
Abstract
Key message
Using combined linkage and association mapping, 26 stable QTL and six stable SNPs were detected across multiple environments for eight ear and grain morphological traits in maize. One QTL, PKS2, might play an important role in maize yield improvement.
Abstract
In the present study, one bi-parental population and an association panel were used to identify quantitative trait loci (QTL) for eight ear and grain morphological traits. A total of 108 QTL related to these traits were detected across four environments using an ultra-high density bin map constructed using recombinant inbred lines (RILs) derived from a cross between Ye478 and Qi319, and 26 QTL were identified in more than two environments. Furthermore, 64 single nucleotide polymorphisms (SNPs) were found to be significantly associated with the eight ear and grain morphological traits (−log10(P) > 4) in an association panel of 240 maize inbred lines. Combining the two mapping populations, a total of 17 pleiotropic QTL/SNPs (pQTL/SNPs) were associated with various traits across multiple environments. PKS2, a stable locus influencing kernel shape identified on chromosome 2 in a genome-wide association study (GWAS), was within the QTL confidence interval defined by the RILs. The candidate region harbored a short 13-Kb LD block encompassing four SNPs (SYN11386, PHM14783.16, SYN11392, and SYN11378). In the association panel, 13 lines derived from the hybrid PI78599 possessed the same allele as Qi319 at the PHM14783.16 (GG) locus, with an average value of 0.21 for KS, significantly lower than that of the 34 lines derived from Ye478 that carried a different allele (0.25, P < 0.05). Therefore, further fine mapping of PKS2 will provide valuable information for understanding the genetic components of grain yield and improving molecular marker-assisted selection (MAS) in maize.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Maize (Zea mays L.) is one of the most widely cultivated cereals in the world in terms of harvested area and production. Improving maize yield is of primary concern for food security (Prado et al. 2014). Grain yield (GY) is a quantitative trait with a complex genetic basis (Messmer et al. 2009). Maize yield is determined by several yield components, including effective ear number, kernel number per ear and kernel weight. Kernel size, a key factor determining kernel weight, is usually evaluated by kernel length (KL), kernel width (KW), kernel thickness (KT), kernel shape (KS), and kernel volume (KV) (Li et al. 2013; Liu et al. 2014, 2016a, b; Peng et al. 2011). Grain yield has proven to be highly positively correlated with kernel size parameters, especially KL (Li et al. 2013; Peng et al. 2011). Kernel number per ear can be further subdivided into two components: kernel row number (KRN) and kernel number per row (KNPR). These traits exhibit higher heritability and better stability across environments compared with grain yield (Li et al. 2013; Messmer et al. 2009; Raihan et al. 2016). Therefore, dissection of QTL for yield components will facilitate a better understanding of the genetic architecture of yield.
Dissection of the genetic architecture of complex quantitative traits primarily depends on the isolation of genes underlying QTL or association loci with the aid of molecular genetic markers. For instance, in rice, approximately 400 QTL associated with kernel traits distributed across the entire genome (http://archive.gramene.org/qtl/) have been identified. Numerous genes controlling rice kernel traits, such as GW2 (Song et al. 2007), GS3 (Fan et al. 2006; Mao et al. 2010), qSW5/GW5 (Shomura et al. 2008; Weng et al. 2008), GS5 (Li et al. 2011a, b), GL7 (Wang et al. 2015b), GW7 (Wang et al. 2015a), GLW7 (Si et al. 2016), and GW8 (Wang et al. 2012) have been isolated and functionally characterized using map-based cloning and GWAS strategies. These genes have been verified to control kernel traits through activation of the cell cycle machinery to promote cell division and cell expansion (Si et al. 2016). Functional characterization of these genes has enriched knowledge of the molecular mechanisms underlying grain yield in rice. In maize, several mutant genes are involved in key pathways associated with ear architecture traits, especially KRN, including td1 (Bommert et al. 2005), fea2 and fea3 (Bommert et al. 2013b; Je et al. 2016), ct2 (Bommert et al. 2013a), the ramosa genes (ra1, ra2, and ra3) (McSteen 2006), Cg1 (Chuck et al. 2007), tsh4 (Chuck et al. 2010), ZFL1 and ZFL2 (Bomblies and Doebley 2006), ub2 and ub3 (Chuck et al. 2014), and KRN4 (Liu et al. 2015b). However, the cloning of maize genes that control kernel size and weight has lagged behind such progress in rice. However, several genes involved in kernel development, such as o2 (Zhou et al. 2016a), rgf1 (Maitz et al. 2000), dek1 (Lid et al. 2002), sh1 and sh2 (Thevenot et al. 2005), and gln1-4 (Martin et al. 2006) have so far been isolated using maize mutants. In addition, ZmGW2 and ZmGS3, which influence maize kernel weight and size and are orthologous to the rice genes GW2 and GS3, respectively, were isolated using homology-based cloning (Li et al. 2010a, b). Although numerous QTL that control yield-related traits have been identified in maize, our understanding of the genetic architecture of and the molecular mechanisms underlying quantitative variation in grain yield remains rudimentary.
QTL identification in plants generally depends on bi-parental population-based linkage analysis or association panel-based linkage disequilibrium (LD) analysis (Lu et al. 2010; Qin et al. 2016). Classical linkage analysis is an effective method for identifying QTL with large effects on yield-related traits in maize (Chen et al. 2016; Liu et al. 2014; Peng et al. 2011; Prado et al. 2014; Qin et al. 2016). Recently, using an immortalized F2 population, three major QTL, qKL3, qKWI6, and qKW10b were identified for KL, KW, and kernel weight in maize, respectively (Zhang et al. 2014). However, with low-density genetic maps based on bi-parental populations, map resolution is relatively low and identifying QTL with small effects is more challenging (Holland 2007). Next-generation sequencing technologies have significant advantages for genotyping large populations and constructing high-density genetic maps to elucidate the genetic basis of complex agronomic traits (Chen et al. 2014; Xiao et al. 2015; Zhou et al. 2016b). In addition, the development of high-throughput sequencing technologies and associated statistical methods has enabled GWAS based on LD analysis. GWAS has been performed to examine natural variation and identify novel genes controlling complex quantitative traits in many species, such as Arabidopsis thaliana (Atwell et al. 2010; Horton et al. 2012), rice (Si et al. 2016; Zhao et al. 2011), sorghum (Morris et al. 2013), soybean (Hwang et al. 2014; Zhao et al. 2015), and maize (Hu et al. 2016; Yang et al. 2014). Unlike mapping with bi-parental populations, GWAS offers the ability to detect nearly all recombination events in the evolutionary history of a population, and allows for increased mapping resolution to simultaneously evaluate the varying effects of many alleles (Li et al. 2010c; Rafalski 2010). However, the key limiting factors for GWAS are population substructure and low-frequency functional alleles that severely limit the power of detecting their phenotypic effects (Lu et al. 2010; Si et al. 2016). Joint linkage and GWAS effectively overcome some of the inherent limitations of both linkage and LD methods. For example, Lu et al. (2010) detected four genomic loci associated with anthesis-silking interval (ASI) in an association panel composed of 305 inbred lines, in addition to nine common ASI QTL in three linkage populations and 12 unique QTL by combined linkage and GWAS mapping. Wu et al. (2016) identified 125 QTL and 965 significant SNPs for male inflorescence size in maize using a nested association mapping (NAM) population by combined linkage and GWAS. Their studies provided extensive information for dissecting the genetic architecture of complicated agronomic traits, which will be helpful for maize yield improvement.
In the present study, phenotypic and genotypic data for 365 maize RILs derived from Ye478 × Qi319 and a set of 240 maize inbred lines were used to (1) identify stable QTL for ear and grain morphological traits in multiple environments with an ultra-high density bin map; (2) detect novel SNPs and loci associated with ear and grain morphological traits by GWAS; and (3) determine consistent QTL and markers flanking favorable alleles by combined linkage and GWAS methods. The present study aims to improve our understanding of the intricate genetic architecture of grain yield and contribute to the development of efficient methods for maize breeding.
Materials and methods
Materials and the phenotypic evaluation
A total of 365 RILs were derived from a cross between two maize elite inbred lines, Ye478 and Qi319, by single-seed descent. There were highly significant differences between these two parents for eight ear and grain morphological traits evaluated in the previous years. These included kernel length (KL), kernel width (KW), kernel thickness (KT), kernel volume (KV), kernel shape (KS), hundred-kernel weight (HKW), kernel row number (KRN), and kernel number per row (KNPR). Ye478, a dent corn, had an average KS of 25.87 g per 100 kernels across multiple environments, with average KL of 10.21 mm, average KW of 8.40 mm, and average KT of 5.74 mm. In contrast, Qi319, a flint corn, had wide, plump kernels weighing an average of 30.00 g per 100 kernels, with correspondingly larger trait averages of 9.05 and 6.27 mm for KW and KT, respectively. For ear architecture traits, Ye478 had more kernel rows than Qi319 (13.13 compared with 12.18), whereas Qi319 had more kernels per row than Ye478 (26.30 compared with 28.33). The RILs and their parents were evaluated in four environments (location/years) in China, including Beijing (N40°13′, E116°34′) and Gongzhuling (N43°52′, E124°82′) in 2013 and 2014. There was an average daily temperature of 16.3 °C and an average annual rainfall of 592.0 mm in Beijing, and 12.3 °C and 594.8 mm in Gongzhuling (http://data.cma.cn). Both these experimental sites are located in the spring maize-growing region of China. These location/year combinations were designated as E1 (Beijing, 2013), E2 (Gongzhuling, 2013), E3 (Beijing, 2014), and E4 (Gongzhuling, 2014).
The association panel consisted of 240 maize inbred lines that belong to the five heterotic groups Lan (Lancaster Sure Crop), LRC (Lvda Red Cob), PB (Partner B), Reid, and SPT (Sipingtou) (Liu et al. 2015a), and included the parental inbred lines of the bi-parental population. These elite inbred lines typically grown in the northeast, southwest, and the Yellow and Huai River valley maize-growing regions in China and had been previously evaluated to exhibit a wide range of phenotypic variation in yield components and tolerance to biotic stress (Liu et al. 2015a). This association panel was evaluated in three environments, including Beijing in 2011 and 2012, and Gongzhuling in 2012, which were designated as E1′, E2′, and E3′, respectively.
All the populations were arranged in the fields in a randomized incomplete block design with two replications per location. Approximately 17 plants were planted in 4 m row plots with 0.6 m row spacing and a final density of 60,000 plants/ha. Field management was performed according to the standard agronomic practices for maize in each location. Each family from the two populations was grown in a single row, allowed to open pollinate and at least 10 well-pollinated ears were harvested per line per replicate. Five well-pollinated ears from each line were then chosen for phenotypic measurements using the standard procedures after air-drying. Three kernel size-related traits, including kernel length (KL, mm), kernel width (KW, mm), and kernel thickness (KT, mm), were examined for each individual by randomly selecting ten kernels from the center of each cob. These traits were measured using an electronic digital caliper with a precision of 0.1 mm. Kernel volume (KV, KL × KW × KT, mm3) and kernel shape (KS, KL:KW:KT) were measured after measuring the three kernel-size-related traits and calculating their averages. Hundred-kernel weight (HKW), which is one of the yield components, was measured as the average weight from three repeated measurements of 100 mixed kernels from five ears weighed in grams using an electronic balance. Kernel row number (KRN) and kernel number per row (KNPR) were determined from five randomly selected ears. The average measured value of each trait across replications in each environment was calculated to represent the trait performance for each family.
Phenotypic data analysis
Broad-sense heritability (H 2) for each trait was estimated as described by Knapp (Knapp et al. 1985) as: H 2 = δ 2 g/(δ 2 g + δ 2 ge/n + δ 2/nr), where δ 2 g, δ 2 ge, and δ 2 are estimates of genetic, G × E and error variances; and n and r are the number of environments and replications per environment, respectively. A combined ANOVA over multiple environments was performed to estimate variance components using the Mixed Linear Model procedure (PROC MLM) in the Statistical Analysis System (SAS) software version 9.2 (SAS Institute, Cary NC, 2009). Pearson correlation coefficients (r) between different traits and environments were calculated using the IBM SPSS Statistics version 21.0 software (IBM, Armonk, NY, 2012).
Linkage mapping
The RIL population was characterized using genotyping-by-sequencing (GBS) technology on an Illumina 2500 platform using methods described in a previous study (Zhou et al. 2016b). A total of 86,257 SNPs were identified, and an ultra-high density linkage map was constructed with 4602 bin markers. The total genetic distance of the resulting map was 1533.72 cM with an average distance of 0.33 cM between markers. QTL controlling the ear and grain in four environments (E1, E2, E3, and E4) were detected using a composition-interval mapping (CIM) method in the R/qtl package (Broman et al. 2003). The threshold of logarithm of the odds (LOD) scores for evaluating the QTL effects at an experimentwise significance level of P = 0.05 were determined using 1000 permutations (Doerge and Churchill 1996). QTL with LOD values larger than the threshold value (threshold = 3.5 after 1000 permutations) were considered further. The proportion of phenotypic variation explained by the identified QTL was determined with the fitqtl function in the R/qtl package. QTL detected for more than two different traits with an overlapping confidence interval of 1.5 LOD were defined as pleiotropic QTL.
Genome-wide association mapping
The association panel was genotyped using the Illumina Maize SNP50 BeadChip (Liu et al. 2015a; Weng et al. 2011). In total, 40,757 SNPs with a minor allele frequency (MAF) of >0.05 in the population were used for association analysis with a compressed mixed linear model (CMLM) in GAPIT (Lipka et al. 2012) with previously defined population parameters (P3D) running in the Rstudio environment (https://www.rstudio.com/). The first three principle components were used as covariants. Because a Bonferroni correction (1/40757 = 2.44E−05) was too conservative, a less stringent threshold of –log10(P) > 4 was used to detect significant association signals. LD analysis within the peak SNP region was performed using the Haploview software (Barrett et al. 2004). A total of 4282 SNPs on maize chromosome 2 were used to estimate π and Tajima’s D in the TASSEL 3.0 software (Weng et al. 2013). Sliding-window analysis of nucleotide diversity was performed with a sliding window size of 500 SNPs and step intervals of ten SNPs. Candidate gene prediction was performed at MaizeGDB (http://www.maizegdb.org) based on the physical positions of significant SNPs.
Results
Phenotypic characteristics of ear and grain morphological traits in RILs and the association panel
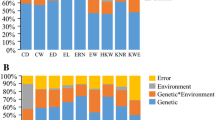
Descriptive statistics for ear and grain morphological traits in the two mapping populations are presented in Tables 1 and 2. Substantial variation in all of the traits was observed in each population. For example, HKW ranged from 14.06 to 36.92 g with a mean of 27.82 g in the RIL population in Beijing in 2014, but ranged from 13.23 to 44.20 g with a mean of 29.40 g in the association panel in Beijing in 2012. KNPR had the highest CV among these traits in the RIL population, whereas in the association panel, KS had the highest CV. The broad-sense heritability (H 2) of the eight ear and grain morphological traits ranged from 0.583 for KT to 0.750 for KV in the RIL population, and from 0.663 (KT) and 0.729 (KNPR) in the association panel (Tables 1, 2). Between these two populations, most traits were continuously and normally distributed, and showed quantitative inheritance. Notably, the phenotypic data of all eight traits exhibited obvious bi-directional transgressive segregation across all environments, indicating their polygenic control (Tables 1, 2). ANOVA revealed highly significant differences (P < 0.001) among genotypes and environments for all traits in the two populations, and the genotype-by-environment interaction was only non-significant for KT in the association panel (Fig. 1a, b). Meanwhile, the genotypic variance was greater than half of the total variance for these traits, except for KRN in the RIL population.

Variation in eight ear and grain morphological traits attributed to genetic and environmental factors. a Variation in ear and grain morphological traits across the RIL population. b Variation in ear and grain morphological traits across the association panel. The different shades of grey in the stacked bar diagram indicate the various factors that explain phenotypic variation. KL kernel length, KW kernel width, KT kernel thickness, KS kernel volume, KV kernel volume, HKW hundred-kernel weight, KRN kernel row number, KNPR kernel number per row
Of the eight ear and grain morphological traits surveyed here, a number of significant pairwise correlations were observed between kernel size and other traits (i.e., HKW, KRN, and KNPR) in the two populations (Table 3, Table S1). For example, across four environments (E1, E2, E3, and E4) in the RIL population, KNPR was consistently significantly positively correlated with KL (r = 0.31, P < 0.01) and KS (r = 0.49, P < 0.01), and significantly negatively correlated with KT (r = −0.51, P < 0.01) and KV (r = −0.27, P < 0.01), suggesting tradeoffs between KNPR and kernel size. Significant positive correlations were also observed between HKW and kernel size (r values were 0.29, 0.60, 0.29, and 0.73 for KL, KW, KT, and KV, respectively), which indicate an important role for kernel size in determining kernel weight. However, HKW was significantly negatively correlated with KRN (r = −0.12, P < 0.05), which suggested tradeoffs between HKW and KRN. In addition, environment correlation analysis indicated that the performance of each of these traits was highly significantly positively correlated across multiple environments, showing that environmental factors had little effect on these traits (Tables S2 and S3).
QTL mapping of ear and grain morphological traits in the RIL population
In a single-environment QTL analysis, 108 QTL distributed over all ten maize chromosomes were identified for these eight traits (Fig. 2; Table 4). The most QTL (18) were detected on chromosome 1 and the fewest on chromosome 5 (3). The proportion of phenotypic variation explained by these individual QTL averaged ~6.15% and ranged from 2.72% (KNPR, qKNPR9-1) to 13.20% (KNPR, qKNPR10) for different traits (Table 4). Among these QTL, over 62.94% had a positive additive effect, indicating that alleles from the parent Qi319 contributed increased phenotypic values in different environments. In addition, the confidence intervals for these QTL spanned physical distances from 0.65 to 32.90 Mb, with an average of 7.17 Mb, compared to the B73 RefGen_v3 genome.

Integrated QTL on ten chromosomes for ear and grain morphological traits across four environments. 1 Distribution of bin markers on ten chromosomes of maize; 2–9 QTL mapping for KL (grey), KW (red), KT (cyan), KS (blue), KV (purple), HKW (orange), KRN (yellow), and KNPR (green). The threshold of LOD scores is 3.5. For each trait, different colors represent different environments: red, 2013 Beijing; blue, 2013 Gongzhuling; green, 2014 Beijing; and yellow, 2014 Gongzhuling. The shaded areas cover the corresponding feature values for each pleiotropic QTL. (Color figure online)
Stable or consistent QTL were those that could be detected under different environments. In the present study, 26 QTL detected in at least two environments were designated as ‘consistent QTL’ (Table 4). Among these, over 73.17% consistent QTL were detected for kernel size. For example, six consistent QTL for KW were found dispersed on chromosomes 1, 3, 4, 6, 7, and 8. These QTL explained between 4.49 and 8.87% of the phenotypic variation in KW, with qKW1-1 (flanked by MK104 and MK138) accounting for the highest percentage. Of the six QTL associated with KW, the positive additive effects of two QTL (qKW3 and qKW4-1) on chromosomes 3 and 4 were consistently contributed by positive alleles from the parent with large kernel width, Qi319. The only consistent QTL detected for KT, qKT10, could explain 8.65% of the total phenotypic variation in KT. The Qi319 alleles had a negative effect on KT. Three consistent QTL influencing HKW were detected on chromosomes 1, 7, and 9. Notably, qHKW7 explained the greatest proportion of phenotypic variation for HKW and co-localized with QTL for most of the kernel size-related traits, such as qKL7, qKW7-2, and qKV7. This result also indicated a close genetic correlation between kernel size and kernel weight, which might result from pleiotropy. Moreover, two consistent QTL for KRN and KNPR were identified. A major QTL, qKNPR10 located on chromosome 10 from position 85.2 to 97.7 Mb, had the highest LOD score (9.64) and explained 13.20% of the total phenotypic variance for KNPR. In this region, qKT10, which influences kernel thickness, shared many of the same flanking markers with qKNPR10. The Qi319 alleles had a positive effect on KNPR, increasing the mean for this trait by 3.22 kernels.
GWAS of ear and grain morphological traits
The GWAS using 40,757 SNPs generated from the 240 accession maize panel permitted the elucidation of the genetic architecture of ear and grain. A total of 64 SNPs highly significantly associated with these traits across three environments were identified, including 13, 6, 13, 5, 6, 3, 4, 4, and 10 SNPs on nine maize chromosomes, including 1, 2, 3, 4, 5, 6, 8, 9, and 10, respectively (Fig. 3). In six cases, multiple SNPs from the same genomic region were in complete LD and were linked to the same trait at a comparable level of significance. These included seven SNPs on chromosome 1 for KL, KT, and KS; seven SNPs on chromosome 3 for KW, HKW, and KNPR; four SNPs on chromosome 4 for KT and KS; three SNPs on chromosome 5 for KRN; two SNPs on chromosome 8 for KNPR; and three SNPs on chromosome 10 for KV and HKW. The lengths of the associated genomic regions ranged from 1.48 to 474.22 Kb. The proportion of phenotypic variation explained by peak SNPs at the identified loci averaged ~25.9% and ranged from 18.8 to 34.4% for different traits (Table 5). Three of the traits, KL, KRN, and KNPR, had one or two strong association peaks with relatively large effects. Two traits with high genetic correlations, KV and HKW, were both associated with three SNPs (PZE-110078281, PZE-110084114, and SYN17906) over the less stringent significance threshold that overlapped on chromosome 10 in a single environment (Fig. 3). In addition, six stable SNPs for KT, KS, KV, and HKW could be detected in more than two environments. For example, PZE-101024700 (P = 4.33E−6), located on chromosome 1 at position 14,757,692 bp on the B73 reference genome (B73 RefGen_v3), was significantly associated with KT in all three environments and explained 21.0% of the phenotypic variation in this trait.

Genome-wide association scan for ear and grain morphological traits. Manhattan plots of the compressed MLM for eight ear and grain morphological traits across three environments. Provisional line indicates the genome-wide significance threshold at −log10(P) = 4. E1′ is 2011 Beijing; E2′ is 2012 Gongzhuling; E3′ is 2012 Beijing
Gene linkage or pleiotropy
Between the two populations, a total of 17 pQTL/SNPs showed clear linkage or pleiotropy with eight ear and grain morphological traits across multiple environments (Table 6). Seven of the ten pQTL from the RIL population that co-localized with more than four traits were distributed on chromosomes 1, 2, 4, 7, 8, 9, and 10, while three other pQTL on chromosomes 3, 4, and 6 encompassed QTL for KW and KV (Fig. 2). pQTL2, which is located on chromosome 2 between positions 1.65 and 16.15 Mb, was strongly linked to six of the eight ear and grain morphological traits, except for KW and KNPR. The QTL for HKW was usually detected together with QTL for kernel size-related traits, especially KV. These results were also supported by Pearson correlations between phenotypes measured in all four environments (Table 3). In the association panel, six pSNPs were associated with multiple traits. For example, three pSNPs (PZE-110078281, PZE-110084114, and SYN17906) on chromosome 10 were closely associated with both KV and HKW, and SNPs associated with both KT and KS were identified at 14.7 and 154.3 Mb on chromosomes 1 and 4, respectively. The set of pQTL/SNPs observed in the present study indicated that underlying genetic correlations and pleiotropy might play an important role in influencing ear and grain in maize.
Co-localization of QTL for ear and grain morphological traits by joint linkage mapping and GWAS
GWAS results for the eight ear and grain morphological traits were compared to QTL identified using linkage mapping in segregating populations. Loci identified by the GWAS that overlap with QTL mapped in bi-parental populations are shown in Fig. 4. Only one locus identified in the GWAS, PKS2 on chromosome 2, which influences kernel shape, was within the QTL confidence interval defined by RILs for the compared traits (Fig. 4a, b). Interestingly, this locus was simultaneously associated with a suite of QTL identified for other traits in the bi-parental population, such as KL, KT, KV, HKW, and KRN (Fig. 4b). The peak SNP (PHM14783.16, P = 9.06E−6) in this locus explained 23.3% of the total observed variation in kernel shape. The most significant SNP was located within a short 13-Kb LD block, including PKS2 using pairwise LD correlations (D’ ≥ 0.8). The candidate region was estimated to reside within the region from 9.93 to 9.95 Mb on chromosome 2 encompassed by four SNP markers (Fig. 4a, right). Three annotated candidate genes, GRMZM2G007713, GRMZM2G007453, and GRMZM2G007256, were located in genomic regions linked to the peak SNP (Fig. 4b). In addition, 13 lines directly selected from hybrid PH78599 possess the same Qi319 allele (GG) at the PHM14783.16 locus, and had an average value of 0.21 for KS, which is significantly lower than that of the 34 lines that harbor the Ye478 allele (0.25, P < 0.05) (Fig. 4c). The estimated values of π and Tajima’s D in the PKS2 window were lower than the means of those parameters for most maize lines in the RIL population (Fig. 4d). These results presumably indicate that artificial selection had occurred in this genomic region during maize breeding.

Co-localization of association peak SNPs, QTL, and well-annotated candidate genes. a Significant association signals on chromosome 2 for kernel shape. Dashed line represents the significance threshold at −log10(P) = 4. Arrowhead indicates the LD heatmap (right) surrounding the peak on chromosome 2. b Major kernel shape QTL covering the significant association signals was mapped between 2.05 and 10.5 Mb on chromosome 2 in Ye478 × Qi319 RILs. Dashed lines indicate the candidate region for the peak SNP. c Boxplot of kernel shape distribution at the peak SNP for the lines representing the sub-groups PB and Reid within the 240-line panel derived from Qi319 and Ye478. Differences between the genotypes were analyzed using Student’s t test. d Sliding-window analyses of nucleotide diversity in the association panel on chromosome 2. Black dots, π; Red dots, Tajima’s D. Black dashes represent mean π value, while red dashes represent mean Tajima’s D. (Color figure online)
Discussion
Elucidation of genetic architectures of ear and grain morphological traits by joint linkage and association mapping in maize
Although numerous QTL for yield-related traits have been identified in diverse maize populations, relatively few favorable alleles have been identified (Weng et al. 2013). The usefulness of RIL populations for QTL mapping has been corroborated in rice, wheat, maize, and soybean (Poland et al. 2012; Li et al. 2014; Zhou et al. 2012), and the value of high-density bin maps has also been well documented (Pan et al. 2012; Chen et al. 2014; Zhou et al. 2016b). Compared to other kinds of segregating populations, such as early generation populations, all the homozygous lines of permanent RIL populations can be evaluated in various locations in multiple years, which could increase the accuracy of QTL detection and reduce experimental error. Guo et al. (2014) developed a high-density integrated genetic linkage map for maize consisting of 3148 bin markers with a mean physical distance between adjacent bin markers of 0.64 Mb. Herein, we constructed a high-density bin map with 4602 bin markers with an average distance of 0.45 Mb between adjacent bin markers.
A number of QTL for yield-related traits have been detected in the previous studies. Liu et al. (2016b) detected 26 QTL for KL, KW, KL/KW, and HKW, with a minimum genetic distance of 2.6 cM and an average of 7.08 cM between map positions 90 and 92.6 cM on chromosome 3. Yang et al. (2016) detected 13 QTL for KL, KW, KT, and HKW, with a minimum physical distance of 0.66 Mb and an average of 7.38 Mb between umc1075 and chr8-8098 on chromosome 8. Liu et al. (2014) detected 83 QTL for KL, KW, and HKW, in a minimum physical interval of 1.37 Mb and an average of 17.30 Mb between umc1165 and umc1265 on chromosome 2. In our study, combining the GBS approach with RIL populations across four environments, 108 QTL were detected for different traits in the RIL population, 26 QTL were identified in more than two environments, and 13 were found in at least three environments. For all QTL identified here, qKRN2-2 was mapped in a minimum physical interval of 0.65 Mb and an average of 7.17 Mb between mk876 and mk879 (Table 4), suggesting that this approach identified QTL with high efficiency. In addition, the previous studies have identified some QTL in the same regions as those identified here, such as those for HKW and KNPR in bin 2.03 (Sa et al. 2015; Yang et al. 2015), KW in bin 4.07–4.08 (Li et al. 2013), HKW in bin 7.02–7.03 (Li et al. 2011a, b), KL in bin 9.02–9.03 (Liu et al. 2014), and HKW in bin 9.04 (Liu et al. 2014). In our study, we identified not only these QTL, but also some additional stable QTL, such as qKV1, qKV7, and qKT10. These stable QTL might be further used for fine mapping and gene cloning.
Association analysis is a powerful approach for exploring the molecular basis of phenotypic variation in plants. It has been widely used in many kinds of plants, since it was introduced for use in plant populations in 2001 (Thornsberry et al. 2001; Andersen et al. 2005; Casa et al. 2008; Krill et al. 2010). However, there have been relatively few GWAS of these yield-related traits in maize. In rice, several genes associated with grain length and grain width have been reported. For example, Si et al. (2016) integrated a GWAS approach with analyses of gene expression patterns, genetic variants and T-DNA-derived mutants to functionally characterize a grain shape-related QTL, GLW7. The candidate gene in QTL, GLW7 encodes the plant-specific transcription factor OsSPL13, which regulates cell size in the grain hull, resulting in enhanced rice grain length and yield. In the present study, a total of 64 SNPs were highly significantly associated with eight ear and grain morphological traits across three environments. The length of the associated genomic regions ranged from 1.48 to 474.22 Kb. Six SNPs could be consistently detected in more than two environments. The GWAS results were used to mutually validate the results of linkage mapping in RIL populations and as an effective method for gene mapping.
Recently, genetic improvement of grain yield was mostly focused on increasing stress tolerance of hybrids, which was selected for yield stability across target environments. However, QTL-by-environment interaction (QEI) often results in change of magnitude of significant QTL effect under different environments and affected the application of MAS. Peng et al. (2011) and Li et al. (2014) reported that QTL for kernel-related traits were more stable across environments than QTL of grain yield influenced by QEI. Therefore, it is often difficult to detect QTL for grain yield per plant and kernel number per plant stably expressed in different environments, especially in stress environments. Yield component traits especially kernel weight displayed more advantages for yield improvement. In the present study, stable and consistent QTL/SNP might be hot spots for the loci that are important for the associated traits. Therefore, a reliable and feasible strategy for QTL/SNP cloning is the fine mapping of these QTL/SNP and the validation of the potential candidate genes.
Gene linkage or pleiotropy associated with multiple ear and grain morphological traits in maize
Many previous studies have indicated that yield-related traits are controlled by a set of QTL, some of which are QTL clusters. The distribution of these QTL clusters can be explained by a pleiotropic QTL or multiple tightly linked QTL. Associated traits are prone to share regions with significant QTL (Li et al. 2007). Domestication has increased the size of maize kernels compared to its progenitor teosinte. In rice, QTL for domestication-related traits tend to occur in clusters that coincide with the regions harboring favorable genes (Cai and Morishima 2002). Among the 108 QTL and 64 SNPs identified here on all ten maize chromosomes (Tables 4, 5), the 17 pQTL/SNPs (Table 6) are each associated with two or more traits. In particular, pQTL2, pQTL4-1 (bin 4.05), and pQTL10 (bin 10.03), which control six traits (KL, KT, KV, KS, HKW, and KRN), four traits (KL, KW, KT, and KS), and five traits (KL, KT, KV, KS, and KNPR), respectively, correspond to physical distances of about 14.5, 13.45, and 33.55 Mb, respectively. Previous reports have shown that bin 4.05 and bin 10.03 are important genomic regions controlling maize yield-related traits, such as KRN, KW, and KL (Veldboom et al. 1994; Doebley et al. 1994; Peng et al. 2011) or KNPR, KW, and KT (Peng et al. 2011), respectively. These reports indicated that these regions might also be required for kernel development. These clustered QTL/SNPs might be closely associated, functionally co-adapted, or related through unknown factors with pleiotropic effects, suggesting that it is possible for QTL/SNPs that control maize yield-related traits to be closely distributed and linked to other loci controlling related characteristics. Therefore, the fine mapping of these QTL/SNPs and validation of the potential candidate genes could be a reliable and feasible strategy for the cloning of QTL/SNP, which might be even more valuable in maize breeding. Our results provide important information for further fine mapping to find genes controlling quantitative traits and may help reveal the molecular mechanisms responsible for these yield-related traits.
Combined linkage and association mapping is a powerful method for QTL detection
Linkage and association analysis are two complementary approaches commonly used to map causal genes and dissect the genetic bases of traits of interest (Lu et al. 2010; Tian et al. 2011; Tao et al. 2013). Association mapping offers higher resolution than traditional QTL mapping and is suitable for mapping multiple traits at once (Yu and Buckler 2006; Yan et al. 2011). In the present study, both GWAS and linkage mapping methods were used to identify loci associated with eight maize ear and grain, including PKS2, a gene on chromosome 2 that influences kernel shape. Using linkage mapping in a bi-parental population, this locus was simultaneously associated with several QTL for other traits, including KL, KT, KV, and HKW. Furthermore, a major QTL for KRN was mapped using RILs to a 14.5 Mb region between 1.65 and 16.15 Mb on chromosome 2, which contains the most significant SNP for PKS2 between 9.93 and 9.95 Mb. According to the maize gene annotation database accessible at MaizeGDB (http://www.maizegdb.org), three annotated candidate genes were located in genomic regions linked to the peak SNP, but only GRMZM2G007713 and GRMZM2G007453 code the definite protein.GRMZM2G007713, a heavy metal transport/detoxification superfamily protein, involved in the metal ion transport in maize endosperm (Sekhon et al. 2011). GRMZM2G007453, accumulation of photosystem (APO) putative expressed encodes a chloroplast localized RNA binding protein that is involved in RNA splicing. It was shown that Arabidopsis apo1 mutants fail to splice the second intron in the ycf3 pre-mRNA and lead to the thylakoid protein deficiencies (Watkins et al. 2011). The consistency between the association loci from the GWAS and the QTL from linkage mapping cross-validates the results of mapping loci for these traits by these two approaches. The combination of these approaches not only reduces false positives, but also increases the efficiency of QTL and candidate gene detection severalfold. These QTL could be important candidate genes related to maize yield, and their further functional verification will improve our understanding of their underlying genetic and molecular mechanisms.
Application of the identified loci in marker-assisted selection in maize breeding
Release of new maize cultivars developed using traditional breeding methods can take 10 or more years (Gasic et al. 2014). However, if the genetic basis of a quantitative trait is greatly clear, genetic improvement of traits by MAS of favorable alleles will greatly accelerate (Dekkers and Hospital 2002; Papst et al. 2004; Ordas et al. 2009). MAS has been successfully used for breeding and selection of many traits, especially yield (Xu et al. 2009), in many crops, such as maize (Widstrom et al. 2003) and wheat (Adhikari et al. 2004) and rice (Wang et al. 2012). In the previous studies, MAS has been used for QTL validation, fine mapping, and gene cloning using backcross populations in maize. In backcross populations, the target QTL becomes the major source of genetic variation in a trait, and the undesired genetic background is then gradually eliminated by continued backcrossing and MAS (Liu et al. 2016a). The subsequent fine mapping of more recombination events within this region can then allow cloning of the candidate gene. In our study, we could consistently detect many QTL/SNPs, including qKL7, qKT10, qKS10, qKV1, qKV7, qKN10, PZE-101024700, and PHM14783.16 in all the test environments, and could also detect 17 pQTL/pSNPs. In future experiments, backcross populations could be developed from particular RIL families and their parents for QTL validation. These stable consistent QTL could also be considered priority candidates for use in MAS. The fine mapping of these QTL and validation of the potential candidate genes could be a reliable and feasible strategy for QTL cloning to isolate loci that could be even more valuable for maize breeding. Our results provide important information for such further fine mapping to find genes controlling quantitative traits and may help reveal the molecular mechanisms responsible for maize yield-related traits. Further analysis of the important genomic regions controlling maize yield-related traits could enable cloning of key genes and reveal the mechanisms of control of these traits, but also provide the basis for MAS, elite inbred line development, and hybrid breeding.
Conclusions
In the present study, a bi-parental population and an association panel were used to identify QTL for eight ear and grain morphological traits (KL, KW, KT, KV, KS, HKW, KRN, and KNPR). A total of 26 stable QTL and six stable SNPs were detected across multiple environments by QTL mapping and GWAS, respectively. Furthermore, 17 QTL/SNPs (pQTL/SNPs) were associated with various traits. Among these, PKS2, a stable locus identified by this combined approach between 9.93 and 9.95 Mb on chromosome 2 might play an important role in the control of grain yield. These results could improve our understanding of the genetic basis of the ear and grain in maize.
Author contribution statement
CZ and ZZ performed the experiments and wrote the paper. HY, XC, HY, ZH, FZ, ML, DZ, and XL performed the experiments and revised the paper. JW and ZW designed the experiments.
Abbreviations
- KL:
-
Kernel length
- KW:
-
Kernel width
- KT:
-
Kernel thickness
- KV:
-
Kernel volume (KL × KW × KT)
- KS:
-
Kernel shape (KL:KW:KT)
- HKW:
-
Hundred-kernel weight
- KRN:
-
Kernel row number
- KNPR:
-
Kernel number per row
- CIM:
-
Composite interval mapping
- QTL:
-
Quantitative trait loci
- SNP:
-
Single nucleotide polymorphisms
- MAS:
-
Marker-assisted selection
- GWAS:
-
Genome-wide association studies
- LD:
-
Linkage disequilibrium
References
Adhikari TB, Yang X, Cavaletto JR, Hu X, Buechley G, Ohm HW, Shaner G, Goodwin SB (2004) Molecular mapping of Stb1, a potentially durable gene for resistance to septoria tritici blotch in wheat. Theor Appl Genet 109:944–953
Andersen JR, Schrag T, Melchinger AE, Zein I, Lübberstedt T (2005) Validation of Dwarf8 polymorphisms associated with flowering time in elite European inbred lines of maize (Zea mays L.). Theor Appl Genet 111:206–217
Atwell S, Huang YS, Vilhjalmsson BJ, Willems G, Horton M, Li Y, Meng DZ, Platt A, Tarone AM, Hu TT, Jiang R, Muliyati NW, Zhang X, Amer MA, Baxter I, Brachi B, Chory J, Dean C, Debieu M, de Meaux J, Ecker JR, Faure N, Kniskern JM, Jones JDG, Michael T, Nemri A, Roux F, Salt DE, Tang CL, Todesco M, Traw MB, Weigel D, Marjoram P, Borevitz JO, Bergelson J, Nordborg M (2010) Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 465:627–631
Barrett JC, Fry B, Maller J, Daly MJ (2004) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–265
Bomblies K, Doebley JF (2006) Pleiotropic effects of the duplicate maize FLORICAULA/LEAFY genes zfl1 and zfl2 on traits under selection during maize domestication. Genetics 172:519–531
Bommert P, Lunde C, Nardmann J, Vollbrecht E, Running M, Jackson D, Hake S, Werr W (2005) Thick tassel dwarf1 encodes a putative maize ortholog of the Arabidopsis CLAVATA1 leucine-rich repeat receptor-like kinase. Development 132:1235–1245
Bommert P, Je BI, Goldshmidt A, Jackson D (2013a) The maize G alpha gene COMPACT PLANT2 functions in CLAVATA signalling to control shoot meristem size. Nature 502:555–558
Bommert P, Nagasawa NS, Jackson D (2013b) Quantitative variation in maize kernel row number is controlled by the FASCIATED EAR2 locus. Nat Genet 45:334–337
Broman KW, Wu H, Sen S, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 17:889–890
Cai H, Morishima H (2002) QTL clusters reflect character associations in wild and cultivated rice. Theor Appl Genet 104:1217–1228
Casa AM, Pressoira G, Brown PJ, Mitchell SE, Rooney WL, Tuinstrac MR, Franks CD, Kresovicha S (2008) Community resources and strategies for association mapping in sorghum. Crop Sci 48:30–40
Chen Z, Wang B, Dong X, Liu H, Ren L, Chen J, Hauck A, Song W, Lai J (2014) An ultra-high density bin-map for rapid QTL mapping for tassel and ear architecture in a large F2 maize population. BMC Genom 15:433
Chen L, Li Y, Li C, Wu X, Qin W, Li X, Jiao F, Zhang X, Zhang D, Shi Y, Song Y, Li Y, Wang T (2016) Fine-mapping of qGW4.05, a major QTL for kernel weight and size in maize. BMC Plant Biol 16:81
Chuck G, Cigan AM, Saeteurn K, Hake S (2007) The heterochronic maize mutant Corngrass1 results from overexpression of a tandem microRNA. Nat Genet 39:544–549
Chuck G, Whipple C, Jackson D, Hake S (2010) The maize SBP-box transcription factor encoded by tasselsheath4 regulates bract development and the establishment of meristem boundaries. Development 137:1243–1250
Chuck GS, Brown PJ, Meeley R, Hake S (2014) Maize SBP-box transcription factors unbranched2 and unbranched3 affect yield traits by regulating the rate of lateral primordia initiation. Proc Natl Acad Sci USA 111:18775–18780
Dekkers JCM, Hospital F (2002) The use of molecular genetics in the improvement of agricultural populations. Nat Rev Genet 3:22–32
Doebley J, Bacigalupo A, Stec A (1994) Inheritance of kernel weight in two maize-teosinte hybrid populations implications for crop evolution. J Hered 85:191–195
Doerge RW, Churchill GA (1996) Permutation tests for multiple loci affecting a quantitative character. Genetics 142:285–294
Fan C, Xing Y, Mao H, Lu T, Han B, Xu C, Li X, Zhang Q (2006) GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor Appl Genet 112:1164–1171
Gasic K, Quick R, Abdelghafar A, Frett T, Rauh B, Reighard G (2014) Marker assisted breeding for red skin coloration in peach. In: Proceedings Plant and Animal Genome XXII Conference, Plant and Animal Genome, 2014
Guo TT, Yang N, Tong H, Pan QC, Yang XH, Tang JH, Wang JK, Li JS, Yan JB (2014) Genetic basis of grain yield heterosis in an “immortalized F2” maize population. Theor Appl Genet 127:2149–2158
Holland JB (2007) Genetic architecture of complex traits in plants. Curr Opin Plant Biol 10:156–161
Horton MW, Hancock AM, Huang YS, Toomajian C, Atwell S, Auton A, Muliyati NW, Platt A, Sperone FG, Vilhjalmsson BJ, Nordborg M, Borevitz JO, Bergelson J (2012) Genome-wide patterns of genetic variation in worldwide Arabidopsis thaliana accessions from the RegMap panel. Nat Genet 44:212–216
Hu H, Schrag TA, Peis R, Unterseer S, Schipprack W, Chen S, Lai J, Yan J, Prasanna BM, Nair SK, Chaikam V, Rotarenco V, Shatskaya OA, Zavalishina A, Scholten S, Schon CC, Melchinger AE (2016) The genetic basis of haploid induction in maize identified with a novel genome-wide association method. Genetics 202:1267–1276
Hwang EY, Song Q, Jia G, Specht JE, Hyten DL, Costa J, Cregan PB (2014) A genome-wide association study of seed protein and oil content in soybean. BMC Genom 15:1
Je BI, Gruel J, Lee YK, Bommert P, Arevalo ED, Eveland AL, Wu Q, Goldshmidt A, Meeley R, Bartlett M, Komatsu M, Sakai H, Jonsson H, Jackson D (2016) Signaling from maize organ primordia via FASCIATED EAR3 regulates stem cell proliferation and yield traits. Nat Genet 48:785–791
Knapp SJ, Stroup WW, Ross WM (1985) Exact confidence intervals for heritability on a progeny mean basis. Crop Sci 25:192–194
Krill AM, Kirst M, Kochian LV, Buckler ES, Hoekenga OA (2010) Association and linkage analysis of aluminum tolerance genes in maize. PLOS One 5:e9958
Li Y, Niu S, Dong Y, Cui D, Wang Y, Liu Y, Wei M (2007) Identification of trait-improving quantitative trait loci for grain yield components from a dent corn inbred line in an advanced backcross BC2F2 population and comparison with its F2:3 population in popcorn. Theor Appl Genet 115:129–140
Li Q, Li I, Yang X, Warburton M, Bai G, Dai J, Li J, Yan J (2010a) Relationship, evolutionary fate and function of two maize co-orthologs of rice GW2 associated with kernel size and weight. BMC Plant Biol 10:143
Li Q, Yang XH, Bai GH, Warburton ML, Mahuku G, Gore M, Dai JR, Li JS, Yan JB (2010b) Cloning and characterization of a putative GS3 ortholog involved in maize kernel development. Theor Appl Genet 120:753–763
Li Y, Huang Y, Bergelson J, Nordborg M, Borevitz JO (2010c) Association mapping of local climate-sensitive quantitative trait loci in Arabidopsis thaliana. Proc Natl Acad Sci USA 107:21199–21204
Li YB, Fan CC, Xing YZ, Jiang YH, Luo LJ, Sun L, Shao D, Xu CJ, Li XH, Xiao JH, He YQ, Zhang QF (2011a) Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat Genet 43:1266–1269
Li ZM, Ding JQ, Wang RX, Chen JF, Sun XD, Chen W (2011b) A new QTL for resistance to Fusarium ear rot in maize. J Appl Genet 52:403–406
Li CH, Li YX, Sun BC, Peng B, Liu C, Liu ZZ, Yang ZZ, Li QC, Tan WW, Zhang Y, Wang D, Shi YS, Song YC, Wang TY, Li Y (2013) Quantitative trait loci mapping for yield components and kernel-related traits in multiple connected RIL populations in maize. Euphytica 193:303–316
Li B, Tian L, Zhang JY, Huang L, Han FX, Yan SR, Wang LZ, Zheng HK, Sun JM (2014) Construction of a high–density genetic map based on large–scale markers developed by specific length amplified fragment sequencing (SLAF–seq) and its application to QTL analysis for isoflavone content in Glycine max. BMC Genom 15:1086
Lid SE, Gruis D, Jung R, Lorentzen JA, Ananiev E, Chamberlin M, Niu XM, Meeley R, Nichols S, Olsen OA (2002) The defective kernel 1 (dek1) gene required for aleurone cell development in the endosperm of maize grains encodes a membrane protein of the calpain gene superfamily. Proc Natl Acad Sci USA 99:5460–5465
Lipka AE, Tian F, Wang Q, Peiffer J, Li M, Bradbury PJ, Gore MA, Buckler ES, Zhang ZW (2012) GAPIT: genome association and prediction integrated tool. Bioinformatics 28:2397–2399
Liu Y, Wang L, Sun C, Zhang Z, Zheng Y, Qiu F (2014) Genetic analysis and major QTL detection for maize kernel size and weight in multi-environments. Theor Appl Genet 127:1019–1037
Liu CL, Hao ZF, Zhang DG, Xie CX, Li MS, Zhang XC, Yong HJ, Zhang SH, Weng JF, Li XH (2015a) Genetic properties of 240 maize inbred lines and identity-by-descent segments revealed by high-density SNP markers. Mol Breed 35:146
Liu L, Du YF, Shen XM, Li MF, Sun W, Huang J, Liu ZJ, Tao YS, Zheng YL, Yan JB, Zhang ZX (2015b) KRN4 controls quantitative variation in maize kernel row number. PLOS Genet 11:e1005670
Liu CL, Hua JG, Liu C, Zhang DG, Hao ZF, Yong HJ, Xie CX, Li MS, Zhang SH, Weng JF, Li XH (2016a) Fine mapping of a quantitative trait locus conferring resistance to maize rough dwarf disease. Theor Appl Genet. doi:10.1007/s00122-016-2770-7
Liu ZB, Garcia A, McMullen MD, Flint-Garcia SA (2016b) Genetic analysis of kernel traits in maize-teosinte introgression populations. G3 (Bethesda) 6:2523–2530
Lu Y, Zhang S, Shah T, Xie C, Hao Z, Li X, Farkhari M, Ribaut JM, Cao M, Rong T, Xu Y (2010) Joint linkage-linkage disequilibrium mapping is a powerful approach to detecting quantitative trait loci underlying drought tolerance in maize. Proc Natl Acad Sci USA 107:19585–19590
Maitz M, Santandrea G, Zhang ZY, Lal S, Hannah LC, Salamini F, Thompson RD (2000) rgf1, a mutation reducing grain filling in maize through effects on basal endosperm and pedicel development. J Plant 23:29–42
Mao H, Sun S, Yao J, Wang C, Yu S, Xu C, Li X, Zhang Q (2010) Linking differential domain functions of the GS3 protein to natural variation of grain size in rice. Proc Natl Acad Sci USA 107:19579–19584
Martin A, Lee J, Kichey T, Gerentes D, Zivy M, Tatout C, Dubois F, Balliau T, Valot B, Davanture M, Terce-Laforgue T, Quillere I, Coque M, Gallais A, Gonzalez-Moro MB, Bethencourt L, Habash DZ, Lea PJ, Charcosset A, Perez P, Murigneux A, Sakakibara H, Edwards KJ, Hirel B (2006) Two cytosolic glutamine synthetase isoforms of maize are specifically involved in the control of grain production. Plant Cell 18:3252–3274
McSteen P (2006) Branching out: The ramosa pathway and the evolution of grass inflorescence morphology. Plant Cell 18:518–522
Messmer R, Fracheboud Y, Banziger M, Vargas M, Stamp P, Ribaut JM (2009) Drought stress and tropical maize: QTL-by-environment interactions and stability of QTLs across environments for yield components and secondary traits. Theor Appl Genet 119:913–930
Morris GP, Ramu P, Deshpande SP, Hash CT, Shah T, Upadhyaya HD, Riera-Lizarazu O, Brown PJ, Acharya CB, Mitchell SE, Harriman J, Glaubitz JC, Buckler ES, Kresovich S (2013) Population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proc Natl Acad Sci USA 110:453–458
Ordas B, Malvar RA, Santiago R, Sandoya G, Romay MC, Butron (2009) A mapping of QTL for resistance to the mediterranean corn borer attack using the intermated B73 × Mo17 (IBM) population of maize. Theor Appl Genet 119:1451–1459
Pan Q, Ali F, Yang X, Li J, Yan J (2012) Exploring the genetic characteristics of two recombinant inbred line populations via high-density SNP markers in maize. PLOS One 7(12):e52777
Papst C, Bohn M, Utz HF, Melchinger AE, Klein D, Elder J (2004) QTL mapping for European corn borer and forage quality traits of testcross progenies in early-maturing European maize (Zea mays L) germplasm. Theor Appl Genet 108:1545–1554
Peng B, Li Y, Wang Y, Liu C, Liu Z, Tan W, Zhang Y, Wang D, Shi Y, Sun B, Song Y, Wang T, Li Y (2011) QTL analysis for yield components and kernel-related traits in maize across multi-environments. Theor Appl Genet 122:1305–1320
Poland JA, Brown PJ, Sorrells ME, Jannink JL (2012) Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLOS One 7(2):e32253
Prado SA, César G, López M, Senior L, Borrás L (2014) The genetic architecture of maize (Zea mays L.) kernel weight determination. G3 (Bethesda) 4:1611–1621
Qin W, Li YX, Wu X, Li X, Chen L, Shi Y, Song Y, Zhang D, Wang T, Li Y (2016) Fine mapping of qKL1.07, a major QTL for kernel length in maize. Mol Breed 36:8
Rafalski JA (2010) Association genetics in crop improvement. Curr Opin Plant Biol 13:174–180
Raihan MS, Liu J, Huang J, Guo H, Pan Q, Yan J (2016) Multi-environment QTL analysis of grain morphology traits and fine mapping of a kernel-width QTL in Zheng58 × SK maize population. Theor Appl Genet 129:1465–1477
Sa KJ, Park JY, Woo SY, Ramekar RV, Jang CS, Lee JK (2015) Mapping of QTL traits in corn using a RIL population derived from a cross of dent corn × waxy corn. Genes Genom 37:1–14
Sekhon RS, Lin H, Childs KL, Hirsch CN, Buell CR, de Leon N, Kaeppler SM (2011) Genome-wide atlas of transcription through maize development. Plant J 66(0):553–562
Shomura A, Izawa T, Ebana K, Ebitani T, Kanegae H, Konishi S, Yano M (2008) Deletion in a gene associated with grain size increased yields during rice domestication. Nat Genet 40:1023–1028
Si L, Chen J, Huang X, Gong H, Luo J, Hou Q, Zhou T, Lu T, Zhu J, Shangguan Y, Chen E, Gong C, Zhao Q, Jing Y, Zhao Y, Li Y, Cui L, Fan D, Lu Y, Weng Q, Wang Y, Zhan Q, Liu K, Wei X, An K, An G, Han B (2016) OsSPL13 controls grain size in cultivated rice. Nat Genet 48:447–456
Song XJ, Huang W, Shi M, Zhu MZ, Lin HX (2007) A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat Genet 39:623–630
Tao Y, Jiang L, Liu Q, Zhang Y, Zhang R, Ingvardsen CR, Frei UK, Wang B, Lai J, Lübberstedt T, Xu M (2013) Combined linkage and association mapping reveals candidates for Scmv1, a major locus involved in resistance to sugarcane mosaic virus (SCMV) in maize. BMC Plant Biol 13:162
Thevenot C, Simond-Cote E, Reyss A, Manicacci D, Trouverie J, Le Guilloux M, Ginhoux V, Sidicina F, Prioul JL (2005) QTLs for enzyme activities and soluble carbohydrates involved in starch accumulation during grain filling in maize. J Exp Bot 56:945–958
Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler ES (2001) Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet 28:286–289
Tian F, Bradbury PJ, Brown PJ, Hung H, Sun Q, Flint-Garcia S, Rocheford TR, McMullen MD, Holland JB, Buckler ES (2011) Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat Genet 43:159–162
Veldboom LR, Lee M, Woodman WL (1994) Molecular marker-facilitated studies in an elite maize population: I. Linkage analysis and determination of QTL for morphological traits. Theor Appl Genet 88:7–16
Wang SK, Wu K, Yuan QB, Liu XY, Liu ZB, Lin XY, Zeng RZ, Zhu HT, Dong GJ, Qian Q, Zhang GQ, Fu XD (2012) Control of grain size, shape and quality by OsSPL16 in rice. Nat Genet 44:950–954
Wang S, Li S, Liu Q, Wu K, Zhang J, Wang Y, Chen X, Zhang Y, Gao C, Wang F, Huang H, Fu X (2015a) The OsSPL16-GW7 regulatory module determines grain shape and simultaneously improves rice yield and grain quality. Nat Genet 47:949–954
Wang Y, Xiong G, Hu J, Jiang L, Yu H, Xu J, Fang Y, Zeng L, Xu E, Ye W, Meng X, Liu R, Chen H, Jing Y, Zhu X, Li J, Qian Q (2015b) Copy number variation at the GL7 locus contributes to grain size diversity in rice. Nat Genet 47:944–948
Watkins KP, Rojas M, Friso G, van Wijk KJ, Meurer J, Barkan A (2011) APO1 promotes the splicing of chloroplast group II introns and harbors a plant-specific zinc-dependent RNA binding domain. Plant Cell 23(3):1082–1092
Weng JF, Gu SH, Wan XY, Gao H, Guo T, Su N, Lei CL, Zhang X, Cheng ZJ, Guo XP, Wang JL, Jiang L, Zhai HQ, Wan JM (2008) Isolation and initial characterization of GW5, a major QTL associated with rice grain width and weight. Cell Res 18:1199–1209
Weng JF, Xie CX, Hao ZF, Wang JJ, Liu CL, Li MS, Zhang DG, Bai L, Zhang SH, Li XH (2011) Genome-wide association study identifies candidate genes that affect plant height in Chinese elite maize (Zea mays L.) inbred lines. PLOS One 6:e29229
Weng JF, Li B, Liu CL, Yang XY, Wang HW, Hao ZF, Li MS, Zhang DG, Ci XK, Li XH, Zhang SH (2013) A non-synonymous SNP within the isopentenyl transferase 2 locus is associated with kernel weight in Chinese maize inbreds (Zea mays L.). BMC Plant Biol 13:98
Widstrom N, Butron A, Guo B, Wilson D, Snook M, Cleveland T, Lynch R (2003) Control of preharvest aflatoxin contamination in maize by pyramiding QTL involved in resistance to ear-feeding insects and invasion by Aspergillus spp. Eur J Agron 19:563–572
Wu X, Li Y, Shi Y, Song Y, Zhang D, Li C, Buckler ES, Li Y, Zhang Z, Wang T (2016) Joint-linkage mapping and GWAS reveal extensive genetic loci that regulate male inflorescence size in maize. Plant Biotechnol J 14(7):1551–1562
Xiao Y, Tong H, Yang X, Xu S, Pan Q, Qiao F, Raihan MS, Luo Y, Liu H, Zhang X, Yang N, Wang X, Deng M, Jin M, Zhao L, Luo X, Zhou Y, Li X, Liu J, Zhan W, Liu N, Wang H, Chen G, Cai Y, Xu G, Wang W, Zheng D, Yan J (2015) Genome-wide dissection of the maize ear genetic architecture using multiple populations. New Phytol 210:1095–1106
Xu Y, Skinner DJ, Wu H, Palacios-Rojas N, Araus JL, Yan J, Gao S, Warburton ML, Crouch JH (2009) Advances in maize genomics and their value for enhancing genetic gains from breeding. Int J Plant Genom 2009:957602
Yan J, Warburton M, Crouch J (2011) Association mapping for enhancing maize (Zea mays L.) genetic improvement. Crop Sci 51:433–449
Yang N, Lu YL, Yang XH, Huang J, Zhou Y, Ali F, Wen WW, Liu J, Li JS, Yan JB (2014) Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PLOS Genet 10:e1004573
Yang C, Tang DG, Zhang L, Liu J, Rong TZ (2015) Identification of QTL for ear row number and two-ranked versus many-ranked ear in maize across four environments. Euphytica 206:33–47
Yang C, Zhang L, Jia A, Rong T (2016) Identification of QTL for maize grain yield and kernel-related traits. J Genet 95:239–247
Yu J, Buckler ES (2006) Genetic association mapping and genome organization of maize. Curr Opin Biotechnol 17:155–160
Zhang Z, Liu Z, Hu Y, Li W, Fu Z, Ding D, Li H, Qiao M, Tang J (2014) QTL analysis of kernel-related traits in maize using an immortalized F2 population. PLOS One 9:e89645
Zhao K, Tung CW, Eizenga GC, Wright MH, Ali ML, Price AH, Norton GJ, Islam MR, Reynolds A, Mezey J, McClung AM, Bustamante CD, McCouch SR (2011) Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat Commun 2:467
Zhao X, Han Y, Li Y, Liu D, Sun M, Zhao Y, Lv C, Li D, Yang Z, Huang L, Teng W, Qiu L, Zheng H, Li W (2015) Loci and candidate gene identification for resistance to Sclerotinia sclerotiorum in soybean (Glycine max L. Merr.) via association and linkage maps. J Plant 82:245–255
Zhou G, Chen Y, Yao W, Zhang C, Xie W, Hua J, Xing Y, Xiao J, Zhang Q (2012) Genetic composition of yield heterosis in an elite rice hybrid. Proc Natl Acad Sci USA 109:15847–15852
Zhou ZQ, Song LY, Zhang XX, Li XH, Yan N, Xia RP, Zhu H, Weng JF, Hao ZF, Zhang DG, Yong HJ, Li MS, Zhang SH (2016a) Introgression of opaque2 into waxy maize causes extensive biochemical and proteomic changes in endosperm. PLOS One 11:e0158971
Zhou ZQ, Zhang CS, Zhou Y, Hao ZF, Wang ZH, Zeng X, Di H, Li MS, Zhang DG, Yong HJ, Zhang SH, Weng JF, Li XH (2016b) Genetic dissection of maize plant architecture with an ultra-high density bin map based on recombinant inbred lines. BMC Genom 17:178
Acknowledgements
This research was jointly funded by the National Basic Research Program of China (2014CB138200), the National Natural Science Foundation of China (31471509), and the Chinese Academy of Agricultural Sciences (CAAS) Innovation Project.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standard
We claim that the experiments described herein comply with the ethical standards in China.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
C. Zhang and Z. Zhou are equal contributors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, C., Zhou, Z., Yong, H. et al. Analysis of the genetic architecture of maize ear and grain morphological traits by combined linkage and association mapping. Theor Appl Genet 130, 1011–1029 (2017). https://doi.org/10.1007/s00122-017-2867-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-017-2867-7