
Abstract
Key message
QTL mapping in multiple families identifies trait-specific and pleiotropic QTL for biomass yield and plant height in triticale.
Abstract
Triticale shows a broad genetic variation for biomass yield which is of interest for a range of purposes, including bioenergy. Plant height is a major contributor to biomass yield and in this study, we investigated the genetic architecture underlying biomass yield and plant height by multiple-line cross QTL mapping. We employed 647 doubled haploid lines from four mapping populations that have been evaluated in four environments and genotyped with 1710 DArT markers. Twelve QTL were identified for plant height and nine for biomass yield which cross-validated explained 59.6 and 38.2 % of the genotypic variance, respectively. A major QTL for both traits was identified on chromosome 5R which likely corresponds to the dominant dwarfing gene Ddw1. In addition, we detected epistatic QTL for plant height and biomass yield which, however, contributed only little to the genetic architecture of the traits. In conclusion, our results demonstrate the potential of genomic approaches for a knowledge-based improvement of biomass yield in triticale.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The small grain cereal triticale (× Triticosecale Wittmack; 2n = 6x = 42), a man-made wheat-rye hybrid, is considered a promising crop due to its high genetic variation for several traits of agronomic importance. It is widely adapted to abiotic stress conditions such as aluminum toxicity, drought, salinity, and acidic or waterlogged soils (Kuleung et al. 2004; Oettler 2005). With regard to biomass yield, triticale is able to produce more biomass for a comparable grain yield than other crops (Pronyk and Mazza 2011). Consequently, biomass production for industrial purposes, such as bioenergy and biofuel, can be increased without aggravating the competition with food production on arable land. Winter triticale is, therefore, ideally suited to diversify crop rotation in regions where maize is the predominant crop grown for biomass production (Gowda et al. 2011).
The development of improved varieties depends on the ability to readily phenotype numerous genotypes in the field. Biomass yield is traditionally evaluated by harvesting yield plots with a field chopper. This destructive analysis, however, has the disadvantage that no further traits can be obtained from these plants and biomass yield is therefore not routinely assessed in plant breeding. Busemeyer et al. (2013a, b) recently reported the development of a multi-sensor platform that enables non-destructive field-based phenotyping of small grain cereals. This precision phenotyping platform has been calibrated to assess biomass yield of triticale which yielded a high prediction accuracy and heritability for the predicted biomass (Busemeyer et al. 2013a).
Results from Gowda et al. (2011) suggested plant height to be a key contributor to biomass yield in triticale while grain yield, heading time, spikes per square meter and 1,000-kernel weight contribute to a lesser extent. This study also revealed a broad genetic variation for biomass yield in triticale germplasm. To date, little is known, however, on the genetics underlying biomass yield in triticale and other small grain cereals. Busemeyer et al. (2013a) reported two major QTL for biomass yield on chromosomes 5A and 5R which were identified by association mapping in multiple families. By contrast, the genetics underlying plant height in cereals are much better understood. Plant height is a complex trait determined by many genes with small effects, but major dwarfing genes resulting in substantial reductions in height are also known and have been intensively used in breeding programs (Banaszak 2011; Börner et al. 1996, 1999a; Flintham et al. 1997; Gale and Youssefian 1985; Ren et al. 2010). The dwarfing genes identified in wheat, rye and barley appear to represent a homoeologous series in these three species (Börner et al. 1996; Ivandic et al. 1999). To the best of our knowledge, no studies on plant height and dwarfing genes are available for hexaploid triticale.
QTL mapping approaches can be employed to dissect the genetic architecture underlying complex traits and to identify QTL for marker-assisted selection programs. The extension of linkage mapping methodology towards multiple segregating families offers several advantages like, for example, an increased QTL-detection power and a higher precision of QTL position estimates (Würschum 2012). Consequently, this multiple-line cross QTL (MC-QTL) mapping approach has recently been applied to a range of diverse crops including maize, sugar beet and rapeseed (Blanc et al. 2006; Liu et al. 2012, 2013; Rebai and Goffinet 1993, 2000; Schwegler et al. 2013; Steinhoff et al. 2011, 2012a, b; Würschum et al. 2012a).
In this study, we used a large mapping population consisting of 647 doubled haploid triticale lines derived from four families evaluated in four environments and genotyped with 1710 DArT markers, to dissect the genetic architecture of biomass yield and plant height by MC-QTL mapping. In particular, the objectives of this study were to (1) identify main-effect QTL for the two traits, (2) assess the precision of QTL position estimates and the predictive power of the identified QTL by cross-validation, and (3) investigate the contribution of epistatic interactions to the genetic architecture of plant height and biomass yield in triticale.
Materials and methods
Plant materials and field experiments
This study was based on 647 doubled haploid (DH) triticale lines (Würschum et al. 2012b) derived from four families: DH06 (131 individuals), DH07 (120), EAW74 (200), and EAW78 (196) which have been described by Alheit et al. (2011). The field experiments and the collection of biomass data have been described by Busemeyer et al. (2013a). In brief, the progenies were evaluated in partially replicated trials (Williams et al. 2011) including common checks with 960 plots per location at two locations, Hohenheim and Bohlingen (both situated in south-west Germany), in the years 2011 and 2012. Six-row plots (5 m2) were machine sown and data recorded for plant height and biomass at the developmental stage BBCH 81 (very early dough development) (Lancashire et al. 1991). Plant height was recorded conventionally, and biomass data was obtained with the precision phenotyping platform and prediction according to a developed calibration model (Busemeyer et al. 2013a, b).
Genotypic and phenotypic analyses
The 647 mapping individuals were genotyped with DArT markers and the map positions of a high-density consensus linkage map incorporating 1,710 markers were used for analyses (Alheit et al. 2011). Phenotypic data were analyzed by ordinary lattice analysis of variance (Cochran and Cox 1957) employing a two-step approach as suggested by Möhring and Piepho (2009). In the first step, the phenotypic data of each environment were analyzed separately based on the statistical model
where \(y_{ijk}\) is the phenotypic performance of the ith entry in the jth replication in the kth incomplete block, μ an intercept term, \(g_{i}\) the genetic effect of the ith genotype, \(r_{j}\) the effect of the jth replication, \(b_{jk}\) the effect of the kth incomplete block in the jth replication, and \(e_{ijk}\) the residual. Except \(b_{jk}\) all effects were treated as fixed. The adjusted entry means of each genotype and one divided by the squared standard error as weighting factor were used in the second step for the analysis across environments with the following model
where \(y_{ij}\) is the adjusted entry mean of the ith genotype at the jth environment estimated in the first step, \(g_{i}\) the genetic effect of the ith genotype, \({\text{env}}_{j}\) the effect of the jth environment, \(g_{i}\!\!:{\text{env}}_{j}\) the genotype by environment interaction, and \(e_{ij}\) the residual. For the analysis of single families, we used dummy variables which indicate the membership of a genotype to the respective family. Variance components were determined by the restricted maximum likelihood (REML) method assuming a random model. Best linear unbiased estimates (BLUEs) were estimated across environments assuming fixed effects for the genotype. Heritability (h 2) on an entry-mean basis was estimated from the variance components as the ratio of genotypic to phenotypic variance (Knapp et al. 1985). All statistical analyses were performed using ASReml 3.0 (Gilmour et al. 2009).
Multiple-line cross QTL mapping
For multiple-line cross QTL (MC-QTL) mapping (Blanc et al. 2006), we applied a model assuming specific QTL effects for every family (disconnected model) as this has recently been shown to perform well (Schwegler et al. 2013, Steinhoff et al. 2011). In brief, the model was
where Y denotes a N × 1 column vector of BLUE values of phenotypic data of N progenies coming from F families, J is a N × F matrix whose elements were 0 or 1 according to whether or not individual i belonged to family f and M is a F × 1 vector of family specific means. \(X_{\text{q}}^{\prime } \left( {X_{\text{c}}^{\prime } } \right)\) denotes an N × F matrix containing the expected number (ranging from 0 to 2) of allele k for each individual in family f at QTL q (cofactor c), and \(B_{\text{q}}^{\prime } \left( {B_{\text{c}}^{\prime } } \right)\) is a F × 1 vector of the expected allele substitution effects of QTL q (cofactor c) in family f. ε denotes the vector of the residuals.
Cofactor selection was performed using PROC GLMSELECT implemented in the statistical software SAS (SAS Institute 2008). Testing for presence of a putative QTL in an interval was performed using a likelihood ratio test with the statistical software R (R Development Core Team 2005). LOD thresholds of 4.7 and 4.6 for plant height and biomass yield, respectively, were used corresponding to an experiment-wise type I error of P < 0.10, based on 2,000 permutations (Doerge and Churchill 1996). QTL were declared as overlapping between both traits if they fell within an arbitrarily defined 10-cM interval surrounding the QTL.
A full two-dimensional scan for pair-wise interactions was performed based on the comparison of a full versus a reduced model (Steinhoff et al. 2012b). In the full model, all cofactors selected plus the main effects of the two loci analyzed were included, whereas in the reduced model the interaction term between the two loci analyzed was excluded. Following the suggestion of Holland et al. (2002), the chosen α level of 0.05 was divided by the number of possible independent pair-wise interactions between chromosome regions, assuming two regions per chromosome (P < 5.8 × 10-5). The circular plots illustrating the epistatic interactions were created with Circos (Krzywinski et al. 2009).
The proportion of the genotypic variance explained by all detected QTL was estimated across the four families as \(R_{\text{adj}}^{2} /h^{2}\), where h 2 refers to the heritability of the trait (Utz et al. 2000). The support interval of a QTL was defined as a LOD fall-off of 1.0 expressed as position on the chromosome in centimorgans (cM) (Lander and Botstein 1989) and cofactors were excluded within a distance to the marker interval under consideration smaller than 10 cM.
Cross-validation
To evaluate the results of QTL detection, a fivefold cross-validation approach with genotypic sampling was performed following Liu et al. (2013). In brief, the data set was randomly subdivided into five subsets, while maintaining the relative contribution of the individual families in each subset. Four of the five genotypic samples were used as estimation set (ES) for QTL detection, localization and estimation of their genetic effects. The remaining lines in the test set (TS) served as an independent sample used for validation of the QTL results from the ES and to obtain unbiased estimates of the genotypic variance explained by the QTL (p G-TS). The random sampling of genotypes into ES and TS was repeated 600 times. The proportion of the genotypic variance of the detected QTL in the ES (p G-ES) was compared to that explained in the TS (p G-TS) to calculate the bias as p G-ES − p G-TS, and the relative bias as 1 − (p G-TS/p G-ES).
Results
For both biomass yield and plant height, we observed significant (P < 0.01) genotypic variances and genotype × environment interaction variances (Table 1). The heritability amounted 0.95 for plant height and 0.85 for biomass yield. The phenotypic correlation between plant height and biomass yield was 0.86 in the entire population and ranged between 0.59 and 0.91 within the four families (all P < 0.001). The means of the parents of the crosses differed to varying degrees for plant height (Δ 2.1–13.6 cm) as well as for biomass yield (Δ 1.3–19.4 dt ha−1) (Fig. 1). Orthogonal contrasts of the means of the parental lines and the means of the populations were not significantly different for both traits. The trait distributions approximately followed a normal distribution except for family EAW78 which especially for plant height showed a bimodal distribution (Fig. 1). For both traits, DH lines showed transgressive segregation compared to the phenotypes of the parental lines. Taken together, the phenotypic data present an excellent basis to dissect the genetics underlying plant height and biomass yield in triticale.

Frequency distributions of phenotypic data for (a) plant height and (b) biomass yield across all genotypes and for the single families. Arrowheads indicate the phenotypic value of the respective parents
The multiple-line cross QTL mapping across the four DH families comprising a total of 647 individuals revealed twelve QTL for plant height and nine QTL for biomass yield (Table 1; Fig. 2). The proportion of genotypic variance explained by these QTL amounted 74.8 % for plant height and 62.8 % for biomass yield. The proportion of genotypic variance explained by individual QTL ranged from 1.0 to 42.0 % for plant height and from 2.2 to 35.6 % for biomass yield (Table 2) and the major QTL for both plant height and biomass yield was located on chromosome 5R. The QTL effects showed considerable variation across families. We employed a fivefold cross-validation approach to obtain unbiased estimates of the predictive power of the QTL for both traits (Table 1). The cross-validated proportion of explained genotypic variance was 59.6 % for plant height and 38.2 % for biomass yield. The relative bias was higher for biomass yield with 33.0 % as compared to 23.4 % for plant height.

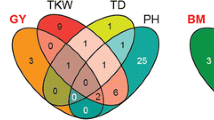
LOD curves from MC-QTL mapping for plant height and biomass yield. The dashed horizontal line indicates the significance threshold. Venn diagram indicates overlapping and trait-specific QTL
The QTL frequency distributions revealed that some of the QTL, including the major QTL on chromosome 5R, were identified in more than 40 % of the runs (Fig. 3). By contrast, few QTL detected with the full data set were only detected in less than 10 % of the runs (e.g., biomass yield QTL on chromosome 4R). In general, the QTL frequency distributions showed narrow peaks at the QTL positions.

Frequency distributions of QTL detected in fivefold cross-validation for plant height and biomass yield. Arrowheads indicate the positions of QTL detected in the full data set
The full two-dimensional genome scan revealed six and four epistatic QTL for plant height and biomass yield, respectively (Fig. 4). For the epistatic QTL affecting biomass yield, involving regions on chromosomes 1A and 4B, the epistatic interaction landscape is exemplarily visualized in Fig. 4. The proportion of genotypic variance explained by these epistatic QTL ranged from 0.6 to 1.1 % for plant height and from 1.3 to 1.7 % for biomass yield.

Epistatic interactions for biomass yield and plant height. The epistatic QTL for biomass yield involving chromosomes 1A and 4B is shown exemplarily as epistatic interaction landscape where red indicates significant −log10 (P values)
Discussion
The combined analysis of multiple biparental families offers a number of advantages as compared to QTL mapping in single populations and has consequently become increasingly popular in recent years (Blanc et al. 2006; Myles et al. 2009; Rebai and Goffinet 1993; Verhoeven et al. 2006; Würschum 2012). In this study, we used data of four partially connected DH families to unravel the genetic architecture of biomass yield and plant height in triticale.
Phenotypic properties of the mapping population
One of the main prerequisites for QTL detection with a high power is the precise estimation of phenotypic values (Liu et al. 2013). Plant height was measured conventionally with a heritability of 0.95 which is in agreement with results from triticale (Herrmann 2007; Oettler et al. 2005), rye (Miedaner et al. 2010, 2012a) and wheat (Khan et al. 2003; Teich 1984). By contrast, biomass yield was obtained based on multi-sensor measurements by a precision phenotyping platform and subsequent prediction with a sensor-fusion calibration model (Busemeyer et al. 2013a). This yielded a high heritability of 0.85 which is in accordance with previous studies on biomass yield in rye (Miedaner et al. 2010, b) and maize (Grieder et al. 2012a, b). Thus, precision phenotyping as applied here is an attractive approach to non-invasively determine biomass yield in small grain cereals with high accuracy and heritability. The major advantage of this non-invasive approach, especially in breeding programs, is that it enables the assessment of additional traits on the same plots.
For plant height, only the parents of family EAW78 differed considerably in their phenotypic values, while for biomass yield substantial differences were found between the parents of DH07 and EAW74 (Fig. 1). However, these differences between the parents were not reflected in the genotypic variances which confirms results of Miedaner et al. (2012a) for complex traits in rye. This can be explained by the observed transgressive segregation for both traits in all families suggesting that the parents carry in part complementary QTL which are newly combined in their progeny. For family EAW78, a bimodal distribution was observed for plant height indicating that a major plant height gene is segregating in this family. In triticale, well-known dwarfing genes, originating from wheat or rye (Börner et al. 1996, 1999b), can result in substantial height differences as observed for the two parents of this cross. The observed high positive correlation between both traits suggests that plant height must be considered as major contributor to biomass yield in triticale which warrants a joint analysis of both traits and a comparison of their underlying QTL.
Detection of main-effect QTL for plant height
Plant height in small grain cereals is a complex trait controlled by many genes (Börner et al. 1996, 1999b; Flintham et al. 1997; Gale and Youssefian 1985; Miedaner et al. 2011, 2012a; Ren et al. 2010). We identified 12 main-effect QTL for plant height which cross-validated explained considerable 59.6 % of the genotypic variance (Table 1). This high proportion of explained genotypic variance can mainly be attributed to two QTL, the QTL on chromosome 6A and the major QTL on chromosome 5R (Table 2). It must be noted, that the latter showed the strongest QTL effect in family EAW78 suggesting that the allele with the strong effect on plant height only segregates in this family. The joint analysis across multiple families as employed here permits the detection of QTL with large effects segregating in a single family, but also of QTL with small effects segregating in several of the families which are only detectable by the combined power across families. The observed bimodal distribution of plant height (Fig. 1) supports the assumption of a major QTL segregating in family EAW78. Furthermore, the parent of this cross, TIW671 or cultivar ‘Pigmej’, is known as very short triticale type, probably as a result of it carrying a dwarfing gene. In rye, this chromosome has been reported to harbor a major plant height QTL at the relative position on the chromosome comparable to the QTL identified here and we, therefore, speculate that the QTL detected on chromosome 5R might represent the dominant dwarfing gene Ddw1 (Börner et al. 1999b; Korzun et al. 1996). Another QTL located on chromosome 5A might represent the dwarfing gene Rht12, a potential homeolog of Ddw1 (Börner et al. 1996), demonstrating the possible combination of major plant height QTL from wheat and rye in triticale. Taken together, our results illustrate the quantitative inheritance of plant height in triticale but also demonstrate the contribution of single major QTL. The confirmation of the identified major QTL as Ddw1 and its fine-mapping warrant further research.
Detection of main-effect QTL for biomass yield
Even though the exploitation of crop biomass holds great potential, little is known until now about the genetics of biomass yield in small grain cereals. We have recently reported the detection of biomass yield QTL in the population underlying this study (Busemeyer et al. 2013a). The biometrical model applied in the previous study is based on identity-by-state and has been show to be rather conservative with regard to QTL detection (Würschum et al. 2012c). Here, we used MC-QTL mapping which is an identity-by-descent approach and the higher proportion of explained genotypic variance realized with the detected QTL illustrates that MC-QTL mapping is well suited for QTL analysis in the underlying data set with four partially connected families. A total of nine QTL were identified for biomass yield explaining 62.8 % of the genotypic variance, thus substantially exceeding the results from the previous approach where the identified QTL explained only 28.5 % of the genotypic variance (Busemeyer et al. 2013a). This corroborates previous findings of Liu et al. (2012) who showed that identity-by-state (association mapping) and identity-by-descent (MC-QTL mapping) approaches each possess specific properties. As expected for complex traits, the cross-validated proportion of genotypic variance was lower as compared to that in the full data set but still amounted to considerable 38.2 %. As for plant height, a major QTL was identified in the same region on chromosome 5R (Table 2). In addition, two further QTL explaining more than 5 % were detected on chromosomes 5A and 6A. Busemeyer et al. (2013a) also reported QTL for biomass yield in triticale, but only on chromosomes 5A and 5R. In summary, this analysis identified several QTL and revealed that biomass yield is mainly controlled by small effect QTL.
Contribution of epistasis
Epistasis refers to interactions between the alleles at two or more genetic loci (Carlborg and Haley 2004) and has recently been shown to contribute to the genetic architecture of complex traits in crops (Buckler et al. 2009; Liu et al. 2012; Reif et al. 2011; Steinhoff et al. 2012b; Würschum et al. 2013). It must be noted that the QTL-detection power for epistatic QTL depends even more on the population size than for main-effect QTL. The comparably large mapping population underlying this study enabled the detection of six epistatic QTL for plant height and four for biomass yield. The proportion of genotypic variance explained by these epistatic QTL averaged 0.9 % for plant height and 1.5 % for biomass yield which is smaller than the average of the respective main-effect QTL (5.9 and 6.9 %, respectively). Thus, epistasis contributes to the genetic architecture of both traits albeit to a minor extent.
Consequences for plant breeding
A recent study has identified grain yield, heading time, spikes per square meter, 1,000-kernel weight, and early plant height as key variables to predict early biomass yield (Gowda et al. 2011). The high correlation between biomass yield and plant height observed in our study, confirms plant height as a major contributor to biomass yield. We consequently observed a high congruency between QTL detected for the two traits with six of the nine biomass yield QTL also being identified as plant height QTL, including the major QTL on chromosome 5R (Fig. 2). Possible reasons for this colocalization of QTL for plant height and biomass yield include: (1) two closely linked genes each affecting a separate trait; (2) one gene with a pleiotropic effect; or (3) one gene with a single function that initiates a sequence of causally related events (Lebreton et al. 1995; Liu et al. 2013).
For marker-assisted selection, the QTL positions must be estimated with high precision to minimize the probability of recombinations between the QTL and the marker used for selection. The QTL frequency distributions revealed narrow peaks for the majority of the identified QTL illustrating the reliability of the QTL position estimates (Fig. 3). Another important criterion to assess the prospects of marker-assisted selection is the proportion of genotypic variance explained by the detected QTL. Cross-validation is an approach to obtain unbiased estimates of this parameter (Liu et al. 2013; Schön et al. 2004). The cross-validated proportion of explained genotypic variance was substantially higher for plant height (59.6 %) than for biomass yield (38.2 %) which is likely due to the higher complexity of the latter. While single QTL explaining a sufficiently high proportion of the genotypic variance may be used in marker-assisted selection, the combined predictive power of all identified QTL was still limited. This is concordant with the quantitative nature of the traits which are controlled by many small effect QTL that escape detection in QTL mapping. Future research should aim at evaluating the performance of genomic selection to improve these traits as this approach also considers QTL with small effects. In summary, our data show that marker-assisted selection based on the QTL detected by MC-QTL mapping, or potentially genomic selection, hold potential for a knowledge-based improvement of biomass yield in triticale.
Conclusions
In this study, we report the first MC-QTL mapping in triticale which identified several QTL for plant height and biomass yield. The high correlation between the two traits confirms plant height as a major contributor to biomass yield and several of the biomass yield QTL have also been identified as plant height QTL. A major plant height QTL was detected on chromosome 5R and is likely to represent the dominant dwarfing gene Ddw1 which is well known from rye breeding. Furthermore, our results showed that epistatic interactions contribute to a minor extent to the genetic architecture of plant height and biomass yield in triticale. In summary, genomic approaches, like MC-QTL mapping applied here, hold the potential for a knowledge-based breeding of triticale cultivars with improved biomass yield.
References
Alheit KV, Reif JC, Maurer HP, Hahn V, Weissmann EA, Miedaner T, Würschum T (2011) Detection of segregation distortion loci in triticale (× Triticosecale Wittmack) based on a high-density DArT marker consensus genetic linkage map. BMC Genomics 12:380
Banaszak Z (2011) Breeding of triticale in DANKO. 61 Tagung der Vereinigung der Pflanzenzüchter und Saatgutkaufleute Österreichs 2010, pp 65–68
Blanc G, Charcosset A, Mangin B, Gallais A, Moreau L (2006) Connected populations for detecting quantitative trait loci and testing for epistasis: an application in maize. Theor Appl Genet 113:206–224
Börner A, Plaschke J, Korzun V, Worland AJ (1996) The relationships between the dwarfing genes of wheat and rye. Euphytica 89:69–75
Börner A, Korzun V, Malyshev S, Ivandic V, Graner A (1999a) Molecular mapping of two dwarfing genes differing in their GA response on chromosome 2H of barley. Theor Appl Genet 99:670–675
Börner A, Korzun V, Voylokov AV, Weber WE (1999b) Detection of quantitative trait loci on chromosome 5R of rye (Secale cereale L.). Theor Appl Genet 98:1087–1090
Buckler ES, Holland JB, Bradbury PJ, Acharya CB, Brown PJ, Browne C, Ersoz E, Flint-Garcia S, Garcia A, Glaubitz JC, Goodman MM, Harjes C, Guill K, Kroon DE, Larsson S, Lepak NK, Li H, Mitchell SE, Pressoir G, Peiffer JA, Rosas MO, Rocheford TR, Romay MC, Romero S, Salvo S, Villeda HS, Da Silva HS, Sun Q, Tian F, Upadyayula N, Ware D, Yates H, Yu J, Zhang Z, Kresovich S, McMullen MD (2009) The genetic architecture of maize flowering time. Science 325:714–718
Busemeyer L, Ruckelshausen A, Möller K, Melchinger AE, Alheit KV, Maurer HP, Hahn V, Weissmann EA, Reif JC, Würschum T (2013a) Precision phenotyping of biomass accumulation in triticale reveals temporal genetic patterns of regulation. Sci Rep 3:2442
Busemeyer L, Mentrup D, Möller K, Wunder E, Alheit K, Hahn V, Maurer HP, Reif JC, Würschum T, Müller J, Rahe F, Ruckelshausen A (2013b) Breedvision—a multi-sensor platform for non-destructive field-based phenotyping in plant breeding. Sensors (Switzerland) 13:2830–2847
Carlborg Ö, Haley CS (2004) Epistasis: too often neglected in complex trait studies? Nat Rev Genet 5:618–625
Cochran WG, Cox GM (1957) Experimental designs. Wiley, New York
Doerge RW, Churchill GA (1996) Permutation tests for multiple loci affecting a quantitative character. Genetics 142:285–294
Flintham JE, Börner A, Worland AJ, Gale MD (1997) Optimizing wheat grain yield: effects of Rht (gibberellin-insensitive) dwarfing genes. J Agric Sci 128:11–25
Gale MD, Youssefian S (1985) Dwarfing genes in wheat. In: Russell GE (ed) Progress in plant breeding 1. Butterworth, London, pp 1–35
Gilmour AR, Gogel BG, Cullis BR, Thompson R (2009) ASReml user guide release 3.0. VSN International Ltd, Hemel Hempstead, HP1 1ES, UK
Gowda M, Hahn V, Reif JC, Longin CFH, Alheit K, Maurer HP (2011) Potential for simultaneous improvement of grain and biomass yield in Central European winter triticale germplasm. Field Crops Res 121:153–157
Grieder C, Dhillon BS, Schipprack W, Melchinger AE (2012a) Breeding maize as biogas substrate in Central Europe: II. Quantitative-genetic parameters for inbred lines and correlations with testcross performance. Theor Appl Genet 124:981–988
Grieder C, Dhillon BS, Schipprack W, Melchinger AE (2012b) Breeding maize as biogas substrate in Central Europe: I. Quantitative-genetic parameters for testcross performance. Theor Appl Genet 124:971–980
Herrmann M (2007) A diallel analysis of various traits in winter triticale. Plant Breed 126:19–23
Holland JB, Portyanko VA, Hoffman DL, Lee M (2002) Genomic regions controlling vernalization and photoperiod responses in oat. Theor Appl Genet 105:113–126
Ivandic V, Malyshev S, Korzun V, Graner A, Börner A (1999) Comparative mapping of a gibberellic acid-insensitive dwarfing gene (Dwf2) on chromosome 4HS in barley. Theor Appl Genet 98:728–731
Khan AS, Salim I, Ali Z (2003) Heritability of various morphological traits in wheat. Int J Agric Biol. doi:1560-8530/2003/05-2-138-140
Knapp SJ, Stroup WW, Ross WM (1985) Exact confidence intervals for heritability on a progeny mean basis. Crop Sci 25:192–194
Korzun V, Melz G, Börner A (1996) RFLP mapping of the dwarfing (Ddw1) and hairy peduncle (Hp) genes on chromosome 5 of rye (Secale cereale L.). Theor Appl Genet 92:1073–1077
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645
Kuleung C, Baenziger PS, Dweikat I (2004) Transferability of SSR markers among wheat, rye, and triticale. Theor Appl Genet 108:1147–1150
Lancashire PD, Bleiholder H, van Boom TD, Langelüddeke P, Stauss R, Weber E, Witzenberger A (1991) A uniform decimal code for growth stages of crops and weeds. Ann Appl Biol 119:561–601
Lander ES, Botstein S (1989) Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121:185
Lebreton C, Lazic-Jancic V, Steed A, Pekic S, Quarrie SA (1995) Identification of QTL for drought responses in maize and their use in testing causal relationships between traits. J Exp Bot 46:853–865
Liu W, Reif JC, Ranc N, Porta GD, Würschum T (2012) Comparison of biometrical approaches for QTL detection in multiple segregating families. Theor Appl Genet 125:987–998
Liu W, Maurer HP, Reif JC, Melchinger AE, Utz HF, Tucker MR, Ranc N, Della Porta G, Würschum T (2013) Optimum design of family structure and allocation of resources in association mapping with lines from multiple crosses. Heredity 110:71–79
Miedaner T, Hübner M, Koch S, Seggl A, Wilde P (2010) Biomass yield of self-incompatible germplasm resources and their testcrosses in winter rye. Plant Breed 129:369–375
Miedaner T, Müller BU, Piepho H-P, Falke KC (2011) Genetic architecture of plant height in winter rye introgression libraries. Plant Breed 130:209–216
Miedaner T, Hübner M, Korzun V, Schmiedchen B, Bauer E, Haseneyer G, Wilde P, Reif JC (2012a) Genetic architecture of complex agronomic traits examined in two testcross populations of rye (Secale cereale L.). BMC Genomics 13:706
Miedaner T, Koch S, Seggl A, Schmiedchen B, Wilde P (2012b) Quantitative genetic parameters for selection of biomass yield in hybrid rye. Plant Breed 131:100–103
Möhring J, Piepho HP (2009) Comparison of weighting in two-stage analysis of plant breeding trials. Crop Sci 49:1977–1988
Myles S, Peiffer J, Brown PJ, Ersoz ES, Zhang Z, Costich DE, Buckler E (2009) Association mapping: critical considerations shift from genotyping to experimental design. Plant Cell 21:2194–2202
Oettler G (2005) The fortune of a botanical curiosity—Triticale: past, present and future. J Agric Sci 143:329–346
Oettler G, Tams SH, Utz HF, Bauer E, Melchinger AE (2005) Prospects for hybrid breeding in winter triticale: I. Heterosis and combining ability for agronomic traits in European elite germplasm. Crop Sci 45:1476–1482
Pronyk C, Mazza G (2011) Optimization of processing conditions for the fractionation of triticale straw using pressurized low polarity water. Bioresour Technol 102:2016–2025
R Development Core Team (2005) R: a language and environment for statistical computing, reference index version 2.x.x. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0. http://www.R-project.org
Rebai A, Goffinet B (1993) Power of tests for QTL detection using replicated progenies derived from a diallel cross. Theor Appl Genet 86:1014–1022
Rebaï A, Goffinet B (2000) More about quantitative trait locus mapping with diallel designs. Genet Res 75:243–247
Reif JC, Maurer HP, Korzun V, Ebmeyer E, Miedaner T, Würschum T (2011) Mapping QTLs with main and epistatic effects underlying grain yield and heading time in soft winter wheat. Theor Appl Genet 123:283–292
Ren X, Sun D, Guan W, Sun G, Li C (2010) Inheritance and identification of molecular markers associated with a novel dwarfing gene in barley. BMC Genet 11:89
SAS Institute (2008) SAS/STAT 9.2 User’s guide. SAS Institute Inc., Cary, NC, USA
Schön CC, Utz HF, Groh S, Truberg B, Openshaw S, Melchinger AE (2004) Quantitative trait locus mapping based on resampling in a vast maize testcross experiment and its relevance to quantitative genetics for complex traits. Genetics 167:485–498
Schwegler DD, Liu W, Gowda M, Würschum T, Schulz B, Reif JC (2013) Multiple-line cross quantitative trait locus mapping in sugar beet (Beta vulgaris L.). Mol Breed 31:279–287
Steinhoff J, Liu W, Maurer HP, Würschum T, Longin CFH, Ranc N, Reif JC (2011) Multiple-line cross quantitative trait locus mapping in European elite maize. Crop Sci 51:2505–2516
Steinhoff J, Liu W, Maurer HP, Würschum T, Longin CFH, Ranc N, Reif JC (2012a) Exploitation of elite maize (Zea mays L.) germplasm across maturity zones. Crop Sci 52:1534–1542
Steinhoff J, Liu W, Reif JC, Porta GD, Ranc N, Würschum T (2012b) Detection of QTL for flowering time in multiple families of elite maize. Theor Appl Genet 125:1539–1551
Teich AH (1984) Heritability of grain yield, plant height and test weight of a population of winter wheat adapted to Southwestern Ontario. Theor Appl Genet 68:21–23
Utz HF, Melchinger AE, Schön CC (2000) Bias and sampling error of the estimated proportion of genotypic variance explained by quantitative trait loci determined from experimental data in maize using cross validation and validation with independent samples. Genetics 154:1839–1849
Verhoeven KJF, Jannink J-L, McIntyre LM (2006) Using mating designs to uncover QTL and the genetic architecture of complex traits. Heredity 96:139–149
Williams E, Piepho H-P, Whitaker D (2011) Augmented p-rep designs. Biometr J 53:19–27
Würschum T (2012) Mapping QTL for Agronomic traits in breeding populations. Theor Appl Genet 125:201–210
Würschum T, Liu W, Maurer HP, Abel S, Reif JC (2012a) Dissecting the genetic architecture of agronomic traits in multiple segregating populations in rapeseed (Brassica napus L.). Theor Appl Genet 124:153–161
Würschum T, Tucker MR, Reif JC, Maurer HP (2012b) Improved efficiency of doubled haploid generation in hexaploid triticale by in vitro chromosome doubling. BMC Plant Biol 12:109
Würschum T, Liu W, Gowda M, Maurer HP, Fischer S, Schechert A, Reif JC (2012c) Comparison of biometrical models for joint linkage association mapping. Heredity 108:332–340
Würschum T, Maurer HP, Dreyer F, Reif JC (2013) Effect of inter- and intragenic epistasis on the heritability of oil content in rapeseed (Brassica napus L.). Theor Appl Genet 126:435–441
Acknowledgments
This research was funded by the German Federal Ministry of Education and Research (BMBF) under the promotional reference 0315414A. This publication reflects the views only of the authors. We acknowledge the handling of the funding by the Project Management Organisation Jülich (PtJ). We thank Angela Harmsen for excellent technical assistance in the laboratory and Agnes Rölfing-Finze, Hans Häge, Jacek Till and Justus von Kittlitz for their outstanding work in the greenhouse and field.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standard
The authors declare that the experiments comply with the current laws of Germany.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by P. Langridge.
K. V. Alheit, L. Busemeyer and W. Liu contributed equally to this work.
Rights and permissions
About this article
Cite this article
Alheit, K.V., Busemeyer, L., Liu, W. et al. Multiple-line cross QTL mapping for biomass yield and plant height in triticale (× Triticosecale Wittmack). Theor Appl Genet 127, 251–260 (2014). https://doi.org/10.1007/s00122-013-2214-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-013-2214-6