
Abstract
Key message
We characterized a novel blast resistance gene Pi50 at the Pi2/9 locus; Pi50 is derived from functional divergence of duplicated genes. The unique features of Pi50 should facilitate its use in rice breeding and improve our understanding of the evolution of resistance specificities.
Abstract
Rice blast disease, caused by the fungal pathogen Magnaporthe oryzae, poses constant, major threats to stable rice production worldwide. The deployment of broad-spectrum resistance (R) genes provides the most effective and economical means for disease control. In this study, we characterize the broad-spectrum R gene Pi50 at the Pi2/9 locus, which is embedded within a tandem cluster of 12 genes encoding proteins with nucleotide-binding site and leucine-rich repeat (NBS–LRR) domains. In contrast with other Pi2/9 locus, the Pi50 cluster contains four duplicated genes (Pi50_NBS4_1 to 4) with extremely high nucleotide sequence similarity. Moreover, these duplicated genes encode two kinds of proteins (Pi50_NBS4_1/2 and Pi50_NBS4_3/4) that differ by four amino acids. Complementation tests and resistance spectrum analyses revealed that Pi50_NBS4_1/2, not Pi50_NBS4_3/4, control the novel resistance specificity as observed in the Pi50 near isogenic line, NIL-e1. Pi50 shares greater than 96 % amino acid sequence identity with each of three other R proteins, i.e., Pi9, Piz-t, and Pi2, and has amino acid changes predominantly within the LRR region. The identification of Pi50 with its novel resistance specificity will facilitate the dissection of mechanisms behind the divergence and evolution of different resistance specificities at the Pi2/9 locus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plants have evolved various mechanisms to protect themselves from diverse pathogens; these mechanisms include pathogen-associated molecular pattern (PAMP)-triggered immunity (PTI) and effector-triggered immunity (ETI) (Jones and Dangl 2006; Chen and Ronald 2011). In the PTI system, plants use pattern-recognition receptors to recognize conserved PAMPs, which can trigger immunity. In the ETI system, plants use surveillance proteins, the so-called resistance (R) proteins, to detect the presence of pathogen effector proteins, the so-called avirulence (Avr) proteins. This recognition leads to host resistance against pathogen invasion usually associated with localized cell death at the infection site, known as the hypersensitive response (HR). The largest family of R proteins includes those with nucleotide-binding site and leucine-rich repeat (NBS–LRR) domains, which account for more than half of known plant R proteins and constitute a unique protein family in land plants, but do not occur in other organisms (Yue et al. 2012; Marone et al. 2013; Liu et al. 2014). The NBS–LRR proteins include two major groups, the TIR–NBS–LRR (TNL) and the non-TIR–NBS–LRR (non-TNL) groups, based on the presence or absence of a Toll/Interleukin-1 receptor domain at the N-terminus. The TNL-subgroup R proteins commonly occur in dicots, as identified through genome-wide analyses (Meyers et al. 2003; Ameline-Torregrosa et al. 2008; Kohler et al. 2008; Jupe et al. 2012). By contrast, they are rare in monocots (Bai et al. 2002; Tan and Wu 2012). Moreover, the N-terminal of non-TNL-subgroup R proteins typically contains a conserved coiled-coil domain in dicots but not necessarily in monocots (Bai et al. 2002; Meyers et al. 2003; Zhou et al. 2004; Marone et al. 2013).
Genome-wide investigations have indicated that NBS–LRR genes occur in clusters in plant genomes. For example, 51 % (263 out of 480) of rice NBS–LRR genes reside in 44 clusters with an average of six genes in each cluster (Zhou et al. 2004). Other genomes have an even higher proportion of clustered NBS–LRR genes, e.g., 73 % in potato and 79.8 % in Medicago truncatula (Ameline-Torregrosa et al. 2008; Jupe et al. 2012). Different genetic events including unequal recombination, tandem duplication, and segmental duplication have been proposed to generate different R gene variants in different clusters with varying sizes, which in turn facilitate rapid evolution of novel resistance specificities (Ellis et al. 1999; Hulbert et al. 2001; Meyers et al. 2003; Leister 2004; Ameline-Torregrosa et al. 2008).
In rice, more than 24 R genes have been molecularly characterized to recognize Avr proteins from the fungal pathogen Magnaporthe oryzae, which causes rice blast, one of the most devastating diseases of rice (Ou 1985, Fukuoka et al. 2012; Xu et al. 2014; Ma et al. 2015). All these isolated R genes encode NBS–LRR proteins, except Pi21 and Pid2, which encode a proline-rich protein and a B-lectin protein kinase, respectively. The large numbers of NBS–LRR genes suggests that they constitute the major reservoir of R genes governing rice immunity against blast (Chen et al. 2006; Fukuoka et al. 2009; Moytri et al. 2012). Of these R genes, some are located in the same locus and each R gene controls resistance to a distinct set of M. oryzae isolates, e.g., Pik, Pikm, Pi1, and Pikp at the Pik locus (Ashikawa et al. 2008; Yuan et al. 2011; Zhai et al. 2011; Hua et al. 2012), Pi2, Pi9, and Piz-t at the Pi2/9 locus (Qu et al. 2006; Zhou et al. 2006), and Pish, Pi35, Pi37, and Pi64 at the Pish locus (Nguyen et al. 2006; Lin et al. 2007; Takahashi et al. 2010; Ma et al. 2015).
The Pi2/9 locus has been reported to contain at least eight R genes in both wild and cultivated rice species (Jiang et al. 2012). Notably, these R genes all confer resistance to diverse isolates, suggesting that they may prove valuable for developing resistant rice varieties in breeding programs (Liu et al. 2002; Deng et al. 2006; Jung et al. 2006). The Pi50 gene identified in a resistant cultivar Er-Ba-Zhan (EBZ) was genetically delimited within the Pi2/9 locus and its derived Pi50 near-isogenic line (NIL-e1) shows resistance to 97.7 % of 523 M. oryzae isolates from different regions of China (Zhu et al. 2012). Moreover, the resistance spectrum of NIL-e1 overlaps, but also differs from those observed, respectively, in Pi2, Pi9, and Piz-t NILs, suggesting that Pi50 controls a novel resistance specificity attributable to its different sequence or structure (Zhu et al. 2012). In this study, we determined the genomic sequence of the Pi50 locus by referring to its allelic sequence in the Pi2-containing cultivar C101A51, which allowed us to identify and functionally validate the candidate genes for Pi50. We also describe the conserved and unique features of the Pi50 gene and its locus. The cloning of Pi50 and other Pi2/9 genes will facilitate the understanding of the evolution of novel resistance specificities and their utilization for rice breeding programs.
Materials and methods
Plant materials and blast inoculation
The resistant near-isogenic line NIL-e1, which carries Pi50 in the genetic background of susceptible cultivar Liangjiangxintuanheigu (LTH), was obtained as previously described (Zhu et al. 2012). The monogenic lines each carrying Pi2, Piz-t, and Pi9 used in this study were kindly provided by the International Rice Research Institute. For functional complementation tests, the japonica cultivar Nipponbare was used for stable transformation. The rice cultures were planted in the greenhouse, and the samples were collected and frozen in liquid nitrogen at the four-leaf stage. The genomic DNA was extracted from frozen leaf materials with the Plant gDNA maxi kit (Biomega, USA). Propagation of M. oryzae isolates, pathogen inoculation, and disease evaluation were performed as described previously (Zhu et al. 2012).
Homology-based cloning for reconstruction of the Pi50 locus
We used the sequence of the Pi2 locus as the reference for the reconstruction of the Pi50 locus in its near-isogenic line NIL-e1 using the homology-based cloning method by overlapped long-range PCR (LR-PCR) (Davies and Gray 2002). Seventeen primer pairs were designed along the NBS–LRR gene cluster at the Pi2 locus using both Primer Premier 5.0 (http://www.premierbiosoft.com) and Fast PCR (http://primerdigital.com/fastpcr.html). Each fragment ranged from approximately 6 to 9 kb in length and had an average 1.3 kb overlap with its adjacent fragments for sequence assembly (Table S1). Each fragment was amplified from the genomic DNA of NIL-e1 using KOD-Plus-Neo (TOYOBO, Japan) by following the recommended method. The resultant PCR products were completely sequenced, with sequence walking as necessary. For cloning the 4 copies of Pi50_NBS4, the primer pair (Pix-F2 and Pix-R5, Table S1) was used for PCR amplification and the resultant PCR products were subsequently cloned into the pBlunt Zero (Transgene, Beijing, China) vector for sequence determination.
Sequencing, gene prediction, and phylogenetic analysis
DNA sequencing was performed by the BGI Corporation (China). The sequence reads were assembled into contigs with Seqman from the Lasergene 7.0 package (http://www.dnastar.com/). The vector sequences of each individual read were removed with VecScreen in NCBI (http://blast.ncbi.nlm.nih.gov/). FGENESH (http://www.softberry.com) was used for gene prediction. BLASTn, BLASTx, and BLASTp (http://www.ncbi.nlm.nih.gov/BLAST/) were used for the homology search. BLAST2 and Matcher were used for pairwise sequence comparison. Multiple sequence alignment was performed using Clustalx in MEGA 4.0 (http://multalin.toulouse.inra.fr/multalin/multalin.html, Thompson et al. 1997). The phylogenetic analysis of the NBS–LRR sequences at Pi2/9 locus in different cultivars was performed with MEGA 4.0 and DNAMAN (http://www.lynnon.com/).
Vector construction, rice transformation, and functional validation
Given that only two protein products were deduced from the four copies of Pi50_NBS4, we selected Pi50_NBS4_1 and 3 as representatives of each subgroup for complementation tests. Moreover, the sequence differences between Pi50_NBS4_1 and 3 and both Pi2 and Piz-t are confined to the LRR regions, which allowed the use of the vector pNBS4-Pi2, described previously (Zhou et al. 2006), as a backbone for the replacement of the fragment of either Pi50_NBS4_1 or 3 containing sequence differences from Pi2. In brief, the construct pNBS4-Pi2 was first digested with EcoRI and BamHI to obtain the linear vector. Second, fragment 1 from pNBS4-Pi2 containing the 5′ portion was released by digestion with EcoRI and NdeI, approximately 5.6 kb in length. Fragment 2 corresponding to the 3′ portion of either Pi50_NBS4_1 or 3 was released from the intermediate clones, i.e., the PCR fragment with primers Pix-F3 and Pix-R5 (Table S1) cloned into the pBlunt Zero vector (Transgene, Beijing, China), by digestion with NdeI and BamHI, approximately 2.8 kb. Lastly, fragments 1 and 2 were ligated with the linear vector, resulting in two vectors designated as pPi50N4_1 and 3, respectively, for Pi50_NBS4_1 and 3.
Both the Pi50_NBS4-derived vectors were transformed into the susceptible cultivar Nipponbare via Agrobacterium-mediated transformation, as described previously (Hiei et al. 1997). All T0 plants were tested for blast resistance reaction using isolates 08-T19. The selected T0 plants were then validated by PCR with primers Pix-F5 and 1305R (Table S1). The transgenic T0 lines containing transgenes were advanced for T1 lines. The derived T1 progeny that segregated with the expected 3:1 ratio for resistant:susceptible plants was further sampled for co-segregation analysis.
Characterization of the race specificity of the transgenic lines
Two T2 lines randomly selected from each pPi50N4_1- and 3-derived transgenic line containing copies of the transgene were used for the race-specificity test. The near-isogenic lines containing different Pi2/9 genes were also used. The pathogen inoculation and disease evaluation were conducted as described (Zhu et al. 2012).
Cloning of transcripts of Pi50_NBS4
Total RNA was isolated from leaves of 3-week-old seedlings of the Pi50 near-isogenic line NIL-e1 using the TRIzol reagent (Invitrogen, Carlsbad, CA), following the manufacturer’s instructions. Reverse transcription PCR (RT-PCR) was performed in two steps. First, 1 μg total RNA was treated with RNase-free DNase I (Promega, Madison, WI) and reverse transcribed by M-MLV (Promega, Madison, WI). Then, the reverse-transcribed products were used to amplify the full-length coding sequence (CDS) of different copies of Pi50_NBS4 using the primer pair NBS4F and NBS2R, as described previously (Zhou et al. 2006). The 5′ and 3′ untranslated sequences were obtained using the SMART RACE cDNA amplification kit (Clontech, Mountain View, CA) with the gene-specific primers Pix-R3 and Pix-F5, respectively, by following the manufacturer’s instructions.
Results
Reconstruction of the Pi50 locus and identification of a central genomic block containing four copies of Pi50_NBS4 in the Pi50 near-isogenic line NIL-e1
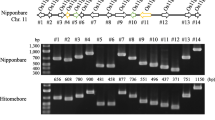
Pi50 was previously mapped to a 53-kb genomic interval of cv. Nipponbare PAC clone P0649C11 (Genbank accession no. AP005659) flanked by the markers GDAP51 and GDAP16 (Zhu et al. 2012). The allelic region of the Pi2 locus (GenBank accession no.: DQ352453) was further delimited to an approximately 85-kb region (position: 39764–124852 bp) mainly due to a large insertion containing Nbs3- and Nbs4-Pi2 (Pi2). Therefore, we used the sequence of the Pi2 locus as the reference for the reconstruction of the Pi50 locus in the Pi50 near-isogenic line NIL-e1 using PCR-based homology cloning. Three sequence contigs including the nitrate-induced protein (NIP) side, the protein kinase (PK) side, and the central block of sequence duplication of the Pi50 locus were obtained (GenBank accession nos. KP985759, KP985760, and KP985761, respectively). Twelve NBS–LRR genes were predicted and named Pi50_NBS1-4 and Pi50_NBS8-12 based on their order in the cluster from the NIP side (Fig. 1a). We note that the central block of sequence duplication contains 4 copies of Pi50_NBS4 named Pi50_NBS4_1 to 4 rather than Pi50_NBS4 to 7, since they are almost identical to each other. The sequences of more than 50 individual clones derived from the PCR-amplified products using the primer pair Pix-F3 and R5, which corresponds to an approximately 4-kb fragment at the 3′ terminal of Pi50_NBS4, clustered into 4 individual clones containing 8 authentic single nucleotide polymorphisms (SNPs) (Fig. 1b). The existence of these SNPs was further confirmed by directly sequencing the PCR products from the genomic DNA (Fig. 1b). Moreover, 6 of those SNPs residing in the coding region were also confirmed by directly sequencing the RT-PCR products (Fig. 1b). Despite our extensive efforts, we failed to decipher the whole sequence covering this central Pi50_NBS4 block due to the limitations of long-PCR technology. However, we obtained two clones from the PCR products using the primer pair Pix-F2 and Pix-R5, which corresponded to Pi50_NBS4_1 and 3, respectively, since each of them have the identical SNPs at the 3′ terminal (GenBank accession nos. KP985761 and KP999983, respectively). Sequence comparison revealed that the whole sequence of Pi50_NBS4_1 is almost identical to Pi50_NBS4_3 (26 nucleotide changes and 3 Indels in 8370 bp). By contrast, significant sequence similarity between Pi50_NBS4_1 and Pi50_NBS2 occurred in only some regions (Fig. S1). Moreover, these homologous regions mainly corresponded to the 1st and 2nd exons and 3′ terminal (Fig. S1). To clarify whether the original Pi50 donors contain duplicated genes, we used the primer pair Pix-F3 and R5 to amplify and sequence the PCR products covering the same region in Er-Ba-Zhan, the original Pi50 donors. Sequences with expected 8 superimposed SNPs further confirmed the existence of 4 copies of Pi50_NBS4 in Pi50-harboring landraces (data not shown). In contrast to the genomic organization observed in the Pi2/9 locus (Zhou et al. 2007), the identification of the genomic block containing 4 copies of Pi50_NBS4 revealed the unique organization of NBS–LRR genes at the Pi50 locus.

The putative genomic organization of the NBS–LRR gene cluster at the Pi50 locus. a A tandem array of 12 NBS–LRR genes at the incompletely determined Pi50 locus, which is composed of three fragments, i.e., the NIP side (GenBank accession no. KP985759), the central genomic block (GenBank accession no. KP985761), and the PK side (GenBank accession no. KP985760). The NBS–LRR genes are indicated by filled arrows, each with a different color, and were named based on their orders in the cluster from the NIP side to the PK side. Due to the incompletely determined sequence, 4 copies of Pi50_NBS4 were stacked and are indicated with numbers for differentiation. The NIP gene is indicated in a square. The figure is drawn to scale. b Distribution of SNPs in 4 copies of Pi50_NBS4 was resolved by sequencing individual clones and PCR products. The portion of the chromatogram file of each sequence read containing the indicated SNPs was retrieved and aligned. The location of the SNP, type of sequence substitution, SNP sequence, and the resultant amino acid residue in one-letter code are listed above the respective position in the released sequence of Pi50_NBS4_1 (GenBank accession no. KP985761). A and S represent non-synonymous and synonymous substitutions, respectively. The one-letter code of each amino acid is as follows, F phenylalanine, S serine, E glutamic, D aspartate, V valine, Y tyrosine, and Q glutamine (color figure online)
To investigate the overall sequence similarity of the regions containing the Pi2 and the Pi50 genes in their respective haplotypes, we generated a pseudomolecule by combining the resolved three contigs (GenBank accession nos. KP985759, KP985760, and KP985761) at the Pi50 locus (approximately 111 kb, neglecting the existence of the three additional Pi50_NBS4 copies) and aligned it to the Pi2 locus. Thus, we identified perfect synteny between the Pi2 and Pi50 regions and these regions showed approximately 91 % nucleotide sequence identity to each other, suggesting that these two regions are significantly conserved with respect to gene order and sequence similarity (Fig. S2). The genes at the Pi50 locus with the same genomic context as those at other Pi2/9 locus are usually clustered together in the same phylogenetic clade, suggesting that orthologs from different haplotypes are more similar than the paralogs in respective haplotypes (Fig. S3). Based on the orthologous relationship of the NBS–LRR genes in different haplotypes, we designated 9 orthologous groups, i.e., Pi50_NBS1-4, Pi50_NBS8-12 (Fig. S2).
Pi50_NBS4_1/2 are responsible for Pi50 function
The reconstruction of the Pi50 locus allowed us to determine the candidate genes responsible for Pi50 function. We first aligned the flanking markers GDAP51 and GDAP16 and delimited the genomic interval ranging from the 3′ part of Pi50_NBS1 to the 3′ terminal of Pi50_NBS12 at the Pi50 locus. Linkage analysis showed that each of these two markers had one recombination with Pi50 (Zhu, et al. 2012). Thus, Pi50_NBS1 and Pi50_NBS12 can be excluded from the Pi50 candidates. We thus speculated that Pi50_NBS2-3, Pi50_NBS4_1-4, and Pi50_NBS8-11 could be the Pi50 candidates. Given that Pi50_NBS2-3 and Pi50_NBS8-11 are pseudogenes, based on their various sequence mutations (see the annotation in GenBank), we further shortlisted Pi50_NBS4_1-4 as the candidates for Pi50.
We found that the 4 copies of Pi50_NBS4 encode two different protein products, resulting in two subgroups, i.e., Pi50_NBS4_1/2 and 3/4 (Figs. 1b, 3b). To test which subgroup is responsible for Pi50 function, we selected Pi50_NBS4_1 and Pi50_NBS4_3 as representatives of their respective subgroups for gene complementation tests. We then tested whether transformation with each representative gene could provide blast resistance to a susceptible cultivar. We generated 59 and 122 independent T0 transgenic lines for Pi50_NBS4_1 and 3, respectively, in the blast-sensitive cultivar Nipponbare. For Pi50_NBS4_1, 48 out of 59 pPi50N4_1-derived transgenic lines were resistant to the Pi50-avirulent isolate 08-T19, which is virulent on Nipponbare. By contrast, for Pi50_NBS4_3, all the 122 pPi50N4_3-derived transgenic plants were susceptible to 08-T19. Moreover, the presence of the transgene segregated with blast resistance in the T1 progeny derived from 9 individual resistant pPi50N4_1 T0 lines, indicating that the resistance to 08-T19 is attributable to the integration of Pi50_NBS4_1 in the transgenic plants (Fig. 2). However, the advanced T1 lines from those pPi50N4_3-derived T0 plants remained susceptible to 08-T19 although they all contained the transgene (Fig. 2).

Gene complementation tests of Pi50. Two T1 lines from each pPi50N4_1- and 3-derived transgenic plant were used for representative samples. Leaves of 4 individual plants from each line at the 7th day after inoculation with the Pi50-avirulent isolate 08-T19 used for photography. The DNA from each respective plant was isolated for molecular validation, which used the primer pair Pix-F5 and 1305R (Table S1) and the product was examined by agarose gel electrophoresis. The Pi50 near-isogenic line NIL-e1, the line transformed with empty vector, and the recipient line for Nipponbare transformation were used as control lines
To validate whether Pi50_NBS4_1 is fully responsible for Pi50 functionality, we selected homozygous lines from the pPi50N4_1 and 3 transgenic lines and tested their resistance to diverse isolates. If Pi50_NBS4_1 is responsible for Pi50 functionality, then transgenic plants should show resistance to the same blast isolates as the Pi50-containing near-isogenic line. Plants from the pPi50N4_1-derived T2 line showed resistance to all 22 blast isolates. By contrast, plants from pPi50N4_3-derived T2 line were susceptible to all isolates except W08-47 and 12-3058 (Table 1). These two isolates are also avirulent on Nipponbare, indicating that the resistance to these two isolates could not be attributable to the transgene. Compared to Pi2, Pi9, and Piz-t NILs, the pPi50N4_1-derived line showed the same reactions as the Pi50 near-isogenic line NIL-e1 to the 20 different isolates, indicating that Pi50_NBS4_1 produces full-spectrum resistance, as does Pi50. Taken together, we concluded that Pi50_NBS4_1, not Pi50_NBS4_3, is responsible for Pi50 function.
Pi50 shares high sequence similarity to other Pi2/9 genes and controls a novel resistance specificity
We obtained full-length cDNAs of Pi50 (Pi50_NBS4_1/2) by a combination of RT- and RACE-RT-PCR, which allowed the determination of the size and position of the introns and exons. The full-length CDS of Pi50 is 3099 bp in length with a 127-bp 5′ UTR and a 505-bp 3′ UTR. It contains 3 exons, 116-bp, 2952-bp, 31-bp, disrupted by two introns, 3839-bp and 128-bp (Fig. 3a). The 1st and 2nd introns are located prior to the predicted NBS and posterior to the LRR domains, respectively (Fig. 3b). Pi50_NBS4_1 and 2, share sequence identity to one another in the coding region, however, they differ in at least two nucleotides from one another; these differences are in the 2nd intron, as deduced from the partially determined sequences (Fig. 1b). Pi50 encodes a 1033-amino acid (aa) protein. Three typical domain/motifs were identified in Pi50, i.e., nT: 68–86 aa, NBS: 153–460 aa, and 17 perfect or imperfect LRR repeats: 535–935 aa (Fig. 3b). As Fig. 3b illustrates, the centralized NBS domain has all the essential motifs: P-loop (193–202 aa), kinase 2 (281–287 aa), RNBS-B (307–315 aa), and GLPL (373–378 aa), as described previously (Bai et al. 2002).

The structure of Pi50 and sequence differences in the Pi2/9-allelic R proteins. a The structure of the transcript of Pi50. Three exons are indicated as filled boxes and two introns and 5′/3′ untranslated regions (UTRs) are indicated as lines. The length of each fragment was based on the sequence of Pi50_NBS4_1 (GenBank accession no. KP985761). The start and stop codons are indicated with ATG and TAG, respectively. b Multiple sequence alignment of deduced proteins of Pi50_NBS4_1/2, 3/4, Pi2, Piz-t, and Pi9. The protein sequence is shown in one-letter code. The positions of two introns in the corresponding genomic DNA sequence are marked by open arrowheads. The non-TIR (nT) motif at the N-terminal is highlighted in bold. The P-loop, Kinase-2, RNBS-B, and GLPL motifs of the NBS domain are underlined. Seventeen imperfect xxLxLxx motifs in the LRR domain are boxed. Dots identical amino acids in Pi50_NBS4_1/2 and other proteins. Dash the single amino acid deletion in all proteins except Piz-t. The sites subject to positive selection with greater than 95 and 99 % confidence are indicated as single and double stars, respectively
Alignment analysis revealed that Pi50 shares 96, 97, and 97 % amino acid sequence identity with Pi9, Pi2, and Piz-t, respectively. Moreover, the variant residues disproportionately occur in the predicted LRR region. For example, 25 out of 31 total amino acid changes between Pi2 and Pi50 occur in the LRR region. Resistance specificity analysis using diverse isolates reciprocally avirulent/virulent to Pi50, Pi2, Pi9, and Piz-t revealed that they each control overlapping, albeit distinct resistance specificities (Fig. 4; Table 1). In this study, we found that Pi9 and Pi50 isogenic lines each control 95.5 % of the resistance spectrum to 22 isolates (Table 1). By contrast, Pi2 and Piz-t isogenic lines control relatively narrower resistance spectra. Taking these results together, we postulate that they differ in the resistance specificities likely due to their sequence differences.

Different resistances controlled by Pi50 and other Pi2/9 allelic R genes. Three isolates (W08-47, 12-3058, 13-856) showing reciprocally avirulent and virulent reactions to Pi50, Pi2, Pi9, and Piz-t were used for inoculation. One leaf representative of each of four near-isogenic lines at the 7th day after the pathogen infection was used for photography. The resistant (R) or susceptible (S) reaction is indicated below each leaf
Pi50_NBS4_3/4 encode a Pi50-variant with four amino acid changes
As described above, Pi50_NBS4_3/4 encode another protein that is not responsible for the function of Pi50. Pi50_NBS4_3 and 4 differ in a single synonymous nucleotide sequence change in the coding region and each encodes a protein without sequence differences (Fig. 1b). The derived protein product Pi50_NBS4_3/4 contains four amino acid differences from Pi50_NBS4_1/2. Of these four amino acid residues, one [Aspartate (D) in Pi50_NBS4_1/2 versus Tyrosine (Y) in Pi50_NBS4_3/4 at 961 aa] is situated outside the LRR repeats at the C terminus (Fig. 3b). The other three residues occur within the predicted xxLxLxx motifs in the LRR domain (Fig. 3b). Interestingly, these four amino acid changes occur within the same hydrophobic/hydrophilic groups with respect to their propensity to contact polar solvent. For example, D961 versus Y961, Glutamic acid (E) in Pi50_NBS4_1/2 versus Glutamine (Q) in Pi50_NBS4_3/4 at 909 aa, Serine (S) in Pi50_NBS4_1/2 versus Y in Pi50_NBS4_3/4 at 861 belong to the hydrophilic group. By contrast, Phenylalanine (F) in Pi50_NBS4_1/2 versus Valine (V) in Pi50_NBS4_3/4 at 859 aa belong to the hydrophobic group. Only Q909 is unique to Pi50_NBS4_3/4 and is not present in either Pi2, Pi9, or Piz-t (Fig. 3b).
The Pi50 and its Pi2/9 homologues are subject to sequence exchange and positive selection
To investigate the evolutionary mechanisms underlying the emergence of different resistance specificities controlled by the Pi2/9 genes, we conducted multiple sequence alignment of the Pi2/9 homologs. As Fig. 5 illustrates, this identified traceable sequence exchanges among different Pi50 homologues from both Pi50_NBS2 and Pi50_NBS4 orthologous groups, resulting in the chimeras with different hinge points. For example, Pi50_NBS2 and AP005659_NBS2 in the Pi50_NBS2 orthologous group have the same informative polymorphism sites (IPS) as Pi50_NBS4_4 in the Pi50_NBS4 orthologous group at 2506, 2555, 2650, and 2874 bp, whereas they do not have the same IPS at 2585 and 2718 bp (Fig. 5).

Chimeric structure of the Pi50 homologues from the Pi50_NBS2 and 4 orthologous groups. Sequence strings containing same informative polymorphism sites (IPS) are indicated without or with different shading. The consensus sequence is shown at the bottom. Dashes identical nucleotides to the consensus sequence
We adopted the pair of maximum likelihood models of codon substitution, M8/M7 for detecting the sites subject to positive selection in the Pi50 homologs (Yang 1997; Yang et al. 2000). This identified 17 amino acid sites with greater than 95 % confidence, indicating these sites as being under positive selection (Fig. 3b). When these sites were plotted along the Pi50 protein, we found that 15 out of 17 sites occurred within the LRR domains (Fig. 3b), with only 1 each at the N-terminal and at C-terminal non-LRR regions. We further found that 10 sites were situated in the putative xxLxLxx motifs (Fig. 3b).
Discussion
Pi50 and its allelic R genes at the Pi2/9 locus control distinct resistance specificities by recognizing unrelated Avr genes in M. oryzae
In plants some R gene loci harbor multiple specificities capable of recognizing related or unrelated pathogen-derived avirulence effectors. The flax L locus consists of 13 L alleles with different specificities (L, L1–L11, and LH) sharing over 90 % amino acid sequence identity to each other (Ellis et al. 1999; Luck et al. 2000). The AvrL567 locus in the flax rust fungus consists of 12 variants, 7 of which can be recognized by L5, L6, and/or L7 through direct protein–protein interaction (Dodds et al. 2006). In the rice/rice blast pathosystem, the Pik locus has maintained multiple alleles with extremely limited sequence differences to recognize different allelic variants at the AvrPik locus in M. oryzae (Kanzaki et al. 2012; Wu et al. 2014). In this study, we describe the cloning and functional characterization of Pi50, the 4th R gene at the Pi2/9 locus, in addition to Pi2, Pi9, and Piz-t (Qu et al. 2006; Zhou et al. 2006). The proteins encoded by these four genes share greater than 96 % amino acid sequence identity to one another and their sequence differences disproportionally occur within the LRR domain (Zhou et al. 2006; this study). Interestingly, their resistance spectra can be clearly differentiated with different sets of M. oryzae isolates, suggesting that these four R genes recognize different Avr effectors (Liu et al. 2002; Li et al. 2009; this study). As described in Table 1, both GUY11 (without AvrPiz-t and AvrPi2, Li et al. 2009) and R01-1 (without AvrPi9, Wu et al. 2015) are avirulent to Pi50 (Table 1), indicating that Pi50-mediated immunity to M. oryzae does not require the existence of AvrPiz-t, AvrPi9, or AvrPi2. Moreover, the reactions of Pi2 to different M. oryzae strains indicate that AvrPi2 is not an allele of either AvrPiz-t or AvrPi9 (Li et al. 2009; Wu et al. 2015). Indeed, AvrPiz-t and AvrPi9 are located in different genomic positions and do not share detectable sequence similarities (Li et al. 2009; Wu et al. 2015). These findings imply that different R genes at the Pi2/9 locus each confer an overlapping, albeit distinct resistance spectrum by recognizing unrelated Avr genes in M. oryzae.
Gene duplication and sequence divergence boost the evolution of Pi50 and its allelic R gene specificities at the Pi2/9 locus
NBS–LRR genes are the largest class of R genes in plants, and tend to be clustered in tandem arrays with varying genomic sizes (Richter and Ronald 2000; Martin et al. 2003). Various genetic events including tandem duplications, intra- and inter-locus R gene sequence exchanges by unequal recombination, sequence mutations, and insertions/excisions of transposable elements have been documented in different NBS–LRR loci (Sun et al. 2001; Zhou et al. 2007; Kuang et al. 2008). The frequency of intra- and inter-locus sequence exchanges between R gene homologs largely determines the evolutionary fate of different R genes, which are classified into Type I and II (Kuang et al. 2004). Type I R genes are associated with frequent sequence exchanges and show significant chimeric relationships among homologs. By contrast, Type II R genes have undetectable sequence exchanges and show significant orthologous/allelic relationships in different haplotypes (Hulbert et al. 2001; Kuang et al. 2004). In the present study, we identified 12 NBS–LRR genes at the Pi50 locus in the Pi50 near-isogenic line NIL-e1. The Pi50 locus shares good synteny with the Pi2/9 locus in other haplotypes (Zhou et al. 2007). Moreover, the different paralogues have significantly limited sequence similarity within the coding sequence, as previously observed in the Pi2/9 locus (Zhou et al. 2007). We speculate that tandem duplications followed by dramatic sequence divergence in the promoter and first intron have contributed significantly to the shape of the extant Pi50 and its allelic complex locus. However, a genomic block containing 4 copies of Pi50_NBS4 with almost identical sequence in both coding and noncoding regions has been identified, representing a unique structure that differs from that of the Pi2/9 locus in other cultivars (Zhou et al. 2007). Coincidently, a similar duplication block was also described at the Pi2/9 locus in the CC subgenome of O. minuta; this block contains four tandem NBS3- and NBS4-OM_CC arrays almost identical in sequence (Dai et al. 2010). We speculate that the tandem duplication resulting in the Pi50_NBS4 duplications could have occurred quite recently. Intriguingly, traceable sequence exchanges among Pi50-NBS2- and Pi50-NBS4-orthologuous groups further imply that frequent unequal recombination could play an important role in generating multiple copies of Pi50_NBS4. Dai et al. (2010) illustrated that the Pi9 gene originally introgressed from O. minuta with the BBCC genome is more related to its homologs in other cultivars than to those in wild rice species, indicating the possibility that Pi9 was introgressed by sequence exchanges rather than authentic introgression of genomic content from the wild relative. Therefore, we postulate that sequence exchanges at the inter- and intra-locus levels provide more variants, allowing for rapid evolution of new resistance specificities at the Pi2/9 locus. To this end, the Pi2/9 homologs can be considered as Type I R genes as characterized previously (Luo et al. 2012). However, we argue that sequence exchanges could be limited within the coding sequences of Pi50_NBS2- and Pi50_NBS4-orthologous groups, for the chimeric structure was not detected among other orthologous groups at the Pi50 and its allelic Pi2/9 locus (Zhou et al. 2007; Dai et al. 2010, and this study). The characterization of Pi50 and its allelic R genes at the Pi2/9 locus permits the delimitation of regions responsible for the recognition specificity. As previously described, differences in eight amino acid residues determine the resistance specificity between Pi2 and Piz-t and most of the amino acid changes between Pi2/z-t and Pi9 are confined within the LRR region, supporting the idea that the resistance specificity is mainly determined by the LRR region (Zhou et al. 2006). In this study, we found a similar scenario displayed in Pi50, which contains more sequence differences in the LRR region compared to other three R genes at the Pi2/9 locus, further supporting the idea that this region plays a critical role in determination of novel resistance specificity.
Plant NBS–LRR-type R genes may be subject to positive selection predominantly confined within the LRR region, in which elevated ratio of non-synonymous (Ka) to synonymous (Ks) nucleotide substitutions is detected (Michelmore and Meyers 1998; Mondragon-Palomino and Gaut 2005; Chen et al. 2010). The positive selection was postulated as the major force driving the diversification of the LRR regions, which play major roles in determination of specific recognition of pathogen-derived avirulence effectors by direct or indirect interactions (Bakker et al. 2006; Ravensdale et al. 2012). Moreover, R genes at complex loci are more divergent than those in singletons, suggesting that diversifying selection acts more prevalently in gene clusters (Luo et al. 2012). In this study, a disproportionate number of sites (16 out of 17) under strong positive selection were confined within the LRR region, further emphasizing the significance of positive selection acting on the Pi2/9 homologs (Zhou et al. 2007; Dai et al. 2010; Wu et al. 2012). We notice that the amino acid difference of glutamine-909 in Pi50_NBS4_3/4 versus glutamic acid-909 in Pi50_NBS4_1/2 is not subject to positive selection. Moreover, the causal nucleotide change (G to C) is only observed in Pi50_NBS4_1/2, suggesting that it could more likely be a de novo mutation rather the result of sequence exchange. It will be interesting to investigate whether this single amino acid is critical for the maintenance of specific recognition of AvrPi50.
Utility of Pi50 and its allelic genes at the Pi2/9 locus for breeding varieties with broad-spectrum resistance to rice blast
Broad-spectrum R gene-mediated resistance does not produce durable resistance in the field. The situation with “broad-spectrum” R genes often occurs when an R gene is newly introduced into a cultivated crop species and the pathogens have not been previously exposed to selection by the so-called “broad-spectrum” R gene. Once strong selection is applied to these genes, previously avirulent pathogen races are likely to evolve virulence to overcome existing R genes (Ellis et al. 2014). Cases corresponding to the R genes at Pi1 and Pi2 loci have been previously reported in South China (Zhu et al. 2008; Wang et al. 2012a). In the interactions between R gene RPS5 alleles in Arabidopsis and the Avr gene avrPphB in Pseudomonas syringae, the R gene polymorphism in the host population may not be maintained through a tightly coupled interaction involving a single coevolved pair of R/Avr genes. More likely, the stable polymorphism is maintained through complex and diffuse community-wide interactions (Karasov et al. 2014). We speculate that introducing only a single, broad-spectrum R gene that is effective against diverse isolates is not sufficient to obtain an elite cultivar with durable resistance. It is, however, reasonable that multiple R genes in a single variety or multiple lines could force the pathogen to evolve new predominant virulent isolates from avirulent ones. Pi50 and its allelic genes at the Pi2/9 locus have been reported to be effective in different rice-growing areas, indicating its high value for rice breeding for blast resistance (Jiang et al. 2012; Jeung et al. 2007; Yang et al. 2008; Wang et al. 2012b; Zhu et al. 2012). Moreover, in contrast to Pi1 and its alleles at the Pik locus, another locus controlling broad-spectrum resistance, which mediates stepwise arms race with different AvrPik genes (Wu et al. 2014), each of the Pi2/9 alleles can recognize distinct Avr gene alleles. We speculate that the utilization of Pi50 and its allelic genes in rice varieties at the Pi2/9 locus should be helpful for maintaining stable polymorphisms of M. oryzae Avr genes, which can in turn help rice to keep relatively stable resistance to the changing pathogen.
Author contribution statement
Jing Su contributed to the construction and the functional analysis of Pi50 and preparation of the manuscript. WenJuan Wang contributed to the long-range PCR-based gene cloning and the transformation with Pi50 candidate genes. JingLuan Han contributed to the sequencing of the Pi50 locus. Shen Chen contributed to the analysis of full-length cDNAs of Pi50 candidate genes. CongYing Wang contributed to the gene expression analysis. LieXian Zeng, AiQing Feng, and Jianyuan Yang contributed to the propagation of blast isolates, pathogen inoculation, and disease evaluation. Bo Zhou contributed to the evolutionary analysis and preparation of the manuscript. XiaoYuan Zhu led the project and wrote the manuscript.
References
Ameline-Torregrosa C, Wang B, O’Bleness M, Deshpande S, Zhu H, Roe B, Young N, Cannon S (2008) Identification and characterization of nucleotide-binding site-leucine-rich repeat genes in the model plant Medicago truncatula. Plant Physiol 146:5–21
Ashikawa I, Hayashi N, Yamane H, Kanamori H, Wu J, Matsumoto T, Ono K, Yano M (2008) Two adjacent nucleotide-binding site-leucine-rich repeat class genes are required to confer Pikm-specific rice blast resistance. Genetics 180:2267–2276
Bai J, Pennill L, Ning J, Lee S, Ramalingam J, Webb C, Zhao B, Sun Q, Nelson J, Leach J, Hulbert S (2002) Diversity in nucleotide binding site-leucine-rich repeat genes in cereals. Genome Res 12:1871–1884
Bakker E, Toomajian C, Kreitman M, Bergelson J (2006) A genome-wide survey of R gene polymorphisms in Arabidopsis. Plant Cell 18:1803–1818
Chen X, Ronald P (2011) Innate immunity in rice. Trends Plant Sci 16:451–459
Chen X, Shang J, Chen D, Lei C, Zou Y, Zhai W, Liu G, Xu J, Ling Z, Cao G, Ma B, Wang Y, Zhao X, Li S, Zhu L (2006) A B-lectin receptor kinase gene conferring rice blast resistance. Plant J 46:794–804
Chen Q, Han Z, Jiang H, Tian D, Yang S (2010) Strong positive selection drives rapid diversification of R-genes in Arabidopsis relatives. J Mol Evol 70:137–148
Dai L, Wu J, Li X, Wang X, Liu X, Jantasuriyarat C, Kudrna D, Yu Y, Wing R, Han B, Zhou B, Wang G (2010) Genomic structure and evolution of the Pi2/9 locus in wild rice species. Theor Appl Genet 121:295–309
Davies PA, Gray G (2002) Long-range PCR. Methods Mol Biol 187:51–55
Deng Y, Zhu X, Shen Y, He Z (2006) Genetic characterization and fine mapping of the blast resistance locus Pigm(t) tightly linked to Pi2 and Pi9 in a broad-spectrum resistant Chinese variety. Theor Appl Genet 113:705–713
Dodds P, Lawrence G, Catanzariti A, Teh T, Wang C, Ayliffe M, Kobe B, Ellis J (2006) Direct protein interaction underlies gene-for-gene specificity and coevolution of the flax resistance genes and flax rust avirulence genes. Proc Natl Acad Sci USA 103:8888–8893
Ellis J, Lawrence G, Luck J, Dodds P (1999) Identification of regions in alleles of the flax rust resistance gene L that determine differences in gene-for-gene specificity. Plant Cell 11:495–506
Ellis J, Lagudah E, Spielmeyer W, Dodds P (2014) The past, present and future of breeding rust resistant wheat. Front Plant Sci 5:641
Fukuoka S, Saka N, Koga H, Ono K, Shimizu T, Ebana K, Hayashi N, Takahashi A, Hirochika H, Okuno K, Yano M (2009) Loss of function of a proline-containing protein confers durable disease resistance in rice. Science 325:998–1001
Fukuoka S, Mizobuchi R, Saka N, Suprun I, Matsumoto T, Okuno K, Yano M (2012) A multiple gene complex on rice chromosome 4 is involved in durable resistance to rice blast. Theor Appl Genet 125:551–559
Hiei Y, Komari T, Kubo T (1997) Transformation of rice mediated by Agrobacterium tumefaciens. Plant Mol Biol 35:205–218
Hua L, Wu J, Chen C, Wu W, He X, Lin F, Wang L, Ashikawa I, Matsumoto T, Wang L, Pan Q (2012) The isolation of Pi1, an allele at the Pik locus which confers broad spectrum resistance to rice blast. Theor Appl Genet 125:1047–1055
Hulbert S, Webb C, Smith S, Sun Q (2001) Resistance gene complexes: evolution and utilization. Annu Rev Phytopathol 39:285–312
Jeung J, Kim B, Cho Y, Han S, Moon H, Lee Y, Jena K (2007) A novel gene, Pi40(t), linked to the DNA markers derived from NBS–LRR motifs confers broad spectrum of blast resistance in rice. Theor Appl Genet 115:1163–1177
Jiang N, Li Z, Wu J, Wang Y, Wu L, Wang S, Wang D, Wen T, Liang Y, Sun P, Liu J, Dai L, Wang Z, Wang C, Luo M, Liu X, Wang G (2012) Molecular mapping of the Pi2/9 allelic gene Pi2-2 conferring broad-spectrum resistance to Magnaporthe oryzae in the rice cultivar Jefferson. Rice 5:29
Jones J, Dangl J (2006) The plant immune system. Nature 444:323–329
Jung Y, Agrawal G, Rakwal R, Kim J, Lee M, Choi P, Kim Y, Kim M, Shibato J, Kim S, Iwahashi H, Jwa N (2006) Functional characterization of OsRacB GTPase–a potentially negative regulator of basal disease resistance in rice. Plant Physiol Biochem 44:68–77
Jupe F, Pritchard L, Etherington G, Mackenzie K, Cock P, Wright F, Sharma S, Bolser D, Bryan G, Jones J, Hein I (2012) Identification and localization of the NB-LRR gene family within the potato genome. BMC Genom 13:75
Kanzaki H, Yoshida K, Saitoh H, Fujisaki K, Hirabuchi A, Alaux L, Fournier E, Tharreau D, Terauchi R (2012) Arms race co-evolution of Magnaporthe oryzae AVR-Pik and rice Pik genes driven by their physical interactions. Plant J 72:894–907
Karasov T, Kniskern J, Gao L, DeYoung B, Ding J, Dubiella U, Lastra R, Nallu S, Roux F, Innes R, Barrett L, Hudson R, Bergelson J (2014) The long-term maintenance of a resistance polymorphism through diffuse interactions. Nature 512:436–440
Kohler A, Rinaldi C, Duplessis S, Baucher M, Geelen D, Duchaussoy F, Meyers B, Boerjan W, Martin F (2008) Genome-wide identification of NBS resistance genes in Populus trichocarpa. Plant Mol Biol 66:619–636
Kuang H, Woo S, Meyers B, Nevo E, Michelmore R (2004) Multiple genetic processes result in heterogeneous rates of evolution within the major cluster disease resistance genes in lettuce. Plant Cell 16:2870–2894
Kuang H, Caldwell K, Meyers B, Michelmore R (2008) Frequent sequence exchanges between homologs of RPP8 in Arabidopsis are not necessarily associated with genomic proximity. Plant J 54:69–80
Leister D (2004) Tandem and segmental gene duplication and recombination in the evolution of plant disease resistance gene. Trends Genet 20:116–122
Li W, Wang B, Wu J, Lu G, Hu Y, Zhang X, Zhang Z, Zhao Q, Feng Q, Zhang H, Wang Z, Wang G, Han B, Wang Z, Zhou B (2009) The Magnaporthe oryzae avirulence gene AvrPiz-t encodes a predicted secreted protein that triggers the immunity in rice mediated by the blast resistance gene Piz-t. Mol Plant Microbe Interact 22:411–420
Lin F, Chen S, Que Z, Wang L, Liu X, Pan Q (2007) The blast resistance gene Pi37 encodes a nucleotide binding site leucine-rich repeat protein and is a member of a resistance gene cluster on rice chromosome 1. Genetics 177:1871–1880
Liu G, Lu G, Zeng L, Wang G (2002) Two broad-spectrum blast resistance genes, Pi9(t) and Pi2(t), are physically linked on rice chromosome 6. Mol Genet Genomics 267:472–480
Liu W, Liu J, Triplett L, Leach J, Wang G (2014) Novel insights into rice innate immunity against bacterial and fungal pathogens. Annu Rev Phytopathol 52:213–241
Luck J, Lawrence G, Dodds P, Shepherd K, Ellis J (2000) Regions outside of the leucine-rich repeats of flax rust resistance proteins play a role in specificity determination. Plant Cell 12:1367–1377
Luo S, Zhang Y, Hu Q, Chen J, Li K, Lu C, Liu H, Wang W, Kuang H (2012) Dynamic nucleotide-binding site and leucine-rich repeat-encoding genes in the grass family. Plant Physiol 159:197–210
Ma J, Lei C, Xu X, Hao K, Wang J, Cheng Z, Ma X, Ma J, Zhou K, Zhang X, Guo X, Wu F, Lin Q, Wang C, Zhai H, Wang H, Wan J (2015) Pi64, encoding a novel CC–NBS–LRR protein, confers resistance to leaf and neck blast in rice. Mol Plant Microbe Interact (Epub ahead of print)
Marone D, Russo M, Laido G, De Leonardis A, Mastrangelo A (2013) Plant nucleotide binding site-leucine-rich repeat (NBS–LRR) genes: active guardians in host defense responses. Int J Mol Sci 14:7302–7326
Martin G, Bogdanove A, Sessa G (2003) Understanding the functions of plant disease resistance proteins. Annu Rev Plant Biol 54:23–61
Meyers B, Kozik A, Griego A, Kuang H, Michelmore R (2003) Genome-wide analysis of NBS–LRR-encoding genes in Arabidopsis. Plant Cell 15:809–834
Michelmore R, Meyers B (1998) Clusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res 8:1113–1130
Mondragon-Palomino M, Gaut B (2005) Gene conversion and the evolution of three leucine-rich repeat gene families in Arabidopsis thaliana. Mol Biol Evol 22:2444–2456
Moytri R, Jia Y, Richard DC (2012) Structure, function, and co-evolution of rice blast resistance genes. Acta Agron Sin 38:381–393
Nguyen T, Koizumi S, La T, Zenbayashi K, Ashizawa T, Yasuda N, Imazaki I, Miyasaka A (2006) Pi35(t), a new gene conferring partial resistance to leaf blast in the rice cultivar Hokkai 188. Theor Appl Genet 113:697–704
Ou S (1985) Rice disease. Second Edition, Commonwealth Mycological Institute, Kew Surrey, The Cambrian News Ltd, UK, pp 109–201
Qu S, Liu G, Zhou B, Bellizzi M, Zeng L, Dai L, Han B, Wang G (2006) The broad-spectrum blast resistance gene Pi9 encodes a nucleotide-binding site-leucine-rich repeat protein and is a member of a multigene family in rice. Genetics 172:1901–1914
Ravensdale M, Bernoux M, Ve T, Kobe B, Thrall P, Ellis J, Dodds P (2012) Intramolecular interaction influences binding of the Flax L5 and L6 resistance proteins to their AvrL567 ligands. PLoS Pathog 8:e1003004
Richter T, Ronald P (2000) The evolution of disease resistance genes. Plant Mol Biol 42:195–204
Sun Q, Collins N, Ayliffe M, Smith S, Drake J, Pryor T, Hulbert S (2001) Recombination between paralogues at the Rp1 rust resistance locus in maize. Genetics 158:423–438
Takahashi A, Hayashi N, Miyao A, Hirochika H (2010) Unique features of the rice blast resistance Pish locus revealed by large scale retrotransposon-tagging. BMC Plant Biol 10:175
Tan S, Wu S (2012) Genome Wide analysis of nucleotide-binding site disease resistance genes in Brachypodium distachyon. Comp Funct Genomics 2012:418208
Thompson J, Gibson T, Plewniak F, Jeanmougin F, Higgins D (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl Acids Res 25:4876–4882
Wang W, Su J, Zhang J, Li Y, Chen S, Zeng L, Yang J, Zhu X (2012a) Pathogenicity analysis of the rice blast fungus isolated from the blast panicles of Yuejingsimiao 2. Guangdong Agric Sin 23:59–61 (in Chinese)
Wang Y, Wang D, Deng X, Liu J, Sun P, Liu Y, Huang H, Jiang N, Kang H, Ning Y, Wang Z, Xiao Y, Liu X, Liu E, Dai L, Wang G (2012b) Molecular mapping of the blast resistance genes Pi2-1 and Pi51(t) in the durably resistant rice ‘Tianjingyeshengdao’. Phytopathology 102:779–786
Wu K, Xu T, Guo C, Zhang X, Yang S (2012) Heterogeneous evolutionary rates of Pi2/9 homologs in rice. BMC Genet 13:73
Wu W, Wang L, Zhang S, Li Z, Zhang Y, Lin F, Pan Q (2014) Stepwise arms race between AvrPik and Pik alleles in the rice blast pathosystem. Mol Plant Microbe Interact 27:759–769
Wu J, Kou Y, Bao J, Li Y, Tang M, Zhu X, Ponaya A, Xiao G, Li J, Li C, Song M, Cumagun C, Deng Q, Lu G, Jeon J, Naqvi N, Zhou B (2015) Comparative genomics identifies the Magnaporthe oryzae avirulence effector AvrPi9 that triggers Pi9-mediated blast resistance in rice. N Phytol. doi:10.1111/nph.13310 (Epub ahead of print)
Xu X, Lv Q, Shang J, Pang Z, Zhou Z, Wang J, Jiang G, Tao Y, Xu Q, Li X, Zhao X, Li S, Xu J, Zhu L (2014) Excavation of Pid3 orthologs with differential resistance spectra to Magnaporthe oryzae in rice resource. PLoS One 9:e93275
Yang Z (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13:555–556
Yang Z, Nielsen R, Goldman N, Pedersen AM (2000) Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449
Yang J, Chen S, Zeng L, Li Y, Chen Z, Li C, Zhu X (2008) Race specificity of major rice blast resistance genes to Magnaporthe grisea isolates collected from indica Rice in Guangdong, China. Rice Sci 15:311–318
Yuan B, Zhai C, Wang W, Zeng X, Xu X, Hu H, Lin F, Wang L, Pan Q (2011) The Pik-p resistance to Magnaporthe oryzae in rice is mediated by a pair of closely linked CC–NBS–LRR genes. Theor Appl Genet 122:1017–1028
Yue J, Meyers B, Chen J, Tian D, Yang S (2012) Tracing the origin and evolutionary history of plant nucleotide-binding site-leucine-rich repeat (NBS–LRR) genes. N Phytol 193:1049–1063
Zhai C, Lin F, Dong Z, He X, Yuan B, Zeng X, Wang L, Pan Q (2011) The isolation and characterization of Pik, a rice blast resistance gene which emerged after rice domestication. N Phytol 189:321–334
Zhou T, Wang Y, Chen J, Araki H, Jing Z, Jiang K, Shen J, Tian D (2004) Genome-wide identification of NBS genes in japonica rice reveals significant expansion of divergent non-TIR NBS–LRR genes. Mol Genet Genomics 271:402–415
Zhou B, Qu S, Liu G, Dolan M, Sakai H, Lu G, Bellizzi M, Wang G (2006) The eight amino-acid differences within three leucine-rich repeats between Pi2 and Piz-t resistance proteins determine the resistance specificity to Magnaporthe grisea. Mol Plant Microbe Interact 19:1216–1228
Zhou B, Dolan M, Sakai H, Wang G (2007) The genomic dynamics and evolutionary mechanism of the Pi2/9 locus in rice. Mol Plant Microbe Interact 20:63–71
Zhu X, Yang J, Chen Y, Yang W, Chen X, Zeng L, Chen S (2008) Race identification and pathogenicity test of the blast fungus causing the resistance breakdown of hybrid rice Tianyou 998. Guangdong Agric Sin 12:84–86 (in Chinese)
Zhu X, Chen S, Yang J, Zhou S, Zeng L, Han J, Su J, Wang L, Pan Q (2012) The identification of Pi50(t), a new member of the rice blast resistance Pi2/Pi9 multigene family. Theor Appl Genet 124:1295–1304
Acknowledgments
We thank Y. L. Peng and S. Q. Wu for providing rice blast isolates. This research is supported by grants from the National Transgenic Research Projects (2014ZX0800904B), the National Natural Science Foundation (31301304, 31461143019), the Guangzhou sciences and technology project (2012J2200066, 2012J4300059), Earmarked Fund for Modern Agro-Industry Technology Research System (CARS-01-24).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Communicated by M. Thomson.
J. Su and W. Wang contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
122_2015_2579_MOESM1_ESM.pdf
Supplementary Fig. S1 Pairwise comparison of Pi50_NBS4_1 respectively with Pi50_NBS4_3 (top panel) and Pi50_NBS2 (lower panel). The sequence alignment was conducted using BLAST2 (http://www.ncbi.nlm.nih.gov) and the graphic summary was captured in scale. The sequence of Pi50_NBS4_1 was used as the subject for each comparison. The promoter/exon/intron are indicated corresponding to their positions
122_2015_2579_MOESM2_ESM.pdf
Supplementary Fig. S2 Overall good synteny with respect to gene order and composition between the Pi2 and Pi50 loci. The X-axis displays the genomic context of NBS–LRR genes at the Pi50 locus and the Y-axis displays the one at the Pi2 locus. The pseudomolecule of the Pi50 locus is composed of three fragments, i.e., the NIP side (GenBank accession no. KP985759), the central genomic block (GenBank accession no. KP985761), and the PK side (GenBank accession no. KP985760) was compared to the sequence of the Pi2 locus (GenBank accession no. DQ352453) using BLAST2. Nine orthologous groups (Pi50_NBS1-4, Pi50_NBS8-12) each indicated in different colors are named only for the Pi50 locus as an example. (PDF 54 kb)
122_2015_2579_MOESM3_ESM.pdf
Supplementary Fig. S3 Phylogenetic analysis of different NBS–LRR genes at the Pi2/9 locus. Pi2_NBS4, Pi9_NBS3, and Piz-t_NBS4 correspond to functional genes Pi2, Pi9, and Piz-t respectively. The Pi2/9 homologues in Nipponbare were named with the clone name AP005659. The tree was constructed using a neighbor-joining algorithm based on the predicted full-length sequence of proteins. Numbers on the branches indicate the percentage of 1000 bootstrap replicates. The unit branch length is equivalent to 0.1 amino acid substitutions per site, as indicated by the bar at the upper left corner. (PDF 24 kb) (PDF 13 kb)
Rights and permissions
About this article

Cite this article
Su, J., Wang, W., Han, J. et al. Functional divergence of duplicated genes results in a novel blast resistance gene Pi50 at the Pi2/9 locus. Theor Appl Genet 128, 2213–2225 (2015). https://doi.org/10.1007/s00122-015-2579-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-015-2579-9