
Abstract
Key message
Considerable genome variation had been incorporated within rapeseed breeding programs over past decades.
Abstract
In past decades, there have been substantial changes in phenotypic properties of rapeseed as a result of extensive breeding effort. Uncovering the underlying patterns of allelic variation in the context of genome organisation would provide knowledge to guide future genetic improvement. We assessed genome-wide genetic changes, including population structure, genetic relatedness, the extent of linkage disequilibrium, nucleotide diversity and genetic differentiation based on F ST outlier detection, for a panel of 472 Brassica napus inbred accessions using a 60 k Brassica Infinium® SNP array. We found genetic diversity varied in different sub-groups. Moreover, the genetic diversity increased from 1950 to 1980 and then remained at a similar level in China and Europe. We also found ~6–10 % genomic regions revealed high F ST values. Some QTLs previously associated with important agronomic traits overlapped with these regions. Overall, the B. napus C genome was found to have more high F ST signals than the A genome, and we concluded that the C genome may contribute more valuable alleles to generate elite traits. The results of this study indicate that considerable genome variation had been incorporated within rapeseed breeding programs over past decades. These results also contribute to understanding the impact of rapeseed improvement on available genome variation and the potential for dissecting complex agronomic traits.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Oilseed rape (canola, rapeseed; Brassica napus L., 2n = 4 × = 38, AACC genomes) is one of the most important oilseed crops worldwide. Because of high oil and protein content in its seed, rapeseed is widely grown in China, Canada, Europe and Australia, with increasing interest in S. America. Rapeseed was grown on 34.3 million ha and produced 64.8 million tons of seed worldwide in 2012 (FAO 2013; http://faostat.fao.org/), it was equal to supply of ~20 million tons plant oil a year. Compared with crops such as wheat, soybean and rice which have a long history of evolution and domestication, rapeseed is a recent domesticated species. It possibly arose as a result of natural hybridization and genome doubling between the diploid species Brassica rapa (2n = 2 × = 20, genome AA) and Brassica oleracea (2n = 2 × = 18, genome CC) along the Mediterranean coastline in Southern Europe at 10,000 years ago (Song and Osborn 1992; UN 1935). However, there is evidence of polyphyletic origins, including in domestication where the widely grown B. rapa and B. oleracea vegetables were cultivated side by side in countries such as Italy (Allender and King 2010). With industrial development of human being, rapeseed was possibly selected as a crop around 300–400 years ago (Morrell et al. 2011). Rapeseed was widely grown in late nineteenth century in northern Europe and valued for high levels of erucic acid required for lubrication of steam engines. Due to the high storage protein in seed, rapeseed was also grown to feed animals, and then improved as an oil crop at the beginning of 1950s.
Although rapeseed has only been subject to a short evolutionary history and period of selection in domestication, there have been substantial changes in phenotypic properties, especially during the past few decades as a result of extensive breeding efforts. Over the past 60 years rapeseed has undergone extensive improvement, affecting a wide range of agronomic traits. The most significant achievement has been a remarkable growth in yield, with seed oil content considerably increased (Abbadi and Leckband 2011). These gains have contributed to higher efficiency of land use, with larger numbers of farmers willing to grow the crop (Abbadi and Leckband 2011). Since the crop has primarily been cultivated for edible oil, seed quality has been improved to make it nutritionally acceptable for human and livestock consumption. The major advance has been in generation of canola quality seed, with ‘double-low’ erucic acid and glucosinolate characteristics and high oleic acid content (Abbadi and Leckband 2011; Shahidi 1990; Stefansson and Hougen 1964). To meet the demands of modern agriculture, additional traits, such as canopy architecture, tolerance to biotic and abiotic stresses, and adaptation of flowering and phenology to different latitudes, have all been improved to some extent (Lefebvre 1989; Rygulla et al. 2007; Wang et al. 2005), although there remains considerable scope for improvement of harvest index. The extensive phenotypic variation present in B. napus suggests the existence of a considerable genotypic variation in this crop species. This raises a number of questions such as how, when and where did these genetic changes occur; which genetic factors or genomic regions control these improved traits. Moreover, since the requirement of more intensive agriculture in future, thus, is there sufficient allelic variation available for future rapeseed breeding? Uncovering the underlying pattern of genetic changes during rapeseed breeding over past decades would answer these questions and provide knowledge to guide future rapeseed improvement.
To unravel the genetic basis of crop trait variation, a number of traditional genome-wide phenotype-to-genotype approaches have been employed including linkage-based QTL mapping and linkage-disequilibrium (LD) association mapping (Duran et al. 2010; Georges 1997; Guo et al. 2013; Lu et al. 2011; McCouch and Doerge 1995; Vaz Patto et al. 2003; Waugh et al. 2010). However, these approaches are limited to phenotypes that are readily measured, and may fail to detect a large portion of the genetic changes associated with plant domestication and improvement (Suzuki 2010). Selection of favorable alleles during domestication and breeding is usually associated with dramatic changes in genetic diversity for a subset of target genes or genomic regions, as a result of linkage drag (Suzuki 2010). This has led tothe development of new genome-wide approaches that reveal the pattern of genetic changes, and enable detection and dissection of regions contributing to complex trait variation without the need for prior trait evaluation. These approaches include genome-wide scans of nucleotide diversity, patterns of LD decay, and outlier genomic selection regions based on genetic differentiation between different populations (Motsinger et al. 2006). A number of studies have successfully investigated genome-wide genetic change during breeding and domestication based on these approaches (Jiao et al. 2012; Lai et al. 2010; Lam et al. 2010; McNally et al. 2009; Morrell et al. 2011). These approaches have also successfully been used to identify candidate genes or genomic regions controlling elite traits. For example, tracking footprints of artificial selection in the dog genome demonstrated that regulatory variation in HAS2 is associated with skin wrinkling of shar-Peis (Waugh et al. 2010). In maize, thousands of genomic regions have been shown to be associated with artificial selection targets during modern breeding and domestication (Jiao et al. 2012; Lai et al. 2010), with genes in such regions most probably representing the key factors controlling traits improved in recent decades (Jiao et al. 2012). Similar studies have also been carried out in wheat, soybean, rice and tomato (Cavanagh et al. 2013; Lam et al. 2010; McNally et al. 2009; Sim et al. 2012).
To understand genetic changes since the recent domestication and breeding selection of rapeseed and to provide a context for future breeding, we assessed genome-wide genetic changes in a large panel of B. napus cultivars and landrace accessions using a 60 k Brassica Infinium® SNP array recently developed by the Brassica Illumina SNP consortium. Most of these accessions represent inbred lines of rapeseed cultivars produced from the 1950s to the 2010s. Our detailed genome-wide analysis reveals that modern breeding effort in rapeseed has played an important role in enlarging the genetic diversity of the crop, and refining elite traits by targeting selected genomic regions. The results of this study contribute to understanding the impact of rapeseed improvement on genome variation and the potential for dissecting complex agronomic traits.
Materials and methods
Plant materials
A panel of 472 worldwide accessions was assembled, including inbred lines of commercial cultivars and landraces produced from 1950s to 2010s. Meta data describing the breeding period, region and growth type were carefully collated and curated based on provenance, recorded pedigree, breeding record and previous field performance (Table S1). DNA was isolated from young leaves of ~10 plants (pooled) for each of the 472 accessions, following a traditional large scale CTAB method (Richards et al. 2001).
Genotyping, quality control and SNP location
SNP genotypes were derived as part of a recently completed study (Li et al. 2014). Briefly, the Brassica 60K Illumina® Infinium SNP array was used to obtain genotypic data using 200 ng sheared DNA from each accession according to the manufacture’s protocol described at (http://www.illumina.com/technology/infinium_hd_assay.ilmn). SNP data were clustered and called automatically using the Illumina BeadStudio genotyping software. SNPs that did not show clearly defined clusters representing any of the three possible genotypes (AA, AB and BB) in the whole panel were excluded.
To control the quality of data used for subsequent analysis, a series of criteria were established for filtering SNP markers. First, SNP markers that had more than 20 % missing genotypes within each group (e.g. accessions in the Europe group), or that had more than 10 % missing genotypes across the whole panel, were discarded from the analysis. Second, to avoid paralogues or homeologues confusing the genotype analysis, four DH (doubled haploid) lines were used as controls. SNP markers that appeared heterozygous within any of these four DH lines were excluded from our analysis. Finally, SNPs that were nearly monomorphic across all accessions (minor allele frequency <0.05; MAF) were discarded from our dataset.
To position the 50,000 SNPs in the context of the 19 B. napus chromosomes, we used the B. napus ‘pseudomolecules’ constructed by integrating high density genetic maps and scaffolds for B. rapa and B. oleracea (Harper et al. 2012). Probe sequences of these SNPs were used to query these scaffolds arranged on 19 ‘pseudomolecules’ corresponding to each of the B. napus chromosomes using blastn (from ftp://ftp.ncbi.nlm.nih.gov/blast/). A custom perl script was used to parse the blast output, and extract the most significant genome hits for each SNP. The chromosomal locations for each probe were then obtained based on their corresponding match positions. SNPs mapping to unanchored scaffolds were excluded from further analysis.
Statistical analysis
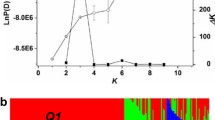
The software package STRUCTURE v2.3.4 implemented with a model-based clustering algorithm (Pritchard et al. 2000) was used to predict the population structure for our 472 accessions. SNPs with minor allele frequency (MAF) larger than 0.1 and with distance to adjacent SNPs larger than the mean distance of all 25,183 SNPs were selected for population structure analysis. The K number was set up from 1 to 10 with the length of burin period and the number of MCMC (Markov Chain Monte Carlo) replications after burin both to 100,000 under the ‘admixturemodel’. Five independent runs were performed for each K number. The most likely number of sub-populations was determined with \(\varDelta k\) method described by Evanno et al. (2005). All the SNPs were used to estimate the genetic relatedness between individuals by principal component analysis (PCA) using the GCTA tool (Yang et al. 2011) and identity by state (IBS) using the plink software (Purcell et al. 2007). Linkage disequilibrium was calculated using SNPs with MAF larger than 0.05 with software TASSEL4.0 (Bradbury et al. 2007).
The statistic \(\pi\) was employed to calculate genome-specific nucleotide diversity (Nei and Miller 1990) for SNPs located on chromosomes A01 to A10 (A genome) or C01 to C09 (C genome). To identify putative loci under artificial selection among different sub-populations, an F ST outlier detection method was employed. Estimation of SNP and population-specific F ST was based on the pure drift model (Nicholson et al. 2002), following the procedure described by Porto-Neto et al. (2013). The sub-populations were classified according to breeding region, growth type and breeding period (Table S1). To avoid abnormal data affecting the final result, outlier regions were detected based on a sliding-window F ST analysis. Briefly, SNP-specific F ST values were calculated within each sub-group, followed by the average F ST in a 0.5 Mb window with a 50 kb sliding bin for each of the 19 chromosomes. The mean F ST was set as zero when the 0.5 Mb windows contained <3 SNP markers and these windows were excluded in further analysis. Subsequently, genomic regions of particularly high F ST (above the first percentile in the sub-population) were regarded as the targets of artificial selection during breeding in the relevant sub-population.
Results
Summary of plant materials
A panel of 472 accessions from a worldwide core collection of B. napus inbred lines produced in the past 60 years was used to represent rapeseed cultivars produced from different regions, growth types and periods. Breeding regions were classified as from Australia (20; incl. New Zealand), China (232), Europe (160), North America (26; Canada and USA) and Northeast Asia (31; Japan, Korea) (Table 1). Three accessions from India and Pakistan were excluded from this classification. We classified accessions as spring (111), semi-winter (199) and winter (162) types based on their vernalization requirement for induction of flowering. We were also able to classify the accessions from China and Europe into five and four groups according to their breeding periods based on year of cultivar release (Table 1).
SNP characteristics, quality and their locations
The Brassica 60K Illumina® Infinium SNP array was used for genotyping. Following quality control, 25,183 SNP markers were used for subsequent analysis. We found that chromosome C03 had the greatest number (2,247) of SNPs; whereas, A02 ranked as the lowest (688). Since an even distribution of SNP markers was critical to our subsequent analysis, we calculated the mean density of SNPs for each of the 19 B. napus chromosomes, which ranged from 1 SNP 17.50 kb−1 on chromosome A10 to 69.42 kb−1 on chromosome C09, with an average density across the whole B. napus genome of 1 SNP 29.20 kb−1.
Genetic relatedness, population structure and linkage disequilibrium for the core collected rapeseed population
To investigate relatedness of the 472 rapeseed accessions, IBS was calculated using all 25,183 SNP markers with the software of plink (Purcell et al. 2007). Totally, these 472 accessions produced 111,156 pairs of comparisons. Six pairs represented 12 unique accessions showed IBS score larger than 0.95. This indicated individuals in these six pairs should be regarded as identical plant materials. Since there were only six overlapped accessions and they distributed evenly in different sub-groups when we classified the 472 core collections with different criterions, thus these accessions were not excluded in subsequent analyses. Of these 111,156 pairs of comparisons, 591, 20,344, 63,446, 25,278, 1,480 and 17 pairs showed IBS score ranged from 0.4 to 0.5, 0.5 to 0.6, 0.6 to 0.7, 0.7 to 0.8, 0.8 to 0.9 and 0.9 to 1, respectively (Fig. 1). The average IBS score was 0.6563 ± 0.0640. Both of distribution and average number of IBS score suggested close genetic relatedness among all of the 472 accessions.

Distribution of IBS scores for the rapeseed core collection
Totally, 7,406 SNPs were selected for population structure analysis using STRUCTURE software (Pritchard et al. 2000). Based on the \(\varDelta k\) method (Fig. 2a), the 472 accessions could be divided into two sub-populations (Fig. 2b). The principle component analysis based on all SNPs showed that the first 20 eigenvalues could explain a total of 37.6 % genetic variance and the first two principle components could explain 9.8 and 5.3 % genetic variance, respectively. Comparison with the classification of these 472 accessions using the first two principle components and using growth type or geographic distribution, there was no very clear correspondence between different classifications (Fig. 3). However, some weak correlations between different classifications could be observed. Figure 3a showed part of semi-winter rapeseed distributed between winter and spring types; similarly, Fig. 3b showed the first two principle components could slightly classify rapeseed from China and Europe. The same results were also observed when compared the classification of these 472 accessions using model-based sub-populations and using growth type, geographic distribution or different released periods (data not shown). All these results suggested that population structure did not strongly contribute to the genetic differentiation between the growth type and different geographic distribution in our core rapeseed population.

Analysis of the population structure of the rapeseed core collection by STRUCTURE. a \(\Delta k\) based on the rate of change of LnP(D) between successive K. b Population structure based on K = 2. Each individual is represented by a vertical bar. Q1 and Q2 are the values of composition belonging to the two sub-populations for a given accession

Principle component analysis for the rapeseed core collection. First three principal components are shown. Each individual is represented by one dot and the color label corresponding to their classification: a different growth type and b different geographic distribution (color figure online)
To investigate the extent of linkage disequilibrium, the r 2 was used to calculate LD in this study. Figure 4 showed the genome-wide LD decay for all the 472 rapeseed accessions. Overall, the average distance over which LD decayed to half of its maximum value in rapeseed was ~21 kb. In addition, when the mean distance between SNP markers is about 0.5 Mb, r 2 nearly decays to 0.2; when the distance is about 1.5 Mb, r 2 nearly decays to 0.1. Figures S1–S3 showed all r 2 versus distance for all 19 chromosomes. Obviously, the LD decay across these 19 chromosomes showed very different. C07 showed almost no LD decay, which is probably due to inaccurate SNP mapping. Except for chromosome C07, A10 showed the fast and C08 showed the lowest LD decay. With a regression analysis to modulate the relationship between r 2 and the distance, r 2 decayed to 0.2 when the distance was 50 kb and to 0.1 when the distance was 480 kb on chromosome A10; whereas, r 2 decayed to 0.2 when the distance was 2.48 Mb and to 0.1 when the distance was 2.49 Mb on chromosome C08 (Table S2). Moreover, the LD decay between the whole A and C genome in rapeseed also showed very different (t test, p value <0.01). r 2 decayed to 0.2 when the average distance for A genome was 210 kb and to 0.1 when the average distance was 980 kb; whereas, r 2 decayed to 0.2 when the average distance for C genome was 810 kb and to 0.1 when the distance was 1.61 Mb (Table S2).

Genome-wide LD decay for all the 472 accessions
Modern breeding has affected rapeseed nucleotide diversity
To identify evidence for recent selection of specific genomic regions or alleles particularly associated with modern breeding efforts, we carefully interrogated the patterns of nucleotide diversity amongst our representative set of 472 rapeseed accessions. Based on the calculated nucleotide diversity for each of the sub-groups based on breeding region, growth type and breeding period, it was apparent that accessions from China had the lowest nucleotide diversity, with \(\pi\) equal to 6.9 × 10−3 (Table 1). In contrast, accessions from Australia showed the highest diversity with \(\pi\) equal to 8.3 × 10−3. Similarly, semi-winter rapeseed had nucleotide diversity of 6.6 × 10−3, compared to higher levels in spring (8.0 × 10−3) and winter (7.8 × 10−3) types.
To test whether the genepool available for rapeseed breeding has varied along time, we also calculated the nucleotide diversity for accessions generated during different periods within the same region. Since there were insufficient accessions sampled from Australia, North America and Northeast Asia, we only performed this analysis for China and Europe. It is clear that the nucleotide diversity increased significantly in both China and Europe during the periods 1950–1970 and 1971–1980 (Table 1; Fig. 5). In China, the mean nucleotide diversity between 1971 and 2011 was relatively stable at around 7.0 × 10−3, compared with 8.0 × 10−3 in Europe. These are significantly higher than the earlier period from 1950 to 1970 (p value <0.01), with 5.8 × 10−3 in China and 5.2 × 10−3 in Europe. The differences in the increase of nucleotide diversity between 1950 and 1980 in China (~14 %) and Europe ~55 % (Table S3) have not been maintained in the subsequent periods. To investigate this further, we interrogated each of the constituent genomes, and found that in China, both A and C genomes contributed nearly equally (15.4 % for A and 16.5 %for C; Table S3), whilst in Europe the C genome contributed much more (87.6 %) than for A (27.2 %) (Table S3).

Boxplot for nucleotide diversity at different breeding periods in China and Europe. Letters of P1 to P5 below the plots correspond to these five periods, 1950–1970, 1971–1980, 1981–1990, 1991–2000 and 2001–2011, respectively. The plots on the top panels show the nucleotide diversity for Chinese cultivars, the plots on the bottom panels show the same statistics for European accessions. a, d Whole rapeseed genome; b, e A genome; c, f C genome
Evidence for putative artificial selection in the rapeseed genome arising from modern breeding
Crop cultivars are required to adapt to specific growth conditions and meet changing human requirements in different periods of plant domestication and crop improvement. This results in some sub-populations having increased variation or frequency of elite alleles within specific genomic regions, whilst some of these alleles may be absent from other sub-populations. This preference for specific loci and alleles in a particular sub-population may be the result of selection.
Recently, several statistics have been used to identify signals for this type of selection, including Tajima’s D test, the long-range haplotype (LRH) test and genetic differentiation (F ST fixation index) between sub-populations (Suzuki 2010). Of these, F ST is not strongly affected by ascertainment bias and so is better suited for analyzing data generated using SNP chips (Albrechtsen et al. 2010). We, therefore, carried out a genome-wide scan for signatures of selection in our set of 472 B. napus accession by estimating Wright’s F ST with a sliding window method. A low F ST (close to 0) usually indicates balancing selection, high levels of gene flow between populations or a low level of diversity across populations, whilst high F ST may arise by genetic drift or artificial selection (Suzuki 2010). Sliding windows of low SNP density were designated as F ST equal to 0, and so we only used the 1st percentile of the empirical distribution of high F ST-windows for detecting putative artificial selection in this study.
When classified by breeding region the mean sub-population specific F ST ranged from 0.029 (China) to 0.055 (Australia) (Table 1), indicating lowest genetic differentiation in accessions from China, consistent with its lowest nucleotide diversity. This also indicated a more restricted genetic relationship amongst rapeseed accessions in China compared with other regions. When classified by growth type, the mean sub-population specific F ST for spring (0.090), semi-winter (0.030) and winter (0.061) types (Table 1) was consistent with the pattern of nucleotide diversity. Thus, semi-winter types had the lowest \(\pi\) and F ST value, whereas spring types had the highest \(\pi\) and F ST.
A considerably larger range of variation in F ST was apparent when accessions from China and Europe were classified by breeding periods. Amongst lines produced in China from 1950 to 2011, we found higher F ST in the sub-populations of 2001–2011 (F ST = 0.072) and 1950–1970 (0.047), whereas the other three sub-populations had a low F ST of ~0.02. A similar pattern was apparent in Europe with highest F ST from 1991–2000 (F ST = 0.081) and 1950–1970 (0.156). More recently, rapeseed breeding has also extended the range of artificial selection, during 1991–2000 in Europe, and in the past decade in China. In contrast, the more uniform and reduced F ST associated with different growth type classifications may indicate fewer loci and alleles under recurrent selection in relation to the flowering time trait.
To track the footprint of selected chromosomal regions during rapeseed modern breeding, we calculated the smoothed F ST for each sub-population. As the distance between SNP markers approached 0.5 Mb, the average r 2 was 0.2. Since this level of linkage disequilibrium (LD) usually indicates limited linkage between markers, we selected a 0.5 Mb sliding window to smooth F ST. The regions subjected to putative artificial selection for all categories are shown in Fig. 6 and Figures S4–S7, with the corresponding detailed information in Tables S4–S6. Seventy-six chromosomal regions had high F ST in at least one sub-population (1st percentile high F ST-window) for breeding regions, with fewer amongst growth types (51 regions), and breeding period in China (68) and Europe (59) (Table 2). The sum of these high F ST chromosomal regions correspond to 73.95 Mb (breeding region), 42.4 Mb (growth types), 69.75 (China) and 61.3 Mb (Europe) (Table 2). This reveals these regions span 6–10 % of the B. napus genome and may be enriched for targets of putative selection. Looking further into these regions, we found that the A genome had considerably high F ST signals than the C genome in all four categories of the collection (t test, p value <0.01), which suggests that the C genome may contribute more valuable alleles to generate elite traits.

Genomic regions showed high F ST values on all 19 rapeseed chromosomes. From the bottom to up, these yellow boxes indicate chromosome 1–19 in order. These four colors, red, green, brown and purple, indicate genomic regions with outlier F ST based on calculation of genetic differentiation among sub-populations on different methods for classification the 472 accessions. The four categories for classification the 472 accessions are listed in the top-right of this figure. Genomic regions with overlying colors indicate overlapped selection signals from different categories (color figure online)
Since a large number of genomic regions showed high F ST, this motivated us to identify which loci and alleles were located on these selected regions. To simplify our data analysis, we only investigated high F ST regions shared by more than two groups, the groups being defined as breeding regions, periods or growth habits. Three genomic regions met these criteria (Table 3). A08:7.20–10.20 mbp and C02:47.20–50.90 clearly show high F ST in three and four of the five sub-populations which were classified according to breeding periods in China. C06:35.95–38.80 showed high F ST in spring and winter sub-populations classified according to growth type.
Comparing with previous and ongoing studies of linkage and linkage-disequilibrium QTL mapping for agronomic traits in rapeseed, we found there were important QTL or candidate genes in all of these three high F ST regions. First, a major QTL for erucic acid and oil content in seeds embedded within the region of A08:7.20–10.20 (Cao et al. 2010; Wang et al. 2008, 2010). BnaA.FAE1 is the candidate gene for this QTL and is located just 120 kb far from the peak position of F ST values according to in silico mapping of homologues from Arabidopsis thaliana onto the B. napus ‘pseudomolecules’ (BnaA.FAE1 locates on A08:9.28 and the peak position is 9.40). Second, according to GWAS analysis in rapeseed for glucosinolate content in seed performed by Harper et al. (2012), loci controlling this trait overlapped with the region C02:47.20–50.90. BnaC.HAG1 is the candidate gene for this QTL, it locates on C02:49.62 and the peak F ST position is 49.35. Third, we previously found a major flowering time QTL located within the region C06:35.8–38.80 (Wang et al. 2009). The location of the candidate gene BnaC.FT for this flowering time QTL was nearly in perfect overlap with the peak position of high F ST (BnaC.FT locates on C06:36.74 and the peak F ST position is 36.75).
Based on our analysis of allele frequencies in these three regions, it was apparent that their distributions among sub-populations also support the hypothesis of strong selection. Major allele frequencies in A08:7.20–10.20 revealed that SNPs in sub-populations 1950–1970, and 2001–2011 are almost fixed (Fig. 7a). Similarly, SNPs in sub-populations 1950–1970, 1971–1980 and 2001–2011 are also nearly fixed in the region C02:47.20–50.90 (Fig. 7b). Careful inspection of major allele frequencies in the region C06:25.8–28.8 indicates that SNPs in the three sub-populations are not only close to fixation, but also showed a nearly identical distribution of major allele frequency to 1 in part of this region simultaneously (Fig. 7c). In the region C06:35.8 to C06:37.3 the winter and semi-winter sub-populations showed 91.7 % SNPs with major allele frequencies nearly equal to 1 simultaneously. Whereas between C06:37.3 to C06:38.8 the spring and semi-winter sub-populations showed 80.6 % SNPs with major allele frequencies nearly equal to 1.

Variation of major allele frequencies within regions on rapeseed genome showing high F ST values. a Major allele frequencies (AF) at region A08:7.20–10.20 within the five sub-populations in China (these five sub-populations are corresponding to accessions produced at periods 1950–1970, 1971–1980, 1981–1990, 1991–2000 and 2001–2011); b major AF at region C02:47.20–50.90 within the five sub-populations in China; c major AF at region C06:35.95–38.80 within the three growth types in China. Black triangles in a–c indicate the positions of genes BnaA.FAE1, BnaC.HAG1 and BnaC.FT, respectively; red triangles in a–c indicates peak positions for putative selection signals (genomic positions with the highest mean F ST values in the showed regions) (color figure online)
Discussion
Until recently the original breeding selection was usually performed without reference to knowledge of the underlying genomic information, but primarily relied on trait performance. These insights into the underlying genomic contributions to selective breeding in the context of agronomic traits will help to extend our understanding of crop improvement and so guide future breeding efforts (Morrell et al. 2011). In this study, our genome-wide analysis 25,000 valid SNP markers revealed that 6–10 % of genomic regions investigated (covering ~40–80 Mb) present high F ST values. These regions represent a list of candidate regions that may have been under strong selection and may be worth further study. Knowing the genomic content of regions with high F ST will prove useful to understand the target of selection and mechanisms at play. This information also provides us with a new approach to dissect complex traits for rapeseed improvement (Morrell et al. 2011).
Genetic diversity is an important parameter for evaluating the ability to identify and sustain generation of new elite cultivars in crop improvement. Low genetic diversity in a given eco-geographical area or special growth type indicates a reduced allelic gene pool available for selection, and thus a bottleneck for crop breeding. In this study, we found that rapeseed in China had the lowest nucleotide diversity based on geographic classification of the 472 accessions (Table 1). As most of the semi-winter rapeseed types were from China, this sub-population also showed the lowest nucleotide diversity based on growth type classification (Table 1). This result was partially in accordance with a previous report that assessed genetic diversity for a panel of 509 accessions with genotypic data from 89 genome-specific SSR primer combinations (Bus et al. 2011). Since rapeseed germplasm in China was isolated from Europe and the rest of the world (Chen et al. 2008), rapeseed population in China or the group of semi-winter type showing lower genetic diversity may be as a result of genetic isolation or bottlenecks.
During crop improvement, a subset of genomic regions, loci or alleles are more likely to be selected as a common genetic resource for many cultivars dependent on region, growth type or the period in which they were developed. The selection for adaptation to similar growth environments may also lead to a reduction of genetic diversity or ‘allelic canalisation’ in modern crops. The phenomenon has been reported for a number of crops, including wheat and soybean (Fu and Somers 2009; Hyten et al. 2006). However, in the case of the amphidiploid B. napus, there is also the opportunity to harness allelic diversity from the two constituent genomes of B. rapa (A) and B. oleracea (C), and so to determine whether canalisation or gene pool enrichment has occurred in B. napus. In modern plant breeding, reduction of genetic diversity due to a reduced gene pool is a very common phenomenon. Moreover, previous studies showed bottlenecks and linkage drag in genomic regions corresponding to loci selected during breeding of modern rapeseed varieties incorporating low oil erucic acid content and low seed glucosinolate content (Becker et al. 1995; Cao et al. 2010; Hasan et al. 2008). These bottlenecks caused by linkage drag also contribute to reduced genetic diversity since the alleles in question only arose from single or few sources (e.g. low erucic acid from cv. Liho, and low glucosinolate from cv. Brownowski) and genomic regions inherited from these ancestors can still be identified in the genomes of rapeseed cultivars (Bancroft et al. 2011). However, we found the genetic diversity increased from 1950 to 1980 and then has been maintained at a similar level since 1981 in both China and Europe (Table 1; Fig. 5). This is partly in contrast to the conclusions from the lower resolution study of Bus et al. (2011) who identified lower genetic diversity within winter rapeseed inbreds of more recent release than for inbreds released in previous decades. By carefully comparing these two results, we find that this apparent contradiction may arise from two factors. First, different plant materials were used in these two studies. Our core collected panel mainly included oil-type rapeseed; whereas a number of forage rapeseed were involved in Bus et al. (2011). Second, different genotyping methods were used in these two studies. SNP array was employed in this study; whereas SSR markers were mainly used in Bus et al. (2011). These different methods for genotyping would produce ascertainment bias. In addition, discrimination between different rapeseed sub-groups by population structure in some previous studies was clear (e.g. Bus et al. 2011); whereas there was only weak correlations between classifications by population structure and sub-groups in our study. This difference may also attribute to these two factors.
Since the specificity of plants in U’ triangle, the genomes of three ancestral species of Brassica (B. rapa, B. oleracea and B. nigra) combined to create three of the common modern vegetables and oilseed crop species (B. napus, B. juncea and B. carinata) (UN 1935), these patterns of initial increases in genetic diversity followed by relative stability should be placed in the context of relationships among species in U’s triangle, particularly through introduction of genetic variation from the A and C genome diploid species B. rapa and B. oleracea. Delourme et al. (2006) concluded that a number of resistance genes/loci had been introduced from B. rapa to the A genome of B. napus. In China genomic components from B. rapa and B. oleracea were also introduced into B. napus rapeseed lines to widen the genetic diversity, and following stabilization have been used as breeding parents (Li et al. 2007; Mei et al. 2011; Qian et al. 2006). We also detected different levels of increased genetic diversity in China and Europe, with the latter having lower genetic diversity between 1950 and 1970, with an increase between 1971 and 1980 (Table S2). This phenomenon may result from particular circumstances. First, in Europe there may have been introduction of more exotic germplasm base from other species in U’s triangle than in China during the period 1971–1980. Second, most of the accessions in our representative collection were oilseed rape cultivars (mainly used for producing oil); with very few fodder types from Europe. Moreover, there was no fodder rapeseed present in China during this period. The introduction of genetic variation (e.g. for resistance to stem canker/blackleg) from fodder rapeseed would have made a more significant contribution to extending genetic diversity in Europe than in China during this period. With a more detailed analysis of the sources of the increased genetic diversity, we found a similar increase in percentage nucleotide diversity from 1950 to 1980 in China for the A and C genomes (15.41 and 17.41 %). In contrast, there was a more than three times increase for C than A genome in Europe (83.64 and 26.41 %). We might be able to attribute these differences to the extensive growth of B. oleracea in Europe and its genetic components were introduced to C genome with a higher possibility in this area.
LD can reflect the fundamental genetic force of recombination, as well as demographic factors, such as population history, artificial selection and inbreeding (Kim et al. 2007). In this study, we calculated the extent of LD for core rapeseed collection. We observed significant LD (r 2 > 0.2) between markers separated by up to 500 kb. This value is smaller than the results obtained by Ecke et al. (2010), who found levels of significant LD over 2 cM, a genetic distance that was equivalent to 1 Mb on the physical map. In their report, 2 cM in genetic distance was equal to 1 Mb in physic genome distance. Moreover, compared with other plants, rapeseed showed a moderate extent of LD decay with an average distance of 21 kb over which LD decayed to half of its maximum value; whereas the average distances for cultivated soybean, wild soybean, maize, wild and cultivated rice and A. thaliana were at ~150, ~75, <1, <1 and ~3–4 kb (Gore et al. 2009; Kim et al. 2007; Lam et al. 2010; Zhu et al. 2007), respectively. This extent of LD decay would provide a moderate resolution for further genome-wide identification of signature of artificial selection in rapeseed.
Our results also highlighted several genomic regions with high F ST. These regions constitute a list of candidate regions of artificial selection which can be of high interest for future breeding programs. Since dramatic changes in allele frequency may arise by chance, especially during bottleneck events, further studies are needed to refine the exact proportion and location of the genome that has been under artificial selection. Interestingly, both sub-genomes do not show the same genetic signal: the C genome shows more loci with high F ST than the A genome (p value <0.01). In addition, the LD decay is lower in the C than in the A genome (Table S2 to S8). Taken together these results support the hypothesis of a higher selection on the C genome. The reason why the rapeseed C genome may harbor genomic regions more attractive to breeders is not immediately apparent. The higher nucleotide diversity (\(\pi\)) in the C genome suggests that the C genome may be a repository for a wider range of loci or alleles contributing to important agronomic traits, such as yield, oil content and biotic and abiotic stress tolerance. This may also be inferred from evidence presented in some recent studies. For example, BnaC.IGMT5.a on chromosome C06 was identified as a candidate gene of the major QTL for resistance to sclerotinia stem rot (Wu et al. 2013). Many QTLs for yield-related traits were detected on the C genome through genome-wide association mapping with 192 inbred lines and linkage mapping with a reconstructed F2 population (Cai et al. 2014; Shi et al. 2009).The functional prominence of one genome may also be in part due to epigenetic imprinting effects, although an early study found dominance of A genomic rRNA genes over the C genome (Chen and Pikaard 1997).
In summary, we have carried out a comprehensive analysis of genetic changes and identified several underlying trends associated with genomic regions subject to selection and the overall range of genetic diversity incorporated in modern rapeseed breeding programs. First, we found a lower level of genetic diversity in Chinese germplasm compared to European samples, indicating that more extensive genetic variability should be introduced in the Chinese one. Second, in contrast to a previous study (Bus et al. 2011), we found that genetic diversity for rapeseed has not undergone significant reduction in recent years compared with cultivars produced in previous decades. Third, our results suggest that the C genome may have been under more extensive selection. Finally, we identify possible targets of selection across the genome, providing several interesting candidate regions for follow-up works. We believe the approach and conclusions from this study provide valuable information to guide ongoing rapeseed breeding.
References
Abbadi A, Leckband G (2011) Rapeseed breeding for oil content, quality, and sustainability. Eur J Lipid Sci Technol 113:1198–1206
Albrechtsen A, Nielsen FC, Nielsen R (2010) Ascertainment biases in SNP chips affect measures of population divergence. Mol Biol Evol 27:2534–2547
Allender CJ, King GJ (2010) Origins of the amphiploid species Brassica napus L. investigated by chloroplast and nuclear molecular markers. BMC Plant Biol 10:54
Bancroft I, Morgan C, Fraser F, Higgins J, Wells R, Clissold L, Baker D, Long Y, Meng J, Wang X, Liu S, Trick M (2011) Dissecting the genome of the polyploid crop oilseed rape by transcriptome sequencing. Nat Biotechnol 29:762–766
Becker HC, Engqvist GM, Karlsson B (1995) Comparison of rapeseed cultivars and resynthesized lines based on allozyme and RFLP markers. Theor Appl Genet 91:62–67
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Bus A, Korber N, Snowdon RJ, Stich B (2011) Patterns of molecular variation in a species-wide germplasm set of Brassica napus. Theor Appl Genet 123:1413–1423
Cai D, Xiao Y, Yang W, Ye W, Wang B, Younas M, Wu J, Liu K (2014) Association mapping of six yield-related traits in rapeseed (Brassica napus L.). Theor Appl Genet 127:85–96
Cao Z, Tian F, Wang N, Jiang C, Lin B, Xia W, Shi J, Long Y, Zhang C, Meng J (2010) Analysis of QTLs for erucic acid and oil content in seeds on A8 chromosome and the linkage drag between the alleles for the two traits in Brassica napus. J Genet Genomics 37:231–240
Cavanagh CR, Chao S, Wang S, Huang BE, Stephen S, Kiani S, Forrest K, Saintenac C, Brown-Guedira GL, Akhunova A, See D, Bai G, Pumphrey M, Tomar L, Wong D, Kong S, Reynolds M, da Silva ML, Bockelman H, Talbert L, Anderson JA, Dreisigacker S, Baenziger S, Carter A, Korzun V, Morrell PL, Dubcovsky J, Morell MK, Sorrells ME, Hayden MJ, Akhunov E (2013) Genome-wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars. Proc Natl Acad Sci USA 110:8057–8062
Chen ZJ, Pikaard CS (1997) Transcriptional analysis of nucleolar dominance in polyploid plants: biased expression/silencing of progenitor rRNA genes is developmentally regulated in Brassica. Proc Natl Acad Sci USA 94:3442–3447
Chen S, Nelson MN, Ghamkhar K, Fu T, Cowling WA (2008) Divergent patterns of allelic diversity from similar origins: the case of oilseed rape (Brassica napus L.) in China and Australia. Genome 51:1–10
Delourme R, Chèvre AM, Brun H, Rouxel T, Balesdent MH, Dias JS, Salisbury P, Renard M, Rimmer SR (2006) Major gene and polygenic resistance to leptosphaeria maculans in oilseed rape (Brassica napus). Eur J Plant Pathol 114:41–52
Duran C, Eales D, Marshall D, Imelfort M, Stiller J, Berkman PJ, Clark T, McKenzie M, Appleby N, Batley J, Basford K, Edwards D (2010) Future tools for association mapping in crop plants. Genome 53:1017–1023
Ecke W, Clemens R, Honsdorf N, Becker HC (2010) Extent and structure of linkage disequilibrium in canola quality winter rapeseed (Brassica napus L.). Theor Appl Genet 120:921–931
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Fu Y-B, Somers DJ (2009) Genome-wide reduction of genetic diversity in wheat breeding. Crop Sci 49:161–168
Georges M (1997) QTL mapping to QTL cloning: mice to the rescue. Genome Res 7:663–665
Gore MA, Chia JM, Elshire RJ, Sun Q, Ersoz ES, Hurwitz BL, Peiffer JA, McMullen MD, Grills GS, Ross-Ibarra J, Ware DH, Buckler ES (2009) A first-generation haplotype map of maize. Science 326:1115–1117
Guo B, Wang D, Guo Z, Beavis WD (2013) Family-based association mapping in crop species. Theor Appl Genet 126:1419–1430
Harper AL, Trick M, Higgins J, Fraser F, Clissold L, Wells R, Hattori C, Werner P, Bancroft I (2012) Associative transcriptomics of traits in the polyploid crop species Brassica napus. Nat Biotechnol 30:798–802
Hasan M, Friedt W, Pons-Kuhnemann J, Freitag NM, Link K, Snowdon RJ (2008) Association of gene-linked SSR markers to seed glucosinolate content in oilseed rape (Brassica napus). Theor Appl Genet 116:1035–1049
Hyten DL, Song Q, Zhu Y, Choi IY, Nelson RL, Costa JM, Specht JE, Shoemaker RC, Cregan PB (2006) Impacts of genetic bottlenecks on soybean genome diversity. Proc Natl Acad Sci USA 103:16666–16671
Jiao Y, Zhao H, Ren L, Song W, Zeng B, Guo J, Wang B, Liu Z, Chen J, Li W, Zhang M, Xie S, Lai J (2012) Genome-wide genetic changes during modern breeding of maize. Nat Genet 44:812–815
Kim S, Plagnol V, Hu TT, Toomajian C, Clark RM, Ossowski S, Ecker JR, Weigel D, Nordborg M (2007) Recombination and linkage disequilibrium in Arabidopsis thaliana. Nat Genet 39:1151–1155
Lai J, Li R, Xu X, Jin W, Xu M, Zhao H, Xiang Z, Song W, Ying K, Zhang M, Jiao Y, Ni P, Zhang J, Li D, Guo X, Ye K, Jian M, Wang B, Zheng H, Liang H, Zhang X, Wang S, Chen S, Li J, Fu Y, Springer NM, Yang H, Wang J, Dai J, Schnable PS, Wang J (2010) Genome-wide patterns of genetic variation among elite maize inbred lines. Nat Genet 42:1027–1030
Lam HM, Xu X, Liu X, Chen W, Yang G, Wong FL, Li MW, He W, Qin N, Wang B, Li J, Jian M, Wang J, Shao G, Wang J, Sun SS, Zhang G (2010) Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat Genet 42:1053–1059
Lefebvre DD (1989) Increased potassium absorption confers resistance to group IA cations in rubidium-selected suspension cells of Brassica napus. Plant Physiol 91:1460–1466
Li M, Liu J, Wang Y, Yu L, Meng J (2007) Production of partial new-typed Brassica napus by introgression of genomic components from B. rapa and B. carinata. J Genet Genomics 34:460–468
Li F, Chen B, Xu K, Wu J, Song W, Bancroft I, Harper AL, Trick M, Liu S, Gao G, Wang N, Yan G, Qiao J, Li J, Li H, Xiao X, Zhang T, Wu X (2014) Genome-wide association study dissects the genetic architecture of seed weight and seed quality in rapeseed (Brassica napus L.). DNA Res. doi:10.1093/dnares/dsu002
Lu HY, Liu XF, Wei SP, Zhang YM (2011) Epistatic association mapping in homozygous crop cultivars. PLoS One 6:e17773
McCouch SR, Doerge RW (1995) QTL mapping in rice. Trends Genet 11:482–487
McNally KL, Childs KL, Bohnert R, Davidson RM, Zhao K, Ulat VJ, Zeller G, Clark RM, Hoen DR, Bureau TE, Stokowski R, Ballinger DG, Frazer KA, Cox DR, Padhukasahasram B, Bustamante CD, Weigel D, Mackill DJ, Bruskiewich RM, Ratsch G, Buell CR, Leung H, Leach JE (2009) Genomewide SNP variation reveals relationships among landraces and modern varieties of rice. Proc Natl Acad Sci USA 106:12273–12278
Mei J, Li Q, Qian L, Fu Y, Li J, Frauen M, Qian W (2011) Genetic investigation of the origination of allopolyploid with virtually synthesized lines: application to the C subgenome of Brassica napus. Heredity 106:955–961
Morrell PL, Buckler ES, Ross-Ibarra J (2011) Crop genomics: advances and applications. Nat Rev Genet 13:85–96
Motsinger AA, Reif DM, Dudek SM, Ritchie MD (2006) Understanding the evolutionary process of grammatical evolution neural networks for feature selection in genetic epidemiology. Proc IEEE Symp Comput Intell Bioinform Comput Biol 2006:1–8
Nei M, Miller JC (1990) A simple method for estimating average number of nucleotide substitutions within and between populations from restriction data. Genetics 125:873–879
Nicholson G, Smith AV, Jónsson F, Gstafsson Ã, Stefánsson K, Donnelly P (2002) Assessing population differentiation and isolation from single-nucleotide polymorphism data. J R Stat Soc 64:695–715
Porto-Neto LR, Lee SH, Lee HK, Gondro C (2013) Detection of signatures of selection using FST. Methods Mol Biol 1019:423–436
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Qian W, Meng J, Li M, Frauen M, Sass O, Noack J, Jung C (2006) Introgression of genomic components from Chinese Brassica rapa contributes to widening the genetic diversity in rapeseed (B. napus L.), with emphasis on the evolution of Chinese rapeseed. Theor Appl Genet 113:49–54
Richards E, Reichardt M, Rogers S (2001) Preparation of genomic DNA from plant tissue. Curr Protoc Mol Biol, chapter 2, unit 2.3
Rygulla W, Snowdon RJ, Eynck C, Koopmann B, von Tiedemann A, Luhs W, Friedt W (2007) Broadening the genetic basis of Verticillium longisporum resistance in Brassica napus by interspecific hybridization. Phytopathology 97:1391–1396
Shahidi F (1990) Canola and rapeseed: production, chemistry, nutrition and processing technology. Van Nostrand Reinhold, New York, pp 1–10
Shi J, Li R, Qiu D, Jiang C, Long Y, Morgan C, Bancroft I, Zhao J, Meng J (2009) Unraveling the complex trait of crop yield with quantitative trait loci mapping in Brassica napus. Genetics 182:851–861
Sim SC, Van Deynze A, Stoffel K, Douches DS, Zarka D, Ganal MW, Chetelat RT, Hutton SF, Scott JW, Gardner RG, Panthee DR, Mutschler M, Myers JR, Francis DM (2012) High-density SNP genotyping of tomato (Solanum lycopersicum L.) reveals patterns of genetic variation due to breeding. PLoS One 7:e45520
Song K, Osborn TC (1992) Polyphyletic origins of Brassica napus: new evidence based on organelle and nuclear RFLP analyses. Genome 35:992–1001
Stefansson BR, Hougen FW (1964) Selection of rape plants (Brassica napus) with seed oil practically free from erucic acid. Can J Plant Sci 44:359–364
Suzuki Y (2010) Statistical methods for detecting natural selection from genomic data. Genes Genet Syst 85:359–376
U N (1935) Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jpn J Bot 7:389–452
Vaz Patto MC, Rubiales D, Martin A, Hernandez P, Lindhout P, Niks RE, Stam P (2003) QTL mapping provides evidence for lack of association of the avoidance of leaf rust in Hordeum chilense with stomata density. Theor Appl Genet 106:1283–1292
Wang J, Chen Z, Du J, Sun Y, Liang A (2005) Novel insect resistance in Brassica napus developed by transformation of chitinase and scorpion toxin genes. Plant Cell Rep 24:549–555
Wang N, Wang Y, Tian F, King GJ, Zhang C, Long Y, Shi L, Meng J (2008) A functional genomics resource for Brassica napus: development of an EMS mutagenized population and discovery of FAE1 point mutations by TILLING. New Phytol 180:751–765
Wang J, Long Y, Wu B, Liu J, Jiang C, Shi L, Zhao J, King GJ, Meng J (2009) The evolution of Brassica napus FLOWERING LOCUS T paralogues in the context of inverted chromosomal duplication blocks. BMC Evol Biol 9:271
Wang N, Shi L, Tian F, Ning H, Wu X, Long Y, Meng J (2010) Assessment of FAE1 polymorphisms in three Brassica species using EcoTILLING and their association with differences in seed erucic acid contents. BMC Plant Biol 10:137
Waugh R, Marshall D, Thomas B, Comadran J, Russell J, Close T, Stein N, Hayes P, Muehlbauer G, Cockram J, O’Sullivan D, Mackay I, Flavell A, Agoueb, BarleyCap, Ramsay L (2010) Whole-genome association mapping in elite inbred crop varieties. Genome 53:967–972
Wu J, Cai G, Tu J, Li L, Liu S, Luo X, Zhou L, Fan C, Zhou Y (2013) Identification of QTLs for resistance to sclerotinia stem rot and BnaC.IGMT5.a as a candidate gene of the major resistant QTL SRC6 in Brassica napus. PLoS One 8:e67740
Yang J, Lee SH, Goddard ME, Visscher PM (2011) GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88:76–82
Zhu Q, Zheng X, Luo J, Gaut BS, Ge S (2007) Multilocus analysis of nucleotide variation of Oryza sativa and its wild relatives: severe bottleneck during domestication of rice. Mol Biol Evol 24:875–888
Acknowledgments
This work was supported by the Chinese National Basic Research and Development Program (2011CB109302), National Science & Technology Pillar Program during the Twelfth Five-year Plan Period (2013BAD01B03 and 2011BAD35B09) and Crop Germplasm Protection Project (NB2011-2130135). GK is supported in part by the Chutian Scholar program of Hubei province.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
The authors declare that the study comply with the current laws of the country in which they were performed.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Michael Gore.
N. Wang and F. Li have contributed equally.
Electronic supplementary material
Below is the link to the electronic supplementary material.
122_2014_2343_MOESM4_ESM.tif
Genomic distribution of putative artificial selection among sub-populations classified by five breeding regions. Signatures of putative artificial selection were measured by FST based on calculation of genetic differentiation among sub-populations five from five breeding regions. From the top to bottom, these five panels are corresponding to sub-populations of Australia, China, Europe, North America and Northeast Asia, respectively. FST was smoothed with a 0.5 Mb window and a 50 kb siding bin (see M&M). The red dash lines indicate value of 1st percentile of the empirical distribution of high FST-windows. Chromosomes were differed by black and gray plots sequentially (TIFF 1588 kb)
122_2014_2343_MOESM5_ESM.tif
Genomic distribution of putative artificial selection among sub-populations classified by three growth types. From the top to bottom, these three panels are corresponding to sub-populations of spring, semi-winter and winter, respectively. All other information is similar with Figure S4 (TIFF 1226 kb)
122_2014_2343_MOESM6_ESM.tif
Genomic distribution of putative artificial selection among sub-populations classified by five breeding periods in China. From the top to the bottom, these five panels are corresponding to sub-populations of 1950-1970, 1971-1980, 1981-1990, 1991-2000 and 2001-2011, respectively. All other information is similar with Figure S4. (TIFF 1723 kb)
122_2014_2343_MOESM7_ESM.tif
Genomic distribution of putative artificial selection among sub-populations classified by four breeding period in Europe. From the top to the bottom, these four panels are corresponding to sub-populations of 1950-1970, 1971-1980, 1981-1990 and 1991-2000, respectively. All other information is similar with Figure S4. (TIFF 1260 kb)
122_2014_2343_MOESM13_ESM.xlsx
Genomic regions with outlier FST windows based on sub-populations classified by different breeding periods in China and Europe (XLSX 17 kb)
Rights and permissions
About this article

Cite this article
Wang, N., Li, F., Chen, B. et al. Genome-wide investigation of genetic changes during modern breeding of Brassica napus . Theor Appl Genet 127, 1817–1829 (2014). https://doi.org/10.1007/s00122-014-2343-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-014-2343-6