
Abstract
Key message
A high-density SNP map was constructed and several novel QTL for branch angle across six environments in Brassica napus were identified.
Abstract
Branch angle is a major determinant for the ideotype of a plant, while the mechanisms underlying this trait in Brassica napus remain elusive. Herein, we developed one doubled haploid population from a cross involving one Capsella bursa-pastoris derived B. napus intertribal introgression line with the compressed branches and wooden stems, and constructed a high-density SNP map covering the genetic distance of 2242.14 cM, with an average marker interval of 0.73 cM. After phenotypic measurements across six environments, the inclusive composite interval mapping algorithm was conducted to analyze the QTL associated with branch angle. In single-environment analysis, a total of 17 QTL were detected and mainly distributed on chromosomes A01, A03, A09 and C03. Of these, three major QTL, qBA.A03-2, qBA.C03-3 and qBA.C03-4 were steadily expressed, each explaining more than 10% of the phenotypic variation in at least two environments. Compared with other results on rapeseed branch angle, these major QTL were newly detected. In QTL by environment interactions (QEI) mapping, 10 QTL were identified, and the QTL average effect and QEI effect were estimated. Of these, 7 QTL were detected in both single-environment analysis and QEI mapping. Based on the physical positions of SNPs and the functional annotation of the Arabidopsis thaliana genome, 27 genes within the QTL regions were selected as candidate genes, including early auxin-responsive genes, small auxin-up RNA, auxin/indoleacetic acid and gretchenhagen-3. These results may pave the way for deciphering the genetic control of branch angle in B. napus.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rapeseed (Brassica napus L., 2n = 38, genome AACC) is a globally important crop, which provides vegetable oil for humans, biodiesel production for industry and fodder for animals (Wang et al. 2011; Prakash et al. 2011). Selection of plants with ideal plant architecture (IPA) is a crucial strategy for crop domestication and improvement (Jiao et al. 2010). Branch angle (BA) is a major determinant for the ideotype of a plant because it influences planting density and further increases biomass yield by affecting photosynthesis efficiency (Zhao et al. 2014). In the field, neither the extreme-spreading nor the compact rice plant type is beneficial to grain production, because the spreading branches occupy too much space, and the compact branches may affect the efficiency of light capture and more susceptible to pathogens (Wang and Li 2008). Particularly, the tightly branching (branch angle < 25°) and lodging resistant cultivars are now eagerly needed for the mechanical harvesting which is quickly developing and gradually replacing the hand harvesting, as the urbanization level increases and young laborers move into cities in China.
Current research showed that the plant branch (tiller) angle size is regulated genetically and hormonally, while very limited genes controlling these processes have been revealed. PROSTRATE GROWTH 1 (PROG1) plays an important role during rice domestication from prostrate to erect growth, which encodes a newly identified zinc-finger nuclear transcription factor that affects tiller angle and tiller number (Tan et al. 2008; Jin et al. 2008). Tiller Angle Control 1 (TAC1) is another major gene controlling the tiller angle and leaf angle in rice and maize, respectively (Yu et al. 2007; Ku et al. 2011). Then the ortholog PpeTAC1 was identified in peach trees (Dardick et al. 2013). A novel gene of TAC3, which greatly controls the tiller angle in rice cultivars, shows a diverse genetic basis for tiller angle between the indica and japonica subpopulations (Dong et al. 2016).
The phytohormone plays a critical role in regulating branch angle. Several genes related to auxin transport were identified. LAZY1 (LA1) was first reported for controlling the tiller angle in rice through asymmetric auxin distribution caused by polar auxin transport (PAT) (Li et al. 2007b; Yoshihara and Iino 2007). OsPIN1 and OsPIN2 genes encode auxin efflux transporters, and over-expression of the two genes leads to increased tiller number and angle during rice growth and development (Xu et al. 2005; Chen et al. 2012). Recently, a gene named BnaYUCCA6, involved in auxin biosynthesis, was identified as the candidate gene for branch angle in B. napus (Wang et al. 2016). In addition, strigolactones (SLs) can rescue the spreading phenotype of la mutant in rice, suggesting that SLs can regulate tiller angle through attenuating shoot gravitropism and inhibiting auxin biosynthesis (Sang et al. 2014).
The values of branch angle present continuous variations, influenced by several polymorphic genes and environmental conditions. Quantitative trait loci (QTL) can affect this variation genetically. With the release of the Illumina Infinium Brassica 60K SNP array and the annotation of B. napus genome (Chalhoub et al. 2014), QTL methodology provides a low-cost, efficient, and powerful way to identify the genomic regions that are associated with quantitative traits in B. napus (Doerge 2002; Liu et al. 2013). This Brassica 60K SNP array has been applied in diverse studies, including molecular karyotyping, germplasm collection characterization, genome-wide association mapping, and biparental QTL mapping (Mason et al. 2017). Despite important progresses in genetics, QTL by environment interactions (QEI) mapping is often conducted in multi-environment trials to verify the QTL detected in single environment and evaluate the QTL average effect and QEI effects (Qi et al. 2015; Zhang et al. 2017b). Combining genotypic and phenotypic data from multiple environments into the model, QEI mapping represents a more powerful and accurate approach to estimate the contribution to variation by the different sources affecting a trait (Zhang et al. 2017a).
Although much attention has been paid to study the plant architecture of B. napus (Chen et al. 2007b; Cai et al. 2016), questions concerning the genetic basis of branch angle remain to be answered. Previous genetic analyses on branch angle were mainly based on genome-wide association study (GWAS) and QTL-seq technology (Wang et al. 2016; Liu et al. 2016; Sun et al. 2016; Li et al. 2017). But the detection power of GWAS is unpredictable and the population structure can cause strong spurious correlations (Aranzana et al. 2005). Cross-based genetic mapping by using suitable parents should enhance the detection of the related loci for particular traits. Previously, we made the intertribal cross between B. napus cv. Zhongyou 821 and Capsella bursa-pastoris (L.) Medic (2n = 4x = 32) (Chen et al. 2007a), and selected an introgression line which showed the plant architecture with compressed branches and rigid stems (Zhang et al. 2013). In this study, we developed one doubled haploid (DH) population from the cross between the introgression line and the genotype ‘Westar’ with larger branch angle and less lignified stem, and then constructed a high-density linkage map, and identified three novel and major QTL across six environments. This dissection improves our understanding of the genetic basis of branch angle in B. napus.
Materials and methods
Plant materials and field trials
A double haploid (DH) population with 208 lines was developed from a cross between ‘Y689’ and ‘Westar’, named as YW-DH population. The parent ‘Y689’ was a derivative of one partial intertribal hybrid between B. napus cv. Zhongyou 821 and Capsella bursa-pastoris (L.) Medic (2n = 4x = 32) (Chen et al. 2007a; Zhang et al. 2013). Zhongyou 821 was one elite cultivar with the merit of high seed yield and good resistance to Sclerotinia sclerotiorum (Zhao et al. 2009), but its branch angles were large and the plant architecture was not very tight. C. bursa-pastoris possessed high resistance to S. sclerotiorum which was probably related to its lignified or wooden stems. Though this hybrid showed some obvious characters of C. bursa-pastoris origin, such as wooden stems, it eliminated most chromosomes from C. bursa-pastoris and also some chromosomes from B. napus (Chen et al. 2007a). After the hybrid was backcrossed twice to Zhongyou 821 as pollen parent and the progenies were self-crossed for six generations and selected for double-low quality and wooden stems, the cytologically stable (2n = 38) and good seed-set lines were obtained. The pure lines of the novel stable introgression was produced by microspore culture method and crossed as female with ‘Westar’ which was highly sensitive to S. sclerotiorum (Zhao et al. 2009) and had larger branch angle.
The YW-DH population, together with the two parents, was planted in the experimental field with a randomized complete block (RCB) design with two replications in six natural environments. Each plot contained two rows of 30 cm apart and 12 plants in each row with 20 cm between plants. Year-location combinations were treated as environments, which were coded as 14WH (Wuhan, 114.33°E, 30.50°N, 2014–2015), 14ND (Weinan, 109.93°E, 34.80°N, 2014–2015), 15WH (Wuhan, 2015–2016), 15CD (Chengdu, 104.15°E, 30.83°N, 2015–2016), 15ER (Ezhou, 114.67°E, 30.08°N, 2015–2016) and 16XN (Xining, 101.77°E, 36.67°N, 2016). The seeds were sown in October in semi winter-type (WH, CD and ER) or winter-type (ND) rapeseed growing area, except in Xining (XN) of spring-type rapeseed growing area, where the sowing was in May. Field management followed conventional agricultural practices. The introgression line ‘Y689’ still kept the semi-winter growing behavior of the donor Zhongyou 821, and ‘Westar’ also showed the semi-winter growing habit in semi-winter regions, Wuhan and Chengdu, though it was the spring type in Canada. So the lines of DH-population derived from them presented semi-winter habit. Then, these lines were able to flower after only about 2 months of planting in Xining.
Trait measurement and statistical analysis
The branch angle (BA) was defined as the angle between the main stem and the branch. For investigating the phenotype, the branch angles for the first three branches from top of the mature plant were measured, and their average was taken as the value of the branch angle for one plant. Five representative plants in the middle of each plot were selected for the phenotype. The experimental data were analyzed using the SPSS 19.0 software. All agronomic traits were measured and the best linear unbiased predictor (BLUP) values from different environments and years were used for the genotypic values. The broad-sense heritability was calculated as \( h^{2} = \sigma_{\text{g}}^{2} /(\sigma_{\text{g}}^{2} + \sigma_{\text{ge}}^{2} /n + \sigma_{\text{e}}^{2} /nr) \), where \( \sigma_{\text{g}}^{2} \) is the variance among DHs, \( \sigma_{\text{ge}}^{2} \) is the interaction variance of the genotype with environment, \( \sigma_{\text{e}}^{2} \) is the error variance, \( n \) is the number of environments, and \( r \) is the number of replications. The estimates of \( \sigma_{\text{g}}^{2} \), \( \sigma_{\text{ge}}^{2} \) and \( \sigma_{\text{e}}^{2} \) were obtained from a two-way analysis of variance (ANOVA) using the general linear model (GLM) procedure in SAS 9.3 (SAS Institute Inc.). On the other hand, we detected the correlations between the angles and other important agronomic traits by Student’s t test at 5 and 1% levels of significance.
SNP marker analysis
Genomic DNA for genotyping was extracted from young leaf tissues by a modified cetyltrimethylammonium bromide method (Hanania et al. 2004). The Brassica 60K Illumina Infinium SNP array with 52,157 SNPs was used to genotype the YW-DH population and the parents. The SNP data were first clustered and called automatically using the Genome Studio software (Illumina Inc., San Diego, CA, USA). A procedure, called bi-filtering analysis was developed to improve the efficiency and accuracy of SNP array data analysis (Cai et al. 2015). Four kinds of probes in this DH population were filtered out consecutively, including SNPs with no polymorphism between the parents, non-parental genotypes, high missing data (> 20%) and distorted segregation (> 5%). Subsequently, all the filtered SNPs were re-genotyped according to their parents and further analysis was proceeded.
Construction of linkage map and QTL mapping
A 60K SNP array containing 52,157 probes for B. napus was used to genotype the YW-DH population. The genetic linkage map was generated using the MSTmap software package with the filtered probes (Wu et al. 2008). The map order was checked manually to ensure the optimal placement of the SNP loci (Clarke et al. 2016). The map distance was calculated using the Kosambi mapping function (Kosambi 1944). Detection of QTL for branch angle was performed in the DH population by inclusive composite interval mapping (ICIM) using the QTL IciMapping v4.1 Software (http://www.isbreeding.net/). The phenotype values of the 208 DH lines in each environment and combined experimental data from all environments were used for single-environment QTL analysis and QEI mapping, using the BIP and MET functionality of the software, respectively (Li et al. 2007a). Two methods of ICIM-ADD and ICIM-EPI in both of the functionalities were used for detecting the additive and epistatic QTL. The walking speed chosen for all QTL was 0.1 cM, with P = 0.001 in stepwise regression. The LOD threshold for detection of significant QTL was set by permutation analysis based on 1000 permutations. In addition, QTL detected in single-environment QTL analysis were termed as identified QTL, and QTL detected in QEI mapping were termed as combined QTL. QTL explained more than 10% of the phenotypic variance in at least two environments were considered as major QTL. It was assumed that two QTL with shared position(s) or with overlapping CI(s) were treated as the same QTL.
Results
Phenotypic performances of the parents and DH lines
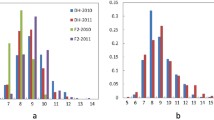
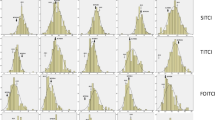
The alien introgression line ‘Y689’ showed the excellent plant architecture with branches being tightly compressed to the main inflorescence, while the branches of ‘Westar’ were not very tight (Fig. 1a). By using phloroglucinol–HCl histochemical staining for lignin, the stems of ‘Y689’ showed the thicker lignified out layer than ‘Westar’, and its pith was also slightly lignified (Fig. 1b). Descriptive statistics for branch angle of the two parents, as well as the DH lines across six environments were presented in Table 1. Significant differences in all of the environments were detected between parents. DH lines showed large variations in branch angle size, ranging from 13.22 to 66.90°, with an average ranging from 25.62 ± 4.63 to 37.51 ± 5.42 (Table 1 and Fig. 2). The frequency distributions of branch angle in the DH lines were shown in Fig. 3. Across 3 years, the DH lines exhibited the broadest variations for average branch angle in Weinan, and the narrowest ones in Xining, the region of spring rapeseed.

Plant architecture of two parents with different depositions of lignin in the stems. a ‘Y689’ has an ideal plant architecture with branches being tightly compressed to the main inflorescence. ‘Westar’ has the larger branch angle than ‘Y689’ (bars = 20 cm). b Fresh hand-cut stem sections of ‘Y689’ and ‘Westar’ stained with phloroglucinol-HCl. The stem of ‘Y689’ had the thicker stained out layer and the pith was also slightly stained, in comparison with ‘Westar’ (bars = 0.2 cm)

Broad phenotypic range of branch angle size in DH lines

Distribution of branch angle in the YW-DH population derived from the cross ‘Y689’ × ‘Westar’. 14WH, 14ND, 15WH, 15CD, 15ER and 16XN represent six environments with different colors. 14WH: Wuhan, 2014–2015; 14ND: Weinan, 2014–2015; 15WH: Wuhan, 2015–2016; 15CD: Chengdu, 2015–2016; 15ER: Ezhou, 2015–2016; 16XN: Xining, 2016. The triangles and rounds indicate ‘Y689’ and ‘Westar’, respectively (color figure online)
ANOVA revealed highly significant differences (P < 0.001) among genotypes, environments, and genotype by environment interactions, indicating that the branch angle was significantly influenced by both genetic and environmental factors (Supplementary Table S1). The broad-sense heritability (h 2) in the YW-DH population across six environments was calculated as 87.33%, demonstrating that despite the environmental effect, branch angle size for a given genotype was fairly stable. The Pearson’s correlation coefficients between the branch angle and other important agronomic traits including branch number (BN), plant height (PH), first branch height (FBH), culm diameter (CD) and acid detergent lignin (ADL) content were detected in Wuhan and Ezhou, in 2015–2016 growing season (Table 2, Supplementary Table S2). The PH was significantly positively correlated with branch angle, which had the highest coefficient (r = 0.37, 15ER), followed by FBH (r = 0.32, 15WH) and BN (r = 0.25, 15ER), while the ADL showed significantly negatively correlated with branch angle (r = −0.30, 15WH, and r = −0.29, 15ER).
High-density SNP map construction
A total of 21,978 SNPs from the array which showed polymorphisms between the parental lines ‘Y689’ and ‘Westar’ were filtered out for further analysis. The final linkage map was summarized in Supplementary Table S3. In present work, 3073 SNPs were used to construct a high-density linkage map with an average distance of 0.73 cM between adjacent markers (Fig. 4, Supplementary Fig. S1). All the SNPs were separated into 19 linkage groups, covering the 19 chromosomes of B. napus. Of these, 1889 (61.5%) SNPs were localized to the A subgenome with a total length of 1176.77 cM and average marker density of 0.62 cM. 1184 (38.5%) SNPs were mapped to the C subgenome with a total length of 1065.38 cM and average marker density of 0.90 cM. The A subgenome had more markers and the higher marker density, indicating that SNP markers on A subgenome were much more polymorphic than that in C subgenome. In addition, all the SNP markers were well distributed throughout the genome, although chromosome A02 exhibited lower marker density and the alignment to physical locations was not satisfied (Fig. 4).

Distribution of SNPs on the linkage map and the QTL information of branch angle trait in six environments. The SNP markers in each linkage group were aligned to their positions in the physical map (from the outermost circle to the second outer circle). The red blocks on the third circle represent the combined QTL identified by QEI mapping. The blue blocks on the fourth circle represent the QTL identified with the phenotype of the best linear unbiased prediction (BLUP) values over six environments. The black blocks on the fifth to the tenth (the innermost) circles represent the QTL identified by single-environment analysis in 16WH, 15ER, 15CD, 15WH, 14ND and 14WH environment, respectively. The ranges of the blocks represent the confidence interval of each QTL (color figure online)
QTL analysis for branch angle
In this study, the branch angles in each environment and the BLUP values across environments were used as a phenotype for the QTL detection. Using the BIP functionality for single-environment analysis, a total of 17 additive QTL were detected for branch angle (LOD threshold = 3.06), distributed on chromosomes A01 (3 QTL), A02 (1 QTL), A03 (3 QTL), A09 (2 QTL) and C03 (8 QTL) (Table 3 and Fig. 4). These identified QTL explained 5.10–21.73% of the phenotypic variance (mean PVE = 10.63%, LOD: 3.06–14.58). Six of these QTL were repeatedly detected in different experiments, and three of them were major QTL which explained more than 10% percentage phenotypic variance (PVE) in two experiments, including qBA.A03-2, qBA.C03-3 and qBA.C03-4. One of the major QTL, qBA.C03-3, explained up to 21.73% phenotypic variance (mean PVE = 14.82%, with LOD: 3.46–14.58), while qBA.A03-2 and qBA.C03-4 explained 10.21–13.21 and 14.04–17.21% phenotypic variance, respectively. Detailed information about the identified QTL was summarized in Table 3. In addition, no significant two-locus epistasis was found between any of the QTL under a default threshold of LOD = 5.0.
To verify the QTL identified in single-environment analysis and evaluate the potential QTL by environment interactions, QEI mapping was implemented using the MET functionality in QTL IciMapping 4.1 software. As a result, a total of ten combined QTL were identified (LOD threshold = 5.98), and seven of them were also detected by single-environment analysis, including two major QTL, qBA.A03-2 (corresponding to cqBA.A03-1) and qBA.C03-4 (corresponding to cqBA.C03-2). But three QTL (cqBA.A01-1, cqBA.A07-1 and cqBA.A09-2) were only detected by QEI mapping. Of the ten combined QTL, some showed strong QEI, such as cqBA.C03-1, whose LOD (A) value was 3.63 and LOD (A by E) was 9.78. Some showed weak QEI, such as cqBA.C03-3, with the LOD (A) and LOD (A by E) values of 8.85 and 2.32, respectively (Table 4 and Fig. 4).
We then compared the QTL detected in single-environment analysis and QEI mapping with those from other reports on branch angle in B. napus (Liu et al. 2016; Sun et al. 2016; Li et al. 2017). Among 105 SNPs significantly associated with branch angle, only 4 SNPs (3.8%) were located within the QTL regions of our study (Table 5, Supplementary Table S4). However, none of our major QTL was colocalized with other SNPs, indicating that they were of novel origin.
Candidate genes mining for branch angle
The proximity of candidate genes was inferred based on functional annotation of the A. thaliana genome and the physical positions of SNPs/candidate genes on reference-sequenced genome of B. napus (Chalhoub et al. 2014). We selected 27 genes from all QTL regions as candidate genes (Supplementary Table S5). Briefly, sixteen (59.3%) candidate genes were early auxin-responsive genes, including small auxin-up RNA (SAUR), auxin/indoleacetic acid (Aux/IAA), and gretchenhagen-3 (GH3) (Hagen and Guilfoyle 2002). Six genes (22.2%) were involved in auxin synthesis, auxin response, and auxin efflux/influx transportation. The remaining candidate genes (18.5%) were TPR2, TPR4 and YUCCA1 (Supplementary Table S5).
Four candidate genes were detected in major QTL regions, two of them were SAUR genes, including BnaC03g14890D and BnaC03g16420D, which were orthologues to Arabidopsis SAUR30 and SAUR55. The other two genes, BnaA03g10890D and BnaA03g11170D, were related to auxin biosynthesis and transport, and were orthologues to Arabidopsis NIT1 and D6PK, respectively (Supplementary Table S5).
Discussion
In this study, we developed a YW-DH population, and constructed a high-density linkage map with 3073 available SNPs, covering a length of 2242.14 cM and with an average marker interval of 0.73 cM (Supplementary Table S3). After alignment to the reference genome, this YW map showed fine collinearity except for the A02 and C02 (Fig. 4), due to high homology between the two chromosomes. Though the density of markers could not be fortified merely by increasing the number of markers because of co-segregating markers (Liu et al. 2013), our linkage map captured a large amount of recombination. To take advantage of the densely distributed 60K SNPs efficiently, a large-sized population of 208 lines were planted with a RCB design over six environments. Though enlarged population size (PS) could enhance the QTL detection power, the component of environment error would increase, because the blocks in RCB design were too large and the soil heterogeneity effect also increase. Even for a highly heritable trait, other more appropriate field designs should be recommended than RCB design with more than 200 entries, such as a commonly used incomplete block design of alpha lattice (Horn et al. 2015; Milner et al. 2016; Sakiroglu and Brummer 2017). Based on this high-density SNP map and large-sized YW-DH population, QTL for other agronomic traits might be identified in B. napus.
The phenotype of branch angle was measured in six environments, and 17 QTL were identified by single-environment analysis. Of these, 3 QTL with higher PVE were referred as major QTL (Table 3). A total of 10 QTL were identified by QEI mapping, and some of the QTL presented strong QEI effect. As a result, 7 QTL were detected both by single-environment analysis and QEI mapping. Compared with the QTL detected by single-environment analysis, QTL detected by QEI mapping might be better ones for the application of marker-assisted selection (MAS) in breeding programs, and their confidence intervals were possibly narrowed down. Taking cqBA.C03-3 as an example, this QTL was stably detected in two experimental environments and BULP analysis. Though its confidence interval (CI) (87.5–96.5, 87.5–97.5 and 87.5–93.5 cM) was slightly different in single-environment analysis, the calculated CI of 87.5–88.5 cM might be closer to the authentic position of QTL, because all data across environments were utilized to the model for QEI mapping (Tables 3, 4). It is worth mentioning that two major QTL qBA.C03-3 and qBA.C03-4, were distributed on positions 30 and 32 cM of chromosome C03 in single-environment analysis, while only qBA.C03-4 (the same position and CI as cqBA.C03-2) was detected in QEI mapping. This two QTL with very adjacent QTL positions in single-environment analysis and individually located in QEI mapping might be the same QTL, though further experimental proof should be provided. All the QTL, especially the major QTL, facilitated fine mapping and map-based cloning of the target gene. To our knowledge, this traditional QTL mapping approach was firstly used in dissecting the genetic control of branch angle trait in B. napus. The robust QTL indicated that traditional linkage mapping was still a powerful method in deciphering the genetic basis of complex traits.
By using the genome-wide association study method to unravel the genetic control of branch angle in B. napus, lots of loci (QTL) significantly associated with the branch angle trait were identified in several recent studies (Wang et al. 2016; Liu et al. 2016; Sun et al. 2016; Li et al. 2017). As the parental ‘Y689’ for our DH population was an intertribal introgressant with C. bursa-pastoris and contained the reorganized genome of B. napus (Chen et al. 2007a; Zhang et al. 2013), few loci were found to be colocalized with those of other studies (Table 5). The character of highly lignified stems from C. bursa-pastoris was expressed by the intertribal partial hybrid. Then the selection for its progenies of successive generations might be responsible for the compressed branches and wooden stems of the introgression ‘Y689’, as the negative correlation between the stem lignin content and branch angle was significant (Table 2, Supplementary Table S2). The alien chromosomal segments and those in the recipient genome might constitute the physical basis of the QTL detected. Further analysis should be proceeded by map-based cloning or other efficient strategy to uncover the genetic mechanisms controlling branch angle.
Phytohormones such as auxin, cytokinin, and strigolactone influenced the shoot branching by a complicated network (Wang et al. 2010). Three gene families were rapidly and transiently induced in response to auxin: the SAUR, Aux/IAA and GH3 family (Hagen and Guilfoyle 2002). SAURs were the largest family of early auxin-response genes, which played a key role in plant growth and development. For example, transgenic rice plants overexpressing the SAUR39 gene resulted in lower shoot and root growth, increased leaf and branch (tiller) angle compared with wild-type plants (Kant et al. 2009). Recently, SAUR10 was discovered to be repressed by Arabidopsis MADS-domain factor FRUITFULL (FUL) in stems and inflorescence branches. Influenced by auxin, brassinosteroids, light conditions and FUL, SAUR10 had an effect on branch angle (Bemer et al. 2017). In this study, two SAUR-like early auxin-response genes, BnaC03g14890D (SAUR30) and BnaC03g16420D (SAUR55) were considered as key candidate genes controlling branch angle in B. napus (Supplementary Table S5). The Aux/IAA proteins, together with ARF proteins, mediated transcriptional regulation by the form of homo- and hetero-oligomers (Chapman and Estelle 2009; Han et al. 2014). The Aux/IAA genes played a vital role in maintaining plant gravitropic setpoint angle (GSA) via the Aux/IAA-ARF-dependent auxin signaling (Roychoudhry et al. 2013). For instance, OsIAA4 (LOC_Os01g18360) was an Aux/IAA protein gene, and overexpressing OsIAA4 exhibited dwarfism and increased tiller angle in rice (Song and Xu 2013). In our study, three IAA homologous genes IAA17, IAA19, IAA29, and two ARF genes ARF4 and ARF16 were identified as candidate genes (Supplementary Table S5). GH3 family was also a member of early auxin-responsive genes. Several GH3 family genes were found as candidate genes in the present study, such as BnaA07g08630D and BnaC03g46000D, orthologues to Arabidopsis GH3.17 and GH3.10, respectively (Supplementary Table S5). From the two functionally characterized SAUR genes OsSAUR39 and OsSAUR45 in rice (Kant et al. 2009; Xu et al. 2017), and the newly reported SAUR gene SAUR10 in Arabidopsis (Bemer et al. 2017), we speculated that the candidate gene(s) also likely influenced auxin synthesis and transport, leading to auxin redistribution and thus regulating the branch angle in B. napus.
In conclusion, by using the excellent B. napus introgression line derived from the intertribal hybridization with C. bursa-pastoris as one parent for the development of DH population and high-density SNP map, several novel and major QTL for branch angle were detected, which provided the new insight into the genetic control of this important trait for the plant architecture. The genetic and developmental relationships between compact branching and stem lignification in this introgression remain to be clarified. Furthermore, together with the tight branching, the lignification-associated high resistance to S. sclerotiorum and lodging of the alien introgression made it an elite germplasm for breeding the varieties suitable for mechanical harvesting (Chen et al. 2007b; Cai et al. 2016).
Author contribution statement
ZL conceived the experiment. YS performed the research. YS, YY, and EX contributed to phenotypic measurements. YS, XG, and YX contributed to data analysis. YS and ZL wrote the manuscript. All authors reviewed and approved this submission.
References
Aranzana MJ, Kim S, Zhao K et al (2005) Genome-wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes. PLoS Genet 1:e60
Bemer M, van Mourik H, Muiño JM et al (2017) FRUITFULL controls SAUR10 expression and regulates Arabidopsis growth and architecture. J Exp Bot. doi:10.1093/jxb/erx184
Cai G, Yang Q, Yi B et al (2015) A bi-filtering method for processing single nucleotide polymorphism array data improves the quality of genetic map and accuracy of quantitative trait locus mapping in doubled haploid populations of polyploid Brassica napus. BMC Genom 16:409
Cai G, Yang Q, Chen H et al (2016) Genetic dissection of plant architecture and yield-related traits in Brassica napus. Sci Rep 6:21625
Chalhoub B, Denoeud F, Liu S et al (2014) Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345:950–953
Chapman EJ, Estelle M (2009) Mechanism of auxin-regulated gene expression in plants. Annu Rev Genet 43:265–285
Chen HF, Wang H, Li ZY (2007a) Production and genetic analysis of partial hybrids in intertribal crosses between Brassica species (B. rapa, B. napus) and Capsella bursa-pastoris. Plant Cell Rep 26:1791–1800
Chen X, Qi C, Pu H et al (2007b) Evaluation of lodging resistance in rapeseed (Brassica napus L.) and relationship between plant architecture and lodging resistance. Chin J Oil Crop Sci 29:54–57
Chen Y, Fan X, Song W et al (2012) Over-expression of OsPIN2 leads to increased tiller numbers, angle and shorter plant height through suppression of OsLAZY1. Plant Biotechnol J 10:139–149
Clarke WE, Higgins EE, Plieske J et al (2016) A high-density SNP genotyping array for Brassica napus and its ancestral diploid species based on optimised selection of single-locus markers in the allotetraploid genome. Theor Appl Genet 129:1887–1899
Dardick C, Callahan A, Horn R et al (2013) PpeTAC1 promotes the horizontal growth of branches in peach trees and is a member of a functionally conserved gene family found in diverse plants species. Plant J 75:618–630
Doerge RW (2002) Mapping and analysis of quantitative trait loci in experimental populations. Nat Rev Genet 3:43–52
Dong H, Zhao H, Xie W et al (2016) A novel tiller angle gene, TAC3, together with TAC1 and D2 largely determine the natural variation of tiller angle in rice cultivars. PLoS Genet 12:e1006412
Hagen G, Guilfoyle T (2002) Auxin-responsive gene expression: genes, promoters and regulatory factors. Plant Mol Biol 49:373–385
Han M, Park Y, Kim I et al (2014) Structural basis for the auxin-induced transcriptional regulation by Aux/IAA17. Proc Natl Acad Sci 111:18613–18618
Hanania U, Velcheva M, Sahar N, Perl A (2004) An improved method for isolating high-quality DNA from Vitis vinifera nuclei. Plant Mol Biol Rep 22:173–177
Horn F, Habekuß A, Stich B (2015) Linkage mapping of Barley yellow dwarf virus resistance in connected populations of maize. BMC Plant Biol 15:29
Jiao Y, Wang Y, Xue D et al (2010) Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat Genet 42:541–544
Jin J, Huang W, Gao J-P et al (2008) Genetic control of rice plant architecture under domestication. Nat Genet 40:1365–1369
Kant S, Bi Y-M, Zhu T, Rothstein SJ (2009) SAUR39, a small auxin-up RNA gene, acts as a negative regulator of auxin synthesis and transport in rice. Plant Physiol 151:691–701
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Ku L, Wei X, Zhang S et al (2011) Cloning and characterization of a putative TAC1 ortholog associated with leaf angle in maize (Zea mays L.). PLoS One 6:e20621
Li H, Ye G, Wang J (2007a) A modified algorithm for the improvement of composite interval mapping. Genetics 175:361–374
Li P, Wang Y, Qian Q et al (2007b) LAZY1 controls rice shoot gravitropism through regulating polar auxin transport. Cell Res 17:402–410
Li H, Zhang L, Hu J et al (2017) Genome-wide association mapping reveals the genetic control underlying branch angle in rapeseed (Brassica napus L.). Front Plant Sci. doi:10.3389/fpls.2017.01054
Liu L, Qu C, Wittkop B et al (2013) A high-density SNP map for accurate mapping of seed fibre QTL in Brassica napus L. PLoS One 8:e83052
Liu J, Wang W, Mei D et al (2016) Characterizing variation of branch angle and genome-wide association mapping in rapeseed (Brassica napus L.). Front Plant Sci 7:1–10
Mason AS, Higgins EE, Snowdon RJ et al (2017) A user guide to the Brassica 60K Illumina Infinium™ SNP genotyping array. Theor Appl Genet 130:621–633
Milner SG, Maccaferri M, Huang BE et al (2016) A multiparental cross population for mapping QTL for agronomic traits in durum wheat (Triticum turgidum ssp. durum). Plant Biotechnol J 14:735–748
Prakash S, Wu X, Bhat SR (2011) History, evolution, and domestication of Brassica crops. Plant Breed Rev 35:19–84
Qi T, Cao Y, Cao L et al (2015) Dissecting genetic architecture underlying seed traits in multiple environments. Genetics 199:61–71
Roychoudhry S, Del Bianco M, Kieffer M, Kepinski S (2013) Auxin controls gravitropic setpoint angle in higher plant lateral branches. Curr Biol 23:1497–1504
Sakiroglu M, Brummer EC (2017) Identification of loci controlling forage yield and nutritive value in diploid alfalfa using GBS-GWAS. Theor Appl Genet 130:261–268
Sang D, Chen D, Liu G et al (2014) Strigolactones regulate rice tiller angle by attenuating shoot gravitropism through inhibiting auxin biosynthesis. Proc Natl Acad Sci 111:11199–11204
Song Y, Xu ZF (2013) Ectopic overexpression of an AUXIN/INDOLE-3-ACETIC ACID (Aux/IAA) gene OsIAA4 in rice induces morphological changes and reduces responsiveness to auxin. Int J Mol Sci 14:13645–13656
Sun C, Wang B, Wang X et al (2016) Genome-wide association study dissecting the genetic architecture underlying the branch angle trait in rapeseed (Brassica napus L.). Sci Rep 6:33673
Tan L, Li X, Liu F et al (2008) Control of a key transition from prostrate to erect growth in rice domestication. Nat Genet 40:1360–1364
Wang Y, Li J (2008) Rice, rising. Nat Genet 40:1273–1275
Wang L, Mai Y-X, Zhang Y-C et al (2010) MicroRNA171c-targeted SCL6-II, SCL6-III, and SCL6-IV genes regulate shoot branching in Arabidopsis. Mol Plant 3:794–806
Wang N, Qian W, Suppanz I et al (2011) Flowering time variation in oilseed rape (Brassica napus L.) is associated with allelic variation in the FRIGIDA homologue BnaA.FRI.a. J Exp Bot 62:5641–5658
Wang H, Cheng H, Wang W et al (2016) Identification of BnaYUCCA6 as a candidate gene for branch angle in Brassica napus by QTL-seq. Sci Rep 6:38493
Wu Y, Bhat PR, Close TJ, Lonardi S (2008) Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph. PLoS Genet 4:e1000212
Xu M, Zhu L, Shou H, Wu P (2005) A PIN1 family gene, OsPIN1, involved in auxin-dependent adventitious root emergence and tillering in rice. Plant Cell Physiol 46:1674–1681
Xu YX, Xiao MZ, Liu Y et al (2017) The small auxin-up RNA OsSAUR45 affects auxin synthesis and transport in rice. Plant Mol Biol. doi:10.1007/s11103-017-0595-7
Yoshihara T, Iino M (2007) Identification of the gravitropism-related rice gene LAZY1 and elucidation of LAZY1-dependent and -independent gravity signaling pathways. Plant Cell Physiol 48:678–688
Yu B, Lin Z, Li H et al (2007) TAC1, a major quantitative trait locus controlling tiller angle in rice. Plant J 52:891–898
Zhang X, Ge X, Shao Y et al (2013) Genomic change, retrotransposon mobilization and extensive cytosine methylation alteration in Brassica napus introgressions from two intertribal hybridizations. PLoS One 8:e56346
Zhang L, Gezan SA, Eduardo Vallejos C et al (2017a) Development of a QTL-environment-based predictive model for node addition rate in common bean. Theor Appl Genet 130:1065–1079
Zhang N, Fan X, Cui F et al (2017b) Characterization of the temporal and spatial expression of wheat (Triticum aestivum L.) plant height at the QTL level and their influence on yield-related traits. Theor Appl Genet 130:1235–1252
Zhao J, Buchwaldt L, Rimmer SR et al (2009) Patterns of differential gene expression in Brassica napus cultivars infected with Sclerotinia sclerotiorum. Mol Plant Pathol 10:635–649
Zhao H, Huai Z, Xiao Y et al (2014) Natural variation and genetic analysis of the tiller angle gene MsTAC1 in Miscanthus sinensis. Planta 240:161–175
Acknowledgements
We are grateful for Drs. Liangcai Jiang, Cheng Cui from Crop Research Institute, Sichuan Academy of Agricultural Sciences, and Profs. Dianrong Li, Yonghong Li, Hybrid Rapeseed Research Center of Shanxi Province, for arranging the field experiments in Chengdu and Weinan. The study was supported by National Sci-Tech Support Plan (2013BAD01B03), Hubei Provincial Sci-Tech Support Plan (201604051006045), Science and Technology Special Project of Guizhou Academy of Agricultural Sciences (no. [2017] 08).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethical standards
The authors declare that the experiments complied with current laws of China.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Benjamin Stich.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Shen, Y., Yang, Y., Xu, E. et al. Novel and major QTL for branch angle detected by using DH population from an exotic introgression in rapeseed (Brassica napus L.). Theor Appl Genet 131, 67–78 (2018). https://doi.org/10.1007/s00122-017-2986-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-017-2986-1