
Abstract
Main stem height (MSH) and the first lateral branch length (LBL) of peanut (Arachis hypogaea L.) are important traits affecting plant shape and yield. Identification of quantitative trait loci (QTLs) related to these two traits, the prediction and cloning of candidate genes, and identification of plant height-related molecular markers are the basis for analysis of the molecular genetic mechanism of plant shape in peanut. In this study, a population of 151 recombinant inbred lines from a single seed, derived from a cross between variety 79266 (P1) and its variant progeny D893 (P2), was used to construct a peanut genetic map. The map consisted of 231 simple sequence repeat markers in 23 linkage groups, had a total length of 905.18 cM with average and minimum marker intervals of 3.92 and 0.1 cM, respectively. There were 11 and 16 QTLs detected in six environments for MSH and LBH with 6.26–22.53 and 5.89–21.63% phenotypic variation explained (PVE), respectively. Seven QTLs were detected in two or more environments: 3 QTLs for MSH (including Qmsh-14-3) with 7.66–22.53% PVE, and 4 QTLs for LBL (including Qllb-11-1) with 6.12–21.63% PVE. Qmsh-14-3 was steadily detected in five environments, localized between two markers, ARS376 and SEQ4G02, exhibited a genetic distance of 0.59–2.59 and 4.11–6.11 cM from the two markers, respectively. Qllb-11-1 was steadily detected in five environments, localized between two markers, ARS203 and AHS1413, exhibited a genetic distance of 1.06–3.06 and 0.23–2.23 cM from the two markers, respectively. There were 220 germplasm accessions used to detect the relationship between genotype and phenotypic values of traits at the marker loci ARS376 and SEQ4G02, average values of MSH and LBL were significantly higher for germplasm with the P1 compared to the P2 genotype. Determination of the marker loci ARS203 and AHS1413 indicated that average values of LBL were greater in germplasm with the P1 than the P2 genotype. The results provide references for fine mapping of QTLs for MSH and LBL, as well as breeding optimum plant-types in peanut.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Peanut (Arachis hypogaea L.), also known as groundnut, is a grain legume and oilseed crop widely cultivated in tropical and subtropical regions (annual production of ~46 million tonnes) (David et al. 2016). Main stem height (MSH) and the first lateral branch length (LBL) of peanut are important agricultural traits affecting plant shape and yield. Moderate plant height, well-developed branches and appropriate bunch density are ideal goals for high-yield peanut breeding. Lan et al. (2011) used the gray relational analysis grade method to study the correlation between peanut yield and main agricultural traits, and found that MSH and LBL were some of the leading factors affecting peanut yield. Correlation and cluster analyses of main agricultural traits in 18 varieties showed that MSH and LBL were highly correlated with peanut yield (Yin et al. 2006). The identification of quantitative trait loci (QTLs) related to MSH and LBL, the prediction and cloning of candidate genes, as well as the identification of molecular markers related to plant height are the research basis for analyzing the molecular genetic mechanism of plant shape, and are important for optimum plant-type breeding in peanut.
The construction of a genetic map is an important basis for QTL identification. Cultivated peanut is an allotetraploid (2n = 4X = 40, AABB) with closely related subgenomes of a total size of ~2.7 Gb (David et al. 2016). The peanut subgenomes are closely related (Moretzsohn et al. 2013; Nielen et al. 2012), which to some extent increase the difficulties of molecular marker genetic map construction and molecular marker development. Since Halward et al. (1993) crossed two diploid wild types of Arachis stenosperma and Arachis cardenasii, and constructed the first genetic linkage map of peanut using an F2 population and amplified fragment length polymorphism markers, studies on peanut genetic linkage maps have made great progress. Especially in recent years, due to the maturity of simple sequence repeat (SSR) marker technology, the construction of genetic linkage maps in cultivated peanut has rapidly developed (Hong et al. 2009, 2010; Li et al. 2015; Qin et al. 2012; Varshney et al. 2009; Wang et al. 2013, 2015). Shirasawa et al. (2013) used 15 genetic linkage maps to integrate and construct a genetic map consisting of 3693 SSR markers in 20 lineage groups, which is currently the peanut integrated genetic map with the highest SSR marker density. David et al. (2016) completed the genome sequencing of diploid ancestors Arachis duranensis and Arachis ipaensis of cultivated varieties, and obtained sequence coverage of 96% of the genomes of the diploid wild species (the website for inquiry of genome sequence information is http://peanutbase.org ). The genome sequence information of the diploid wild species provided important references for distinguishing the two subgenomes (A and B) in cultivated varieties, the development of molecular markers, increasing the density of the molecular genetic linkage map and searching for the physical identification of functional genes in chromosomes.
MSH and LBL in peanut are quantitative traits. Recently, several QTLs related to the above traits were identified. Shirasawa et al. (2012) identified 3 QTLs for MSH with 4.8, 19.2 and 15.7% phenotypic variation explained (PVE) respectively, and detected 2 QTLs for LBL with 21.1 and 14.2% PVE, respectively, on the genetic map of cultivated peanut. Cheng et al. (2015) constructed a genetic map consisting of 234 markers using an F2 population from crossing between cultivated varieties, and identified six stable QTLs for MSH in two environments. Therein, qMHA091.1 and qMHA062.1 with highest PVE of 9.43 and 8.95%, respectively, were separately mapped in the A09 and A06 linkage groups on the integrated map constructed by Shirasawa et al. (2013). Li et al. (2015) identified 3 QTLs for MSH with 8.31, 6.12 and 8.90% PVE, respectively.
In this study, a recombinant inbred lines (RILs) population, constructed from the generations of a cross between the cultivated variety 79266, widely cultivated by farmers in China, and its dwarf mutant line D893, and 220 germplasm accessions of peanut were used to identify QTLs and markers related to MSH and LBL in peanut. The results will be used to identify more QTLs related to plant height and shape, develop more molecular markers and provide references for genetic improvement in future breeding in peanut.
Materials and methods
Materials and field planting
The female parent, 79266 (P1), was a peanut variety bred by the Shandong Peanut Research Institute with characteristics of early maturity and high yield. The male parent, D893 (P2), had characteristics of dwarf and super-large pods, and was developed by our research laboratory. It was derived from the variant progeny by introduction of Sophora japonica Linn DNA into 79266 after pollination via the pollen tube pathway (Zhou 1992). There were significant differences in MSH and LBL between parents (Table 1). The cross 79266 × D893 was performed at Tai’an, Shandong in 2005, false hybrids were removed at F1 generation, single-seed descent method was applied from F2 generation to F6. The F6:10 RIL population consisting of 151 stable families was obtained after successive inbreeding.
A randomized block design with two replications were applied in the field experiment, with planting conducted under six environmental conditions at Tai’an/Shandong of China from 2011 to 2015 and at Ji’ning/Shandong in 2013 (the environments were named E1–E6, respectively). Each material was planted in a row with length of 280 cM and row spacing of 40 cM, and each row had 14 holes. Each hole was reduced to a single plant after germination, and management was the same as for field production.
There were 220 germplasm accessions, collected from China and abroad, planted in in China at Tai’an,Shandong in 2013 and 2014 as a validation population. These were applied to the validation of linkage markers for identified QTLs.
Trait measurement and data analysis
The plants on edges of each row were removed during harvest. Five single plants in succession were selected from 12 plants in the middle and used to measure MSH and LBL according to the standard procedures previously described by Jiang et al. (2006).
SPSS 20.0 (IBM Company, Armonk, NY, USA) was used for analysis of variance, genetic variation coefficient analysis and descriptive statistics for the data.
DNA extraction and SSR genotyping
Genomic DNA was extracted from young leaves collected from each line using a modified cetyltrimethyl ammonium bromide method (Paterson et al. 1993). The quality and quantity of DNA were evaluated using a Nano Drop 2000 spectrophotometer (Nano Drop Technologies, Wilmington, DE, USA).
PCR reactions followed the protocol of Chen et al. (2008). The PCR products were separated through 8% non-denaturing polyacrylamide gel electrophoresis under constant voltage of 150 V for 2 h, silver stained and photographed for preservation after film development.
A total of 4438 SSR primer pairs from different resources were used to screen for polymorphism between the two parental genotypes. The SSR primers were derived from the following: Bertioli et al. (2009); Budiman et al. 2006; Cuc et al. (2008); Ferguson et al. (2004); Gimenes et al. (2007); Guo et al. 2008; He et al. (2003); Hopkins et al. (1999); Koilkonda et al. 2012; Moretzsohn et al. (2005); Nagy et al. (2010); Naito et al. 2008; Proite et al. (2007); Shirasawa et al. (2012); and Wang et al. 2007. All primers were synthesized by Shanghai Sangon Biotech Co. Ltd. The Taq polymerase, dNTP and other reagents for PCR reaction were purchased from Beijing TransGen Biotech Co. Ltd. Acrylamide, ammonium persulfate, TEMED and other reagents for gel electrophoresis were purchased from Taian Keshang Biotech Co. Ltd.
Construction of genetic linkage map and QTL analysis
The results of the experiment banding pattern were digitized. In the mapping population, the families with the same banding pattern as the female parent were recorded as ‘a’, those with the same banding pattern as the male parent were recorded as ‘b’, those with a hybridization banding pattern were recorded as ‘h’ and those without or with unclear banding pattern were recorded as ‘−’. Join Map 4.0 (Van 2006) was used to construct the genetic linkage map. QTL IciMapping V4.0 was used to identify QTLs by Inclusive Composite Interval Mapping (ICIM) (Li et al. 2008).
Validation of QTL linkage markers in the germplasm population
The MSH and LBL of 220 germplasm accessions were measured during the harvest period, using the same methods of measurement and statistics as for the RIL population. The detected QTL linkage markers were validated in the germplasm population and statistical analysis of banding patterns conducted. The individuals with different banding patterns were compared and analyzed to validate the detected markers.
Results
Characterization of phenotypic distribution of parents and RIL population
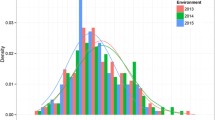
The results for the six planting environments showed significant differences in MSH and LBL between the two parents. There was a greater variation range of characteristics in the RIL population, showing transgressive segregation. The standard deviations (SDs) and coefficient of variations (CVs) indicated abundant population variation, with all CVs >20% (Table 1). The population distribution histogram (Fig. 1) showed continuous variation with a normal distribution for MSH and LBL, which are typical characteristics of quantitative traits. Therefore, this population was suitable for the construction of the genetic linkage map and detection of QTLs.

Distribution of the main stem height and length of lateral branch in the RIL population. The horizontal axis shows the values of main stem height (cM) or lateral branch length (cM): E1, E2, E3, E4 and E5 are Tai’an 2011, 2012, 2013, 2014 and 2015, respectively; and E6 is Ji’ning 2013
Construction of the genetic linkage map
Of the 4438 SSR primer pairs, there were 252 detected polymorphic DNA fragments between the parents 79266 and D893. These 252 primer pairs were used to genotype the 151 RILs. Using Join Map 4.0 with the minimum logarithm of odds (LOD) of 3.0, a genetic linkage map for cultivated peanut was constructed consisting of 231 markers in 23 linkage groups. The map had a total length of 905.18 cM with an average marker interval of 3.92 cM and minimum marker interval of 0.1 cM. The average length of each linkage group was 39.56 cM with 2–63 markers (Table 2).
Identification of QTLs for main stem height and lateral branch length in peanut
Based on the constructed linkage map, combined with the phenotypic data of six environments, QTL identification of MSH and LBL in peanut was conducted using QTL IciMapping V4.0 software and the ICIM method. A total of 27 QTL sites were detected using LOD ≥2.5.
A total of 11 QTLs for MSH were detected in six environments (Table 3; Fig. 2) and distributed on six linkage groups, with PVE of 6.26–22.53% and the additive effects of 3.48–5.29. Of these, 8 QTLs were detected in only one environment. Two QTLs, Qmsh-14-2 and Qmsh-21-1, were detected in two environments. The QTL Qmsh-14-3 was detected in all environments except for E5 and showed positive additive effects, with PVE >10%. The largest PVE and additive effect values of Qmsh-14-3 were 22.53 and 6.27, respectively. Thus, Qmsh-14-3 was considered a stable and main-effect QTL site, being insensitive to environment, for MSH in peanut.

Peanut genetic linkage map and QTLs for the main stem height and length of lateral branch detected in the RILs E1, E2, E3, E4 and E5 are Tai’an 2011, 2012, 2013, 2014 and 2015, respectively; and E6 is Ji’ning 2013
A total of 16 QTLs for LBL were detected in six environments (Table 4; Fig. 2) and distributed on eight linkage groups, with PVE of 5.89–21.63% and additive effects of −8.71 to 12.23. Of these, 12 QTLs were detected in only one environment, Qllb-7-3 and Qllb-14-1 were detected in two environments, and Qllb-14-4 was detected in three environments. Qllb-11-1 was detected in all environments except for E1, with PVEs of 12.18–15.05% and additive effect of 5.24–12.17. Qllb-11-1 was considered a stable and main-effect QTL for lateral branch length in peanut.
Qmsh-14-3 and Qllb-14-4 were simultaneously detected in three environments (E1, E3 and E6), with map distances of 2, 1 and 1 cM, respectively, indicating that these two sites might be pleiotropic.
Prediction of corresponding physical map positions for marker intervals of QTLs Qmsh-14-3 and Qllb-11-1.
In this study, the located site Qmsh-14-3 related to MSH was steadily detected in five environments, comparing with the statistics on QTLs for MSH of Shirasawa et al. (2012) and the Legume Information System (LIS, http://legumeinfo.org/cmap/feature_search). The left marker ARS376 was the same as qML-12-1, which was located by Zhang (2011). The flanking markers of Qmsh-14-3 were ARS376 (27.4) and SEQ4G02 (34.1). ARS376 did not occur in the integration map (Shirasawa et al. 2013), but GM849 outside ARS376 of the map constructed in this study occurred in the integration map. Therefore, the marker regions at both ends of Qmsh-14-3, ARS376 (27.4) and SEQ4G02 (34.1), were detected in the regions of GM849 (74.92)–SEQ4G02 (71.98) in A09 linkage group on the integration map (Shirasawa et al. 2013). The DNA sequence corresponding to marker ARS376 (He et al. 2003) was Aradu.A09: 104,573,697–104,577, 899 bp in the genome physical map of A. duranensis (http://peanutbase.org ). The DNA sequence corresponding to marker SEQ4G02 (Ferguson et al. 2004) was Aradu.A09: 111,380,567–111, 444,791 bp in the genome physical map of A. duranensis. It was concluded that the physical map position corresponding to the marker region of Qmsh-14-3 was Aradu.A09: 104,573,697–111,444,791 bp with DNA length of 6,871,094 bp. The Qllb-14-4 related to LBL, which was steadily detected in three environments, was also detected in this region. The study provides a reference sequence for the fine mapping of Qmsh-14-3 and Qllb-14-4.
In this study, the located Qllb-11-1 for LBL was steadily detected in five environments, and was considered a novel site by comparison with the statistics on QTLs for LBL from previous research (Huang et al. 2015; Liu et al. 2013) and LIS (http://legumeinfo.org/cmap/feature_search). The flanking markers of Qllb-11-1 were ARS203 (1.9) and AHS1413 (5.2). The DNA sequence, corresponding to marker ARS203 (He et al. 2003), was Aradu.A06: 110,497,201–110,498,111 bp in the genome physical map of A. duranensis; and that for marker AHS1413 (Koilkonda et al. 2012) was Aradu.A06:110,540,970–110,541,873 bp. It was concluded that the physical map position corresponding to the marker region of Qllb-11-1 was Aradu.A06: 110,497,201–110,541,873 bp with DNA length of 44,672 bp, which provided a reference sequence for the fine mapping of Qllb-11-1 and prediction of functional genes.
Analysis of Qmsh-14-3 and Qllb-11-1 in the germplasm population
There were 220 germplasm accessions of peanut used for the phenotypic value investigation of MSH and LBL in 2013 and 2014. Phenotypic frequency showed continuous variation and an approximately normal distribution (Fig. 3).

Phenotypic variation of main stem height and lateral branch length in germplasm population. The horizontal axis shows the values of main stem height (cM) or lateral branch length (cM)
The linkage markers of Qmsh-14-3 for MSH, ARS376 and SEQ4G02, were applied for genotypic testing in the germplasm population (Fig. 4). Correlation analysis was conducted with the genotypic data and the phenotypic variation in 2013 and 2014 (Table 5).

The PCR amplification profiles of marker ARS376 (left) and SEQ4G02 (right) primers in the germplasm population M, DL1000 DNA marker; P1, 79266 banding pattern; P2, 04D893 banding pattern; 157–163, the number of accessions in the germplasm population
For marker ARS376, there were 28 accessions with the P1 genotype, representing 12.7% of the population, with MSH averages of 70.16 cM (2013) and 62.85 cM (2014). There were 190 accessions with the P2 genotype, representing 86.4% of the population, with MSH averages of 67.39 cM (2013) and 56.94 cM (2014). The averages of MSH in accessions with the P1 genotype were 4.11% (2013) and 10.04% (2014) higher than those with the P2 genotype. For marker SEQ4G02, there were 86 accessions with the P1 genotype, accounting for 39.1% of the population, with MSH averages of 68.26 cM (2013) and 58.91 cM (2014). Meanwhile, 130 accessions with the P2 genotype accounted for 59.1% of the population, with MSH averages of 67.41 cM (2013) and 56.94 cM (2014). The averages of MSH in accessions with P1 genotype were 1.26% (2013) and 3.46% (2014) higher than those with the P2 genotype.
The main-effect QTL, Qllb-14-4 for LBL was simultaneously detected in the region of ARS376 and SEQ4G02. The averages of LBL in accessions with the P1 genotype for marker ARS376 were 8.67% (2013) and 9.03% (2014) higher than those with the P2 genotype. The averages of LBL in accessions with the P1 genotype for marker SEQ4G02 were 4.02% (2013) and 6.57% (2014) higher than those with the P2 genotype (Table 5). This indicated that ARS376 had stronger linkage with Qmsh-14-3 and Qllb-14-4 than SEQ4G02.
The linkage markers ARS203 and AHS1413 of the main-effect QTL site Qllb-11-1 for LBL were applied for genotypic testing in the germplasm population (Fig. 5). Correlation analysis was conducted with the genotypic data and the phenotypic variation in 2013 and 2014 (Table 5). For marker ARS203, 31 accessions with the P1 genotype accounted for 14.1% of the population, with LBL averages of 77.10 cM (2013) and 68.75 cM (2014). There were 179 accessions with the P2 genotype, accounting for 81.4% of the population, with LBL averages of 72.92 cM (2013) and 64.60 cM (2014). The averages of LBL in accessions with the P1 genotype were 5.73% (2013) and 6.42% (2014) higher than those with the P2 genotype. For marker AHS1413, 33 accessions with the P1 genotype accounted for 15% of the population, with LBL averages of 77.3 cM (2013) and 68.77 cM (2014). Meanwhile, there were 186 accessions with the P2 genotype, accounting for 84.5% of the population, with LBL averages of 72.82 cM (2013) and 64.47 cM (2014). The averages of LBL in accessions with the P1 genotype were 6.15% (2013) and 6.67% (2014) higher than those with the P2 genotype (Table 5). In the germplasm population, ARS203 and AHS1413 showed close linkage, and only four accessions showed recombination between the two markers, accounting for 1.81% of the population.

The PCR amplification profiles of marker ARS203 (left) and AHS1413 (right) primers in the germplasm population M, DL1000 DNA marker; P1, 79266 banding pattern; P2, 04D893 banding pattern; 81–88 and 173–179, the number of accessions in the germplasm population
Discussion
Polymorphism analysis of markers between parents
Most previous peanut mapping populations were generated from crosses between two parents, such as between two wild species, a wild species and a cultivar, or two cultivated varieties with quite different genetic backgrounds. Abundant allelic differences were easily generated as a result of the distant phylogenetic relationships. In the present study, the RIL population derived from the cross between 79266 and D893 was applied for genetic linkage map construction. Because of the close phylogenetic relationship between the two parents, the polymorphic rate of markers between parents was relatively low and accounted for 5.67% of the 4438 SSRs in the study, which was lower than that in general mapping populations. However, the significant differences in phenotypic values of plant shape and pod traits between the parents were convenient for targeted research on the QTLs for traits.
Comparison with integration map
Of the 231 SSR markers on the genetic map constructed in this study, 205 markers were the same as for the integrated consensus map developed by Shirasawa et al. (2013). There were 149 markers of the linkage map identified as having corresponding markers in the 20 chromosomes of the integrated consensus map (Fig. 6), with the corresponding rate of 71.2%. In addition, the 3693 SSR markers on the integration map were used to detect the DNA of two parents in this experiment, and only 205 markers showed polymorphism. The constructed genetic map covered only 16.81% of the integration map, indicating that changes in genome DNA occurred in local regions of the chromosome after the exogenous DNA was introduced into the recipient cells. The mechanism of the genetic change in multiple chromosomes of the mutant line D893, resulting from the exogenous DNA introduction, is not yet clear and requires more research.

The alignment markers of the linkage map (LG01–LG21) in this study and the integrated consensus map (A01–A10 and B01–B10) developed by Shirasawa et al. (2013)
Analysis of multiple-effect QTLs
Clustering of QTLs for two traits (MSH and LBL) were mapped in the same region of linkage groups (Fig. 2). Qmsh-14-3 related to MSH and Qllb-14-4 controlling LBL were steadily detected in the region of ARS376-SEQ4G02 of LG14. It was considered that there were multiple-effect QTLs between markers ARS376 (27.4) and SEQ4G02 (34.1). Simultaneously, multiple-effect QTLs for the above two traits were also detected in LG10, LG13, LG14 and LG21, respectively. These results support the attribution of the correlation of MSH and LBL to the effect of pleiotropy or to the very close linkage of genes.
Validation of molecular markers in peanut germplasm population
The detection result of identified markers ARS376 and SEQ4G02 in 220 accessions indicated that, the averages of MSH and LBL were significantly higher in varieties with genotype 79266 than D893. In the markers ARS203 and AHS1413 on both sides of the QTL Qllb-11-1 related to LBL, the average LBL was significantly higher in varieties with genotype 79266 than D893. The above four markers had correlations with the corresponding traits; however, analysis showed that the differences were not significant (P > 0.05). The possible reason was that both MSH and LBL were quantitative traits and controlled by multiple genes. In the RIL population of this experiment, the phylogenetic relationship between two parents was relatively close, so with the exception of the detected site in the experiment, the other genetic loci had high similarity. In the germplasm population with complex genetic background, there may be other main-effect genetic loci with effects on the phenotypic values of MSH and LBL.
Conclusions
A genetic map of cultivated peanut was constructed, consisting of 231 markers with total length of 905.18 cM and average marker interval of 3.92 cM. A total of 27 QTLs were detected in six environments: 11 QTLs were related to MSH and 16 QTLs were related to LBL, with PVE of 5.89–22.53%. A total of 7 QTLs were detected in two or more environments, including 3 QTLs for MSH and 4 QTLs for LBL. Two main-effect QTLs with stable heredity and environment insensitivity, Qmsh-14-3/Qllb-14-4 and Qllb-11-1, were identified. The physical map positions corresponding to the marker regions of Qmsh-14-3/Qllb-14-4 and Qllb-11-1 were predicted using the genome physical map of A. duranensis. The effects of flanking markers of main-effect QTLs, Qmsh-14-3/Qllb-14-4 and Qllb-11-1, on MSH and LBL were validated in the germplasm population of peanut. The results provide references for fine mapping of QTLs for MSH and LBL, as well as breeding optimum plant-types in peanut.
Abbreviations
- QTL:
-
Quantitative trait locus/loci
- RIL:
-
Recombinant inbred line
- SSR:
-
Simple sequence repeat
- MSH:
-
Main stem height
- LBL:
-
The first lateral branch length
References
Bertioli DJ, Moretzsohn MC, Madsen LH, Sandal N, Leal-Bertioli S, Guimarães PM, Hougaard BK, Fredslund J, Schauser L, Nielsen AM, Sato S, Tabata S, Cannon S, Stougaard J (2009) An analysis of synteny of Arachis with Lotus and Medicago sheds new light on the structure, stability and evolution of legume genomes. BMC Genom 10(1):45
Budiman MA, Jones JIT, Citek RW, Warek U, Bedell JA, Knapp SJ (2006) Methylation-filtered and shotgun genomic sequences for diploid and tetraploid peanut taxa. GenBank Available at http://www.ncbi.nlm.nih.gov
Chen BY, Jiang HF, Liao BS, Ren XP, Huang JQ, Lei Y, Wang SY (2008) Genetic diversity analysis of Arachis gerplasm by SSR. J Trop Subtrop Bot 16(4):296–303
Cheng LQ, Tang M, Ren XP, Huang L, Chen WG, Li ZD, Zhou XJ, Chen YN, Liao BS, Jiang HF (2015) Construction of genetic map and QTL analysis for main stem height and total branch number in peanut (Arachis hypogaea L.). Acta Agronomica Sinica 41(6):979–987
Cuc LM, Mace ES, Crouch JH, Quang VD, Long TD, Varshney RK (2008) Isolation and characterization of novel microsatellite markers and their application for diversity assessment in cultivated groundnut (Arachis hypogaea). BMC Plant Biol 8(1):1
David JB, Steven BC, Lutz F, Huang GD, Andrew DF, Ethalinda KSC, Liu X, Gao DY, Josh C, Sudhansu D, Ren LH, Márcio CM, Kenta S, Huang W, Bruna V, Brian A, Chu Y, Chad EN, Pooja U, Ana CGA, Alexander K, Kyung DK, Mark DB, Rajeev KV, Wang XJ, Zhang XY, Noelle B, Patrícia MG, Sachiko I, Guo BZ, Liao BS, Thomas HS, Robert JS, Brian ES, Soraya CML-B, Xu X, Scott AJ, Richard M, Peggy OA (2016) The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat Genet. doi:10.1038/ng.3517
Ferguson ME, Burow MD, Schultze SR, Bramel PJ, Paterson AH, Kresovich S, Mitchell S (2004) Microsatellite identification and characterization in peanut (A. hypogaea L.). Theor Appl Genet 108:1064–1070
Gimenes MA, Hosino AA, Barbosa AV, Palmieri DA, Lopes CR (2007) Characterization and transferability of microsatellite markers of cultivated peanut (Arachis hypogaea). BMC Plant Biol 7:9
Guo BZ, Chen X, Dang P, Scully BT, Liang X, Holbrook CC, Yu J, Culbreath AK (2008) Peanut gene expression profiling in developing seeds at different reproduction stages during Aspergillus parasiticus infection. BMC Dev Biol 8(1):12
Halward T, Stalker HT, Kochert G (1993) Development of an RFLP linkage map in diploid peanut species. Theor Appl Genet 87:379–384
He G, Meng R, Newman M, Gao G, Pittman RN, Prakash CS (2003) Microsatellites as DNA markers in cultivated peanut (Arachis hypogaea L.). BMC Plant Biol 3:3
Hong YB, Liang XQ, Chen XP, Liu HY, Zhou GY, Li SX, Wen SJ (2009) Construction of genetic linkage map in peanut (Arachis hypogaea L.) Cultivars. Acta Agronomica Sinica 35(3):395–402
Hong YB, Chen XP, Liang XQ, Liu HY, Zhou GY, Li SX, Guo B (2010) A SSR based composite genetic linkage map for the cultivated peanut (Arachis hypogaea L.) genome. BMC Plant Biol 10:17
Hopkins MS, Casa AM, Wang T, Mitchell SE, Dean RE, Kochert GD, Kresovich S (1999) Discovery and characterization of polymorphic simple sequence repeats (SSRs) in peanut. Crop Sci 39(4):1243–1247
Huang L, He HY, Chen WG, Ren XP, Chen YN, Zhou XJ, Xia YL, Wang XL, Jiang XG, Liao BS, Jiang HF (2015) Quantitative trait locus analysis of agronomic and quality-related traits in cultivated peanut (Arachis hypogaea L.). Theor Appl Genet 128(6):1103–1115
Jiang HF, Duan N, Ren XP (2006) Descriptors and data standard for peanut (Arachis spp.). China Agriculture Press, Beijing
Koilkonda P, Sato S, Tabata S, Shirasawa K, Hirakawa H, Sakai H, Sasamoto S, Watanabe A, Wada T, Kishida Y, Tsuruoka H, Fujishiro T, Yamada M, Kohara M, Suzuki S, Hasegawa M, Kiyoshima H, Isobe S (2012) Large-scale development of expressed sequence tag-derived simple sequence repeat markers and diversity analysis in Arachis spp. Mol Breed 30(1):125–138
Lan XL, Tang ZX, Xu R (2011) Analysis of gray gorrelation between yield and major agronomic traits of peanut in Fujian Province. Acta Agric Jiangxi. 23(8):61–63
Li H, Ribaut J, Li Z, Wang J (2008) Inclusive composite interval mapping (ICIM) for digenic epistasis of quantitative traits in biparental populations. Theor Appl Genet 116(2):243–260
Li QB, Yang CY, Huang L, Zhou XJ, Jiang HF, Ren XP, Wang HM, Chen YN, Lei Y, Yan LY, Liao BS (2015) Genetic linkage map and QTL analysis of resistance to Aspergillus flavus in Arachis hypogaea L. Chin J Oil Crop Sci 37(5):596–604
Liu H, Zhang XY, Han SY, Yan M, Xu J, Dong WZ, Sun ZQ (2013) Inheritance analysis and QTL mapping of main stem height and lateral branch length in peanut (Arachis hypogaea L.). Chin J Oil Crop Sci 35(5):508–514
Moretzsohn MC, Leoi L, Proite K, Guimares PM, Leal-Bertioli SC, Gimenes MA, Martins WS, Valls JFM, Grattapaglia D, Bertioli DJ (2005) A microsatellite based, gene-rich linkage map for the AA genome of Arachis (Fabaceae). Theor Appl Genet 111(6):1060–1071
Moretzsohn MC, Gouvea EG, Inglis PW, Leal-Bertioli SC, Valls JF, Bertioli DJ (2013) A study of the relationships of cultivated peanut (Arachis hypogaea) and its most closely related wild species using intron sequences and microsatellite markers. Ann Bot 111(1):113–126
Nagy ED, Chu Y, Guo Y, Khanal S, Tang S, Lee Y, Dong W, Timper P, Taylor C, Ozias-Akins P, Holbrook C, Beilinson V, Nelson N, Stalker T, Knapp S (2010) Recombination is suppressed in an alien introgression in peanut harbouring Rma, a dominant root-knot nematode resistance gene. Mol Breed 26(2):357–370
Naito Y, Suzuki S, Iwata Y, Kuboyama T (2008) Genetic diversity and relationship analysis of peanut germplasm using SSR markers. Breeding Science 58(3):293–300
Nielen S, Vidigal BS, Leal-Bertioli SC, Ratnaparkhe M, Paterson AH, Garsmeur O, Bertioli DJ (2012) Matita, a new retro element from peanut: characterization and evolutionary context in the light of the Arachis A-B genome divergence. Mol Genet Genom 287(1):21–38
Paterson AH, Brubaker CL, Wendel JF (1993) A rapid method for extraction of cotton (Gossypium spp.) genomic DNA suitable for RFLP or PCR analysis. Plant Mol Biol Rep 11(2):122–127
Proite K, Leal-Bertioli SC, Bertioli DJ, Moretzsohn MC, Silva FR, Martins NF, Guimaraes PM (2007) ESTs from a wild Arachis species for gene discovery and marker development. BMC Plant Biol 7(1):1
Qin HD, Feng SP, Chen C, Guo YF, Knapp S, Culbreath A, He GH, Wang ML, Zhang XY, Horlbrook CC, Ozias-Akins P, Guo BZ (2012) An integrated genetic linkage map of cultivated peanut (Arachis hypogaea L.) constructed from two RIL populations. Theor Appl Genet 124(4):653–664
Shirasawa K, Koillconda P, Koillconda P, Aoki K, Hirakawa H, Tabata S, Watanabe M, Hasegawa M, Kiyoshima H, Suzuki S, Kuwata C, Naito K, Kuboyama T, Nakaya A, Sasamoto S, Watanabe A, Kato M, Kawashima K, Kishida K, Kohara M, Kurabayashi A, Takahashi C, Tsuruoka H, Wada T, Isobe S (2012) In silico polymorphism analysis for the development of simple sequence repeat and transposon markers and construction of linkage map in cultivated peanut. BMC Plant Biol 12(1):80
Shirasawa K, Bertioli DJ, Varshney RK, Moretzsohn MC, Leal-Bertiol SC, Thudi M, Pandey MK, Rami JF, Fonceka D, Gowda MV, Qin HD, Guo BZ, Hong YB, Liang XQ, Hirakawa H, Tabata S, Isobe S (2013) Integrated consensus map of cultivated peanut and wild relatives reveals structures of the A and B genomes of Arachis and divergence of the legume genomes. DNA Res 20:173–184
Van Oijen JW (2006) Join Map Version 4.0: Software for the Calculation of Genetic Linkage Maps in experimental populations. The Netherlands Plant Research International, Wageningen
Varshney RK, Bertioli DJ, Moretzohn MC, Vadez V, Krishnamurthy L, Aruna R, Nigam SN, Ravi K, He G, Knapp SJ, Hoisington DA (2009) The first SSR based genetic linkage map for cultivated groundnut (Arachis hypogaea L.). Theor Appl Genet 118(4):729–739
Wang CT, Yang XD, Chen DX, Yu SL, Liu GZ, Tang YY, Xu JZ (2007) Isolation of simple sequence repeats from groundnut. Electron J Biotechnol 10(3):473–480
Wang H, Pandey MK, Qiao LX, Qin HD, Culbreath AK, He GH, Varshney RK, Guo BZ (2013) Genetic mapping and quantitative trait loci analysis for disease resistance using F2 and F5 generation-based genetic maps derived from ‘Tifrunner’ x ‘GT-C20’ in Peanut (Arachis hypogaea L.). Plant Genome 6(3):1–28
Wang ML, Khera P, Pandey MK, Wang H, Qiao L, Feng S, Culbreath AK (2015) Genetic Mapping of QTLs Controlling Fatty Acids Provided Insights into the Genetic Control of Fatty Acid Synthesis Pathway in Peanut (Arachis hypogaea L.). PLoS ONE 10(4):e0119454
Yin DM, Shang MZ, Cui DQ (2006) Studies on genetic analysis of major agronomic characters in peanut. Chin Agric Sci Bull 22(7):261–265
Zhang XY. (2011) Inheritance of main traits related to yield, quality and disease resistance and their QTLs mapping in peanut (Arachis hypogaea L.). Doctoral dissertation. Zhejiang University, Hangzhou, China
Zhou GY (1992) Introduction of exogenous DNA into plants after pollination via the pollen tube pathway. In: Ottaviano E, Mulcahy DL, Sari Gorla M, Bergamini Mulcahy G (eds) Angiosperm pollen and ovules. Springer, New York, pp 336–339
Acknowledgements
This research was supported by the Natural Science Foundation of China (NSFC) (31571711), the Earmarked Fund for China Agriculture Research System (CARS-14), the Peanut Seed Industry Project in Shandong Province of China, the Earmarked Fund for Agriculture Research System in Shandong Province of China (SDAIT-04-03) and the Fund for Independent Innovation of Agricultural Sciences in Jiangsu Province (Grant No. CX(12)5076).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Li, Y., Li, L., Zhang, X. et al. QTL mapping and marker analysis of main stem height and the first lateral branch length in peanut (Arachis hypogaea L.). Euphytica 213, 57 (2017). https://doi.org/10.1007/s10681-017-1847-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-017-1847-y