype analysis; SP, EA, RS, EL, LH and KVF edited and revised the manuscript.
References
Aoun M, Breiland M, Turner MK, Loladze A, Chao S, Xu SS, Ammar K, Anderson JA, Kolmer JA, Acevedo M (2016) Genome-wide association mapping of leaf rust response in a durum wheat worldwide germplasm collection. Plant Genome 9. doi:10.3835/plantgenome2016.01.0008
Asseng S, Ewert F, Martre P, Rotter RP, Lobell DB, Cammarano D, Kimball BA, Ottman MJ, Wall GW, White JW, Reynolds MP, Alderman PD, Prasad PVV, Aggarwal PK, Anothai J, Basso B, Biernath C, Challinor AJ, De Sanctis G, Doltra J, Fereres E, Garcia-Vila M, Gayler S, Hoogenboom G, Hunt LA, Izaurralde RC, Jabloun M, Jones CD, Kersebaum KC, Koehler AK, Muller C, Naresh Kumar S, Nendel C, O’Leary G, Olesen JE, Palosuo T, Priesack E, Eyshi Rezaei E, Ruane AC, Semenov MA, Shcherbak I, Stockle C, Stratonovitch P, Streck T, Supit I, Tao F, Thorburn PJ, Waha K, Wang E, Wallach D, Wolf J, Zhao Z, Zhu Y (2015) Rising temperatures reduce global wheat production. Nat Clim Change 5:143–147
Aulchenko YS, Ripke S, Isaacs A, van Duijn CM (2007) GenABEL: an R library for genome-wide association analysis. Bioinformatics 23:1294–1296
Bansal UK, Forrest KL, Hayden MJ, Miah H, Singh D, Bariana HS (2011) Characterisation of a new stripe rust resistance gene Yr47 and its genetic association with the leaf rust resistance gene Lr52. Theor Appl Genet 122:1461–1466
Bansal UK, Arief VN, DeLacy IH, Bariana HS (2013) Exploring wheat landraces for rust resistance using a single marker scan. Euphytica 194:219–233
Bariana HS, Miah H, Brown GN, Willey N, Lehmensiek A (2007) Molecular mapping of durable rust resistance in wheat and its implication in breeding. In: Buck HT, Nisi JE, Salomón N (eds) Developments in plant breeding: Wheat production in stressed environments. Springer, Dordrecht, pp 723–728
Basnet BR, Singh RP, Ibrahim AMH, Herrera-Foessel SA, Huerta-Espino J, Lan C, Rudd JC (2014) Characterization of Yr54 and other genes associated with adult plant resistance to yellow rust and leaf rust in common wheat Quaiu 3. Mol Breed 33:385–399
Bolton MD, Kolmer JA, Garvin DF (2008) Wheat leaf rust caused by Puccinia triticina. Mol Plant Pathol 9:563–575
Brown JK (2015) Durable resistance of crops to disease: a Darwinian perspective. Annu Rev Phytopathol 53:513–539
Burdon JJ, Barrett LG, Rebetzke G, Thrall PH (2014) Guiding deployment of resistance in cereals using evolutionary principles. Evol Appl 7:609–624
Cavanagh CR, Chao S, Wang S, Huang BE, Stephen S, Kiani S, Forrest K, Saintenac C, Brown-Guedira GL, Akhunova A (2013) Genome-wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars. Proc Natl Acad Sci USA 110:8057–8062
Chaves MS, Martinelli JA, Wesp-Guterres C, Graichen FAS, Brammer SP, Scagliusi SM, da Silva PR, Wiethölter P, Torres GAM, Lau EY, Consoli L, Chaves ALS (2013) The importance for food security of maintaining rust resistance in wheat. Food Secur 5:157–176
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Cloutier S, McCallum BD, Loutre C, Banks TW, Wicker T, Feuillet C, Keller B, Jordan MC (2007) Leaf rust resistance gene Lr1, isolated from bread wheat (Triticum aestivum L.) is a member of the large psr567 gene family. Plant Mol Biol 65:93–106
Consortium REX (2016) Combining selective pressures to enhance the durability of disease resistance genes. Front Plant Sci. doi:10.3389/fpls.2016.01916
Cuddy W, Park R, Bariana H, Bansal U, Singh D, Roake J, Platz G (2016) Expected responses of Australian wheat, triticale and barley varieties to the cereal rust diseases and genotypic data for oat varieties. Cereal Rust Rep 14:1–8
Daetwyler HD, Bansal UK, Bariana HS, Hayden MJ, Hayes BJ (2014) Genomic prediction for rust resistance in diverse wheat landraces. Theor Appl Genet 127(8):1795–1803
Diaz A, Fergany M, Formisano G, Ziarsolo P, Blanca J, Fei Z, Staub JE, Zalapa JE, Cuevas HE, Dace G, Oliver M, Boissot N, Dogimont C, Pitrat M, Hofstede R, van Koert P, Harel-Beja R, Tzuri G, Portnoy V, Cohen S, Schaffer A, Katzir N, Xu Y, Zhang H, Fukino N, Matsumoto S, Garcia-Mas J, Monforte AJ (2011) A consensus linkage map for molecular markers and quantitative trait loci associated with economically important traits in melon (Cucumis melo L.). BMC Plant Biol. doi:10.1186/1471-2229-11-111
Dinglasan E, Godwin ID, Mortlock MY, Hickey LT (2016) Resistance to yellow spot in wheat grown under accelerated growth conditions. Euphytica 209:693–707
Dyck P, Samborski D (1970) The genetics of two alleles for leaf rust resistance at the Lr14 locus in wheat. Can Genet Cytol 12:689–694
Ellis JG, Lagudah ES, Spielmeyer W, Dodds PN (2014) The past, present and future of breeding rust resistant wheat. Front Plant Sci. doi:10.3389/fpls.2014.00641
Feuillet C, Travella S, Stein N, Albar L, Nublat A, Keller B (2003) Map-based isolation of the leaf rust disease resistance gene Lr10 from the hexaploid wheat (Triticum aestivum L.) genome. Proc Nat Acad Sci USA 100:15253–15258
Flor HH (1971) Current status of the gene-for-gene concept. Annu Rev Phytopathol 9:275–296
Friedman AR, Baker BJ (2007) The evolution of resistance genes in multi-protein plant resistance systems. Curr Opin Genet Dev 17:493–499
Gao L, Turner MK, Chao S, Kolmer J, Anderson JA (2016) Genome wide association study of seedling and adult plant leaf rust resistance in elite spring wheat breeding lines. PLoS One. doi:10.1371/journal.pone.0148671
Grassini P, Eskridge KM, Cassman KG (2013) Distinguishing between yield advances and yield plateaus in historical crop production trends. Nat Commun. doi:10.1038/ncomms3918
Gurung S, Mamidi S, Bonman JM, Xiong M, Brown-Guedira G, Adhikari TB (2014) Genome-wide association study reveals novel quantitative trait loci associated with resistance to multiple leaf spot diseases of spring wheat. PLoS One 9(9):e108179. doi:10.1371/journal.pone.0108179
Hall D, Tegström C, Ingvarsson PK (2010) Using association mapping to dissect the genetic basis of complex traits in plants. Brief Funct Genom 9:157–165. doi:10.1093/bfgp/elp048
Hao K, Di X, Cawley S (2007) LdCompare: rapid computation of single-and multiple-marker r 2 and genetic coverage. Bioinformatics 23(2):252–254
Hao C, Wang Y, Hou J, Feuillet C, Balfourier F, Zhang X (2012) Association mapping and haplotype analysis of a 3.1-Mb genomic region involved in Fusarium head blight resistance on wheat chromosome 3BS. PLoS One. doi:10.1371/journal.pone.0046444
Hayes BJ, Chamberlain AJ, McPartlan H, Macleod I, Sethuraman L, Goddard ME (2007) Accuracy of marker-assisted selection with single markers and marker haplotypes in cattle. Genet Res 89:215–220
Hennig C (2014) fpc: Flexible procedures for clustering. R package version 2.1-7. https://cran.r-project.org/web/packages/fpc/index.html
Herrera-Foessel SA, Singh RP, Huerta-Espino J, Rosewarne GM, Periyannan SK, Viccars L, Calvo-Salazar V, Lan C, Lagudah ES (2012) Lr68: a new gene conferring slow rusting resistance to leaf rust in wheat. Theor Appl Genet 124:1475–1486
Hickey LT, Dieters MJ, DeLacy IH, Kravchuk OY, Mares DJ, Banks PM (2009) Grain dormancy in fixed lines of white-grained wheat (Triticum aestivum L.) grown under controlled environmental conditions. Euphytica 168:303–310
Hickey LT, Wilkinson PM, Knight CR, Godwin ID, Kravchuk OY, Aitken EAB (2012) Rapid phenotyping for adult-plant resistance to stripe rust in wheat. Plant Breed 131:54–61
Hiebert CW, Thomas JB, McCallum BD, Gavin Humphreys D, DePauw RM, Hayden MJ (2010) An introgression on wheat chromosome 4DL in RL6077 (Thatcher*6/PI 250413) confers adult plant resistance to stripe rust and leaf rust (Lr67). Theor Appl Genet 121:1083–1091
Huang X, Han B (2014) Natural variations and genome-wide association studies in crop plants. Annu Rev Plant Biol 65:531–551
Huang L, Brooks SA, Li W, Fellers JP, Trick HN, Gill BS (2003) Map-based cloning of leaf rust resistance gene Lr21 from the large and polyploid genome of bread wheat. Genetics 164:655–664
Huang X, Kurata N, Wei X, Wang ZX, Wang A, Zhao Q, Zhao Y, Liu K, Lu H, Li W, Guo Y, Lu Y, Zhou C, Fan D, Weng Q, Zhu C, Huang T, Zhang L, Wang Y, Feng L, Furuumi H, Kubo T, Miyabayashi T, Yuan X, Xu Q, Dong G, Zhan Q, Li C, Fujiyama A, Toyoda A, Lu T, Feng Q, Qian Q, Li J, Han B (2012) A map of rice genome variation reveals the origin of cultivated rice. Nature 490:497–501
Huerta-Espino J, Singh RP, Germán S, McCallum BD, Park RF, Chen WQ, Bhardwaj SC, Goyeau H (2011) Global status of wheat leaf rust caused by Puccinia triticina. Euphytica 179:143–160
Jordan KW, Wang S, Lun Y, Gardiner L-J, MacLachlan R, Hucl P, Wiebe K, Wong D, Forrest KL, Sharpe AG, Sidebottom CH, Hall N, Toomajian C, Close T, Dubcovsky J, Akhunova A, Talbert L, Bansal UK, Bariana HS, Hayden MJ, Pozniak C, Jeddeloh JA, Hall A, Akhunov E (2015) A haplotype map of allohexaploid wheat reveals distinct patterns of selection on homoeologous genomes. Genome Biol 16:48
Juliana P, Singh RP, Singh PK, Crossa J, Huerta-Espino J, Lan C, Bhavani S, Rutkoski JE, Poland JA, Bergstrom GC, Sorrells ME (2017) Genomic and pedigree-based prediction for leaf, stem, and stripe rust resistance in wheat. Theor Appl Genet 130:1415–1430
Kankwatsa P, Singh D, Thomson PC, Babiker EM, Bonman JM, Newcomb M, Park RF (2017) Characterization and genome-wide association mapping of resistance to leaf rust, stem rust and stripe rust in a geographically diverse collection of spring wheat landraces. Mol Breed 37:113
Kersey PJ, Allen JE, Armean I, Boddu S, Bolt BJ, Carvalho-Silva D, Christensen M, Davis P, Falin LJ, Grabmueller C (2016) Ensembl genomes 2016: more genomes, more complexity. Nucleic Acids Res 44:D574–D580
Kertho A, Mamidi S, Bonman JM, McClean PE, Acevedo M (2015) Genome-wide association mapping for resistance to leaf and stripe rust in winter-habit hexaploid wheat landraces. PLoS One. doi:10.1371/journal.pone.0129580
Kollers S, Rodemann B, Ling J, Korzun V, Ebmeyer E, Argillier O, Hinze M, Plieske J, Kulosa D, Ganal MW (2014) Genome-wide association mapping of tan spot resistance (Pyrenophora tritici-repentis) in European winter wheat. Mol Breed 34:363–371
Kolmer J, Mert Z, Akan K, Demir L, Ünsal R, Şermet C, Keser M, Akin B, Morgounov A (2013) Virulence of Puccinia triticina in Turkey and leaf rust resistance in Turkish wheat cultivars. Eur J Plant Pathol 135:703–716
Koornneef M, Alonso-Blanco C, Vreugdenhil D (2004) Naturally occurring genetic variation in Arabidopsis thaliana. Annu Rev Plant Biol 55:141–172
Krattinger SG, Lagudah ES, Spielmeyer W, Singh RP, Huerta-Espino J, McFadden H, Bossolini E, Selter LL, Keller B (2009) A putative ABC transporter confers durable resistance to multiple fungal pathogens in wheat. Science 323:1360–1363
Kumar S, Archak S, Tyagi RK, Kumar J, Vk V, Jacob SR, Srinivasan K, Radhamani J, Parimalan R, Sivaswamy M, Tyagi S, Yadav M, Kumari J, Deepali Sharma S, Bhagat I, Meeta M, Bains NS, Chowdhury AK, Saha BC, Bhattacharya PM, Kumari J, Singh MC, Gangwar OP, Prasad P, Bharadwaj SC, Gogoi R, Sharma JB, Gm SK, Saharan MS, Bag M, Roy A, Prasad TV, Sharma RK, Dutta M, Sharma I, Bansal KC (2016) Evaluation of 19,460 Wheat accessions conserved in the Indian National Genebank to identify new sources of resistance to rust and spot blotch diseases. PLoS One. doi:10.1371/journal.pone.0167702
Lagudah ES (2011) Molecular genetics of race non-specific rust resistance in wheat. Euphytica 179:81–91
Lagudah ES, Krattinger SG, Herrera-Foessel S, Singh RP, Huerta-Espino J, Spielmeyer W, Brown-Guedira G, Selter LL, Keller B (2009) Gene-specific markers for the wheat gene Lr34/Yr18/Pm38 which confers resistance to multiple fungal pathogens. Theor Appl Genet 119:889–898
Leigh JW, Bryant D (2015) Popart: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116
Li Z, Lan C, He Z, Singh RP, Rosewarne GM, Chen X, Xia X (2014) Overview and application of QTL for adult plant resistance to leaf rust and powdery mildew in wheat. Crop Sci 54:1907–1925
Li H, Vikram P, Singh RP, Kilian A, Carling J, Song J, Burgueno-Ferreira JA, Bhavani S, Huerta-Espino J, Payne T, Sehgal D, Wenzl P, Singh S (2015) A high density GBS map of bread wheat and its application for dissecting complex disease resistance traits. BMC Genom 16:1–15
Li G, Xu X, Bai G, Carver BF, Hunger R, Bonman JM, Kolmer J, Dong H (2016) Genome-wide association mapping reveals novel QTL for seedling leaf rust resistance in a worldwide collection of winter wheat. Plant Genome. doi:10.3835/plantgenome2016.06.0051
Liu H, Sørensen AC, Meuwissen THE, Berg P (2014) Allele frequency changes due to hitch-hiking in genomic selection programs. GSE 46:8
Loegering W (1959) Methods for recording cereal rust data. USDA International Spring Wheat Nursery
Lopes MS, El-Basyoni I, Baenziger PS, Singh S, Royo C, Ozbek K, Aktas H, Ozer E, Ozdemir F, Manickavelu A, Ban T, Vikram P (2015) Exploiting genetic diversity from landraces in wheat breeding for adaptation to climate change. J Exp Bot 66:3477–3486
Maccaferri M, Zhang J, Bulli P, Abate Z, Chao S, Cantu D, Bossolini E, Chen X, Pumphrey M, Dubcovsky J (2015) A genome-wide association study of resistance to stripe rust (Puccinia striiformis f. sp. tritici) in a worldwide collection of hexaploid spring wheat (Triticum aestivum L.). G3 Genes Genomes Genet 5:449–465
McCallum B, Hiebert C, Huerta-Espino J, Cloutier S (2012) 3 Wheat Leaf Rust. Dis Resist Wheat 1:33
McIntosh R, Yamazaki Y, Dubcovsky J, Rogers J, Morris C, Xia XC (2017) Catalogue of gene symbols for wheat: 2017 supplement. https://shigen.nig.ac.jp/wheat/komugi/genes/macgene/supplement2017.pdf. Accessed 01 Oct 2017
Mitrofanova OP (2012) Wheat genetic resources in Russia: current status and prebreeding studies. Russ J Genet Appl Res 2:277–285
Mondal S, Rutkoski JE, Velu G, Singh PK, Crespo-Herrera LA, Guzmán C, Bhavani S, Lan C, He X, Singh RP (2016) Harnessing diversity in wheat to enhance grain yield, climate resilience, disease and insect pest resistance and nutrition through conventional and modern breeding approaches. Front Plant Sci. doi:10.3389/fpls.2016.00991
Moore JW, Herrera-Foessel S, Lan C, Schnippenkoetter W, Ayliffe M, Huerta-Espino J, Lillemo M, Viccars L, Milne R, Periyannan S, Kong X, Spielmeyer W, Talbot M, Bariana H, Patrick JW, Dodds P, Singh R, Lagudah E (2015) A recently evolved hexose transporter variant confers resistance to multiple pathogens in wheat. Nat Genet 47:1494–1498
Mujeeb-Kazi A, Kazi AG, Dundas I, Rasheed A, Ogbonnaya F, Kishii M, Bonnett D, Wang RR-C, Xu S, Chen P (2013) Genetic diversity for wheat improvement as a conduit to food security. Adv Agron 122:179–258
Muleta KT, Bulli P, Rynearson S, Chen X, Pumphrey M (2017) Loci associated with resistance to stripe rust (Puccinia striiformis f. sp. tritici) in a core collection of spring wheat (Triticum aestivum). PLoS One. doi:10.1371/journal.pone.0179087
Mundt CC (2014) Durable resistance: a key to sustainable management of pathogens and pests. Infect Genet Evol 27:446–455
Naruoka Y, Garland-Campbell KA, Carter AH (2015) Genome-wide association mapping for stripe rust (Puccinia striiformis f. sp. tritici) in US Pacific Northwest winter wheat (Triticum aestivum L.). Theor Appl Genet 128:1083–1101
Nelson RR (1978) Genetics of horizontal resistance to plant diseases. Annu Rev Phytopathol 16:359–378
Nielsen NH, Backes G, Stougaard J, Andersen SU, Jahoor A (2014) Genetic diversity and population structure analysis of european hexaploid bread wheat (Triticum aestivum L.) varieties. PLoS One. doi:10.1371/journal.pone.0094000
Niks RE, Qi X, Marcel TC (2015) Quantitative resistance to biotrophic filamentous plant pathogens: concepts, misconceptions, and mechanisms. Annu Rev Phytopathol 53:445–470
Ordonez ME, Kolmer JA (2007) Simple sequence repeat diversity of a worldwide collection of Puccinia triticina from durum wheat. Phytopathology 97:574–583
Park RF (2016) The wheat leaf rust pathogen in Australia - pathogenic variation and pathotype designation. Cereal Rust Rep 14:1–5
Pasam RK, Bansal U, Daetwyler HD, Forrest KL, Wong D, Petkowski J, Willey N, Randhawa M, Chhetri M, Miah H, Tibbits J, Bariana H, Hayden MJ (2017) Detection and validation of genomic regions associated with resistance to rust diseases in a worldwide hexaploid wheat landrace collection using BayesR and mixed linear model approaches. Theor Appl Genet 130:777–793
Peterson RF, Campbell A, Hannah A (1948) A diagrammatic scale for estimating rust intensity on leaves and stems of cereals. Can J Res 26:496–500
Reif JC, Zhang P, Dreisigacker S, Warburton ML, van Ginkel M, Hoisington D, Bohn M, Melchinger AE (2005) Wheat genetic diversity trends during domestication and breeding. Theor Appl Genet 110:859–864
Riaz A, Hickey LT (2017) Rapid phenotyping adult plant resistance to stem rust in wheat grown under controlled conditions. In: Periyannan S (ed) Wheat rust diseases: methods and protocols. Springer, New York, pp 183–196
Riaz A, Athiyannan N, Periyannan S, Afanasenko O, Mitrofanova O, Aitken EAB, Lagudah E, Hickey LT (2016a) Mining Vavilov’s treasure chest of wheat diversity for adult plant resistance to Puccinia triticina. Plant Dis 101:317–323
Riaz A, Periyannan S, Aitken E, Hickey L (2016b) A rapid phenotyping method for adult plant resistance to leaf rust in wheat. Plant Methods 12:1–10
Riaz A, Hathorn A, Dinglasan E, Ziems L, Richard C, Singh D, Mitrofanova O, Afanasenko O, Aitken E, Godwin I, Hickey L (2017) Into the vault of the Vavilov wheats: old diversity for new alleles. Genet Resour Crop Evol 64:531–544
Richard CA, Hickey LT, Fletcher S, Jennings R, Chenu K, Christopher JT (2015) High-throughput phenotyping of seminal root traits in wheat. Plant Methods 11:1–13
Rinaldo A, Gilbert B, Boni R, Krattinger SG, Singh D, Park RF, Lagudah E, Ayliffe M (2016) The Lr34 adult plant rust resistance gene provides seedling resistance in durum wheat without senescence. Plant Biotechnol J 15:894–905
Risk JM, Selter LL, Krattinger SG, Viccars LA, Richardson TM, Buesing G, Herren G, Lagudah ES, Keller B (2012) Functional variability of the Lr34 durable resistance gene in transgenic wheat. Plant Biotechnol J 10:477–487
Sadovaya AS, Gultyaeva EI, Mitrofanova OP, Shaidayuk EL, Hakimova AG, Zuev EV (2015) Leaf rust resistance in common wheat varieties and lines from the collection of the Vavilov Plant Industry Institute carrying alien genetic material. Russ J Genet Appl Res 5:233–241
Sehgal D, Vikram P, Sansaloni CP, Ortiz C, Pierre CS, Payne T, Ellis M, Amri A, Petroli CD, Wenzl P, Singh S (2015) Exploring and mobilizing the gene bank biodiversity for wheat improvement. PLoS One. doi:10.1371/journal.pone.0132112
Semagn K, Bjørnstad Å, Xu Y (2010) The genetic dissection of quantitative traits in crops. Electron J Biotechnol 13:16–17
Shewry PR, Hey SJ (2015) The contribution of wheat to human diet and health. Food Energy Secur 4:178–202
Singh RP, Hodson DP, Huerta-Espino J, Jin Y, Bhavani S, Njau P, Herrera-Foessel S, Singh PK, Singh S, Govindan V (2011) The emergence of Ug99 races of the stem rust fungus is a threat to world wheat production. Annu Rev Phytopathol 49:465–481
Singh RP, Herrera-Foessel S, Huerta-Espino J, Singh S, Bhavani S, Lan C, Basnet BR (2014) Progress towards genetics and breeding for minor genes based resistance to Ug99 and other rusts in CIMMYT high-yielding spring wheat. J Integr Agric 13:255–261
Stakman E, Stewart D, Loegering W (1962) Identification of physiologic races of Puccinia graminis var. tritici. USDA-ARS Sci J Ser 4691:1–53
Team RC (2014) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Terracciano I, Maccaferri M, Bassi F, Mantovani P, Sanguineti MC, Salvi S, Šimková H, Doležel J, Massi A, Ammar K (2013) Development of COS-SNP and HRM markers for high-throughput and reliable haplotype-based detection of Lr14a in durum wheat (Triticum durum Desf.). Theor Appl Genet 126:1077–1101
Thind AK, Wicker T, Simkova H, Fossati D, Moullet O, Brabant C, Vrana J, Dolezel J (2017) Rapid cloning of genes in hexaploid wheat using cultivar-specific long-range chromosome assembly. Nat Biotechnol. doi:10.1038/nbt.3877
Thrall PH, Burdon JJ (2000) Effect of resistance variation in a natural plant host-pathogen metapopulation on disease dynamics. Plant Pathol 49:767–773
Turner MK, Kolmer JA, Pumphrey MO, Bulli P, Chao S, Anderson JA (2017) Association mapping of leaf rust resistance loci in a spring wheat core collection. Theor Appl Genet 130:345–361
Van Ooijen G, Mayr G, Kasiem MM, Albrecht M, Cornelissen BJ, Takken FL (2008) Structure-function analysis of the NB-ARC domain of plant disease resistance proteins. J Exp Bot 59:1383–1397
Vikram P, Franco J, Burgueño-Ferreira J, Li H, Sehgal D, Saint Pierre C, Ortiz C, Sneller C, Tattaris M, Guzman C, Sansaloni CP, Ellis M, Fuentes-Davila G, Reynolds M, Sonder K, Singh P, Payne T, Wenzl P, Sharma A, Bains NS, Singh GP, Crossa J, Singh S (2016) Unlocking the genetic diversity of Creole wheats. Sci Rep. doi:10.1038/srep23092
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Voss-Fels K, Snowdon RJ (2016) Understanding and utilizing crop genome diversity via high-resolution genotyping. Plant Biotechnol J 14:1086–1094
Voss-Fels K, Frisch M, Qian L, Kontowski S, Friedt W, Gottwald S, Snowdon RJ (2015) Subgenomic diversity patterns caused by directional selection in bread wheat gene pools. Plant Genome 8(2):1–13
Wang S, Wong D, Forrest K, Allen A, Chao S, Huang BE, Maccaferri M, Salvi S, Milner SG, Cattivelli L, Mastrangelo AM, Whan A, Stephen S, Barker G, Wieseke R, Plieske J, International Wheat Genome Sequencing C, Lillemo M, Mather D, Appels R, Dolferus R, Brown-Guedira G, Korol A, Akhunova AR, Feuillet C, Salse J, Morgante M, Pozniak C, Luo M-C, Dvorak J, Morell M, Dubcovsky J, Ganal M, Tuberosa R, Lawley C, Mikoulitch I, Cavanagh C, Edwards KJ, Hayden M, Akhunov E (2014a) Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant Biotechnol J 12:787–796
Wang X, Mace E, Hunt C, Cruickshank A, Henzell R, Parkes H, Jordan D (2014b) Two distinct classes of QTL determine rust resistance in sorghum. BMC Plant Biol 14:366
Watson A, Ghosh S, Williams M, Cuddy WS, Simmonds J, Rey M-D, Hatta MAM, Hinchliffe A, Steed A, Reynolds D, Adamski N, Breakspear A, Korolev A, Rayner T, Dixon LE, Riaz A, Martin W, Ryan M, Edwards D, Batley J, Raman H, Rogers C, Domoney C, Moore G, Harwood W, Nicholson P, Dieters MJ, DeLacy IH, Zhou J, Uauy C, Boden SA, Park RF, Wulff BBH, Hickey LT (2017) Speed breeding: a powerful tool to accelerate crop research and breeding. bioRxiv. doi:10.1101/161182
Wright SI, Bi IV, Schroeder SG, Yamasaki M, Doebley JF, McMullen MD, Gaut BS (2005) The effects of artificial selection on the maize genome. Science 308:1310–1314
Wulff BBH, Moscou MJ (2014) Strategies for transferring resistance into wheat: from wide crosses to GM cassettes. Front Plant Sci. doi:10.3389/fpls.2014.00692
Xing L, Wang C, Xia X, He Z, Chen W, Liu T, Li Z, Liu D (2014) Molecular mapping of leaf rust resistance gene LrFun in Romanian wheat line Fundulea 900. Mol Breed 33:931–937
Yu J, Buckler ES (2006) Genetic association mapping and genome organization of maize. Curr Opin Biotechnol 17:155–160
Zegeye H, Rasheed A, Makdis F, Badebo A, Ogbonnaya FC (2014) Genome-wide association mapping for seedling and adult plant resistance to stripe rust in synthetic hexaploid wheat. PLoS One. doi:10.1371/journal.pone.0105593
Zeven AC (2002) Traditional maintenance breeding of landraces: 2. Practical and theoretical considerations on maintenance of variation of landraces by farmers and gardeners. Euphytica 123:147–158
Zhang H, Xia X, He Z, Li X, Li Z, Liu D (2011) Molecular mapping of leaf rust resistance gene LrBi16 in Chinese wheat cultivar Bimai 16. Mol Breed 28:527–534
Zhu C, Gore M, Buckler ES, Yu J (2008) Status and prospects of association mapping in plants. Plant Genome 1:5–20
Ziems LA, Hickey LT, Hunt CH, Mace ES, Platz GJ, Franckowiak JD, Jordan DR (2014) Association mapping of resistance to Puccinia hordei in Australian barley breeding germplasm. Theor Appl Genet 127:1199–1212
Acknowledgements
This research was supported by an Early Career Research Grant and a Ph.D. scholarship from the University of Queensland (UQ), Australia. We thank Dr. Laura Ziems and Dr. Dharmendra Singh from UQ for their valuable advice regarding the analyses performed in this study. We also greatly appreciate valuable advice from Prof. Robert McIntosh from the University of Sydney for positioning the catalogued Lr genes in the wheat genome.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by Steven S. Xu.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Riaz, A., Athiyannan, N., Periyannan, S.K. et al. Unlocking new alleles for leaf rust resistance in the Vavilov wheat collection. Theor Appl Genet 131, 127–144 (2018). https://doi.org/10.1007/s00122-017-2990-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-017-2990-5