
Abstract
Detection of QTL in multiple segregating populations is of high interest as it includes more alleles than mapping in a single biparental population. In addition, such populations are routinely generated in applied plant breeding programs and can thus be used to identify QTL which are of direct relevance for a marker-assisted improvement of elite germplasm. Multiple-line cross QTL mapping and joint linkage association mapping were used for QTL detection. We empirically compared these two different biometrical approaches with regard to QTL detection for important agronomic traits in nine segregating populations of elite rapeseed lines. The plants were intensively phenotyped in multi-location field trials and genotyped with 253 SNP markers. Both approaches detected several additive QTL for diverse traits, including flowering time, plant height, protein content, oil content, glucosinolate content, and grain yield. In addition, we identified one epistatic QTL for flowering time. Consequently, both approaches appear suited for QTL detection in multiple segregating populations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Oilseed rape (Brassica napus L.) is grown in many parts of the world and is the most important source of vegetable oil in Europe and second most important worldwide, after soybean. It is used both, for human nutrition and for industrial products. Yield is the most important breeding goal in rapeseed breeding, and high seed oil and protein content are additional breeding goals depending on the country. After the extraction of oil from the seeds, the residual meal is commonly used in livestock feeding and is thus desired to contain high levels of protein. Glucosinolates are secondary plant metabolites that restrict the use of this protein-rich feed supplement, and the goal for plant breeders is to decrease the glucosinolate content. In addition, yield and yield-related traits, such as flowering time and plant height, are also traits under intense selection in breeding programs. Linkage mapping and more recently association mapping approaches have identified QTL for all the above-mentioned quantitative traits (Delourme et al. 2006; Mahmood et al. 2006; Qiu et al. 2006; Hasan et al. 2008; Mei et al. 2009; Chen et al. 2010; Smooker et al. 2011).
B. napus is an amphidiploid (AACC) and originated by the hybridization of B. rapa and B. oleracea (Kimber and McGregor 1995). The genepool of elite oilseed rape has been selected for high yield and oil content and more recently went through a strong genetic bottleneck due to the selection for the desired zero seed erucic acid and low glucosinolate content. Both traits were introduced into the elite breeding material in the 1960s and 1970s and each originate from single genotypes (Becker et al. 1999). This breeding history potentially has implications on the linkage disequilibrium (LD) structure of current elite oilseed rape. In a first study of LD in rapeseed, Ecke et al. (2010) investigated LD in 85 elite rapeseed genotypes and observed that LD is high between closely linked markers and decays within a few centimorgan.
In plant breeding programs, segregating populations are routinely generated and phenotyped. The decreasing costs for molecular marker analyses have paved the way for a routine genotyping of the same plants. Thus, both phenotypic and genotypic data are available and can be exploited to detect QTL segregating in elite germplasm for a later implementation in knowledge-based breeding programs. Different approaches have recently been suggested to dissect the genetic architecture of complex traits in multiple segregating populations. Multiple-line cross QTL (MC-QTL) mapping is based on linkage mapping methodology (Blanc et al. 2006; Steinhoff et al. 2011), whereas joint linkage association mapping (JLAM) is an association mapping approach and is based on LD (Yu et al. 2008; Reif et al. 2010).
The main goal of this study was to use phenotypic and genotypic data from several populations derived from an applied breeding program to dissect the genetic architecture of agronomically important traits in oilseed rape. In particular, the objectives of our study were to use a large population of 391 elite rapeseed lines to (1) examine correlations between agronomic traits, (2) investigate LD structure in this panel of elite lines, (3) identify main and epistatic QTL for important agronomic traits and (4) empirically compare two different biometrical approaches for QTL detection in multiple segregating populations.
Materials and methods
Plant materials and field trials
Our study was based on 391 doubled haploid (DH) rapeseed (Brassica napus L.) progenies, which were derived from nine crosses among 10 parental lines (Supplementary Table 1). The parents were elite breeding lines from the breeding pool of Limagrain bred for release as varieties in Central Europe. The relationship among the parental lines is shown in the principal coordinate plot (Supplementary Figure S1). The number of progenies from each cross ranged from 9 to 93. All material used in this study was provided by the breeding company Limagrain GmbH. The 391 progenies and nine of the ten parents were tested in 4 environments representing the Central European Mega environment. The four environments were RSRS (Germany, 75 m asl, loamy sand texture, mean annual temperature 8.2°C, mean annual precipitation 660 mm), RSBE (Germany, 262 m asl, loamy sand texture, mean annual temperature 9.0°C, mean annual precipitation 630 mm), VRVR (France, 91 m asl, loamy sand texture, mean annual temperature 9.4°C, mean annual precipitation 671 mm), and ROLN (UK, 65 m asl, loamy sand texture, mean annual temperature 9.6°C, mean annual precipitation 599 mm).The experimental design at each environment was an α-lattice design with two replicates per environment. Plant density was 500.000 plants ha−1.
Data were recorded for flowering time (FT visual rating from 1 to 9 at one time point to assess how advanced the plants were in flowering), plant height (PH; cm), protein content (PROT; %), oil content (OIL; %), glucosinolate content (GLU; μmol g−1), dry matter content (DMC%), and grain yield (GY; Mg ha−1). PROT, OIL and GLU were determined by near-infrared reflectance spectroscopy (FOSS XDS XD-1000, calibrations by Limagrain). Two technical replications were done for each measurement.
Phenotypic data analyses
The phenotypic data of each environment were first analyzed separately based on the statistical model y ikl = μ + g i + r k + b lk + e ikl , where y ikl was the phenotypic observation for the ith rapeseed line in the lth incomplete block of the kth replication, μ was an intercept term, g i was the genetic effect of the ith genotype, r k was the effect of the kth replication, b lk was the effect of the lth incomplete block of the kth replication, and e ikl was the residual. Except b lk , all effects were regarded as fixed. We estimated the variance of the residuals in single environments and calculated the average across environments, which was denoted in the following as σ² e .
A combined analysis across locations based on the BLUE values from the above model was performed using the following statistical model Y im = μ + G i + E m + ε im , where Y im was the best linear unbiased estimate (BLUE) for the ith rapeseed line in the mth environment, μ was an intercept term, G i was the genetic effect of the ith genotype, E m was the effect of the mth environment, and ε im was the residual, which equals \( {\text{GE}}_{im} + \bar{e} \). Variance components were determined by the restricted maximum likelihood (REML) method assuming a random model. The estimate of \( \sigma_{\varepsilon }^{2} \) reflects the sum of \( \sigma_{{{\text{G}} \times {\text{E}}}}^{2} \) and \( \sigma_{e}^{2} \) divided by the number of replications, which was two in our study. Variance component due to genotype × environment interactions was, therefore, calculated as \( \sigma_{{{\text{G}} \times {\text{E}}}}^{2} \) = \( \sigma_{\varepsilon }^{2} \)−\( \sigma_{e}^{2} \)/2 following standard procedure (Cochran and Cox 1957). Heritability on an entry-mean basis was calculated as the ratio of genotypic to phenotypic variance according to Melchinger et al. (1998). In addition, BLUEs across environments were estimated assuming fixed genetic effects.
Molecular data
The 10 parents and their 391 progenies were fingerprinted with 253 single nucleotide polymorphism (SNP) markers. These markers were validated single-locus markers which were randomly distributed across the rapeseed genome with an average marker distance of 11.6 cM (Supplementary Figure S2). 62% of the adjacent markers had a genetic map distance of <10 cM, 50% had a distance <5 cM, and 33% of <2 cM. We used the accepted chromosome nomenclature A1–A10 and C1–C9 (http://www.brassica.info) and map positions of all markers were based on the linkage map of Limagrain (unpublished data). Associations among the 10 parents and their 391 genotypes were analyzed by applying principal coordinate analysis (PCoA) (Gower 1966) based on the modified Rogers’ distances of the individuals (Wright 1978). Extent of LD between all pairs of loci was determined estimating r² as described by Hill and Robertson (1968). LD analyses and PCoA were performed using software Plabsoft (Maurer et al. 2008).
Multiple-line cross QTL mapping
QTL mapping was performed using the connected model suggested by Blanc et al. (2006), which accounts for relationships between the parents. Details are given in Steinhoff et al. (2011), in brief the model used was
where Y was an N × 1 column vector of BLUE values across environments of N progenies (here N = 391), l was an N × 1 column vector of 1, μ was the intercept, \( X_{p} \) was an N × P matrix whose elements were 0 or 1 according to whether or not progeny i belonged to population p, \( M_{P} \) was a P × 1 vector of population effects, \( {\text{X}}_{q}^{ * } \left( {X_{c}^{ * } } \right) \) was an N × K matrix containing the number of parental allele k at marker q (cofactor c) given the marker data for each progeny i, \( B_{q}^{ * } \left( {B_{c}^{ * } } \right) \) was a K × 1 vector of the allele effects of marker q (cofactor c), and ε was the vector of the residual errors.
Cofactor selection was performed using the Schwarz (1978) Bayesian Criterion (SBC) implemented in PROC GLMSELECT implemented in the statistical software SAS (SAS Institute Inc 2008). Testing for the presence of a putative QTL in an interval was performed using a likelihood ratio test using statistical software R. The experiment-wise type I error was determined to be P e < 0.10, using 2,000 permutation runs (Churchill and Doerge 1994). Support intervals for the detected QTL were calculated based on a 1.5 LOD drop. The proportion of genotypic variance (p G) explained by the detected QTL was calculated by fitting all QTL simultaneously in a linear model to obtain R 2adj . The ratio p G = R 2adj /h 2 yielded the proportion of genotypic variance (Utz et al. 2000).
Joint linkage association mapping
Joint linkage association mapping was done with a biometrical model which previously performed well in a comparison of different statistical approaches for JLAM (Würschum et al. 2011a). In brief, an additive genetic model was chosen for the progenies as described by Utz et al. (2000). We applied a two-step procedure for QTL detection. In a first step, stepwise multiple linear regression was used to select a set of cofactors based on the Schwarz (1978) Bayesian Criterion (SBC). Cofactor selection was performed using Proc GLMSELECT implemented in the statistical software SAS (SAS Institute Inc 2008). In the second step, we calculated a P value for the association of each marker with the phenotypic value for the F test with a full model (with marker effect) against a reduced model (without marker effect) (for details, see Reif et al. 2010 and Liu et al. 2011). The applied model includes a population effect, cofactors, and SNP effect across populations. The Bonferroni–Holm procedure (Holm 1979) was used to detect markers with significant (P < 0.05) main effects.
The proportion of the genotypic variance explained by the detected QTL was calculated as described above. The α effect was calculated in the linear model and could only be calculated for the JLAM approach due to the colinearity of the parental alleles modeled in MC-QTL mapping.
Results
The genotypic variances and the genotype-by-environment interaction variances estimated in the population of 391 elite rapeseed lines were significantly larger than zero (P < 0.01) for all six traits (Table 1). Heritability ranged from 0.78 for grain yield to 0.93 for glucosinolate content. Trait distributions approximately followed the expected normal distribution (Fig. 1). Absolute values of phenotypic correlations among the six traits were minimum between oil content and flowering time (0.02) and maximum between flowering time and plant height (0.68) (Supplementary Figure S3).

Trait distribution and parental performance. Histograms of the best linear unbiased estimates (BLUEs) of the six traits analyzed in this study. The performance of the parents from the populations is indicated by arrows
The first two principal coordinates together explained 26.8% of the total variation. Principal coordinate analysis (PCoA) revealed that the progenies within segregating populations cluster together and that the nine populations show different degrees of relatedness with progenies having a common parent being more closely clustered (Supplementary Figure S1). To obtain an estimate for the mapping resolution of JLAM for the population underlying our study, we analyzed the extent of LD. The estimated r 2 values decreased with increasing map distance between marker pairs (Fig. 2).

Extent of linkage disequilibrium. Distribution of linkage disequilibrium assessed between the parental lines. The LD between linked marker pairs is shown for different genetic map distances between these pairs of markers (genetic map distance <1, 1–5, 5–10, and >10 cM). In addition, the LD between unlinked markers is shown. The boxplots show the variation of r 2 values for each of these genetic map distances. Circles indicate outlier which are outside the extreme of the upper whisker
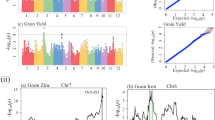
The full genome scan for main effect QTL with MC-QTL mapping identified two QTL for flowering time, two for plant height, one for protein content, two for glucosinolate content, and one for grain yield (Table 2; Fig. 3). The estimated support intervals for the detected QTL ranged from 1 to 61.1 cM. The explained proportion of genotypic variance of single QTL was lowest for a plant height QTL with 3.7% and highest for a flowering time QTL with 14.1%. The total proportion of explained genotypic variance by all detected QTL for one trait was 20.6% for flowering time, 9.6% for plant height, 9.5% for protein content, 15.3% for glucosinolate content, and 7.6% for grain yield.

Comparison of JLAM and MC-QTL results. LOD score profiles from MC-QTL mapping and P values from JLAM as vertical bars for the chromosomes on which QTL were detected. The dotted line indicates the threshold for MC-QTL mapping which was determined by permutation tests and the dashed line indicates the significance threshold for JLAM (P < 0.05 Bonferroni corrected). The positions on the chromosomes are given in centimorgan
The JLAM approach identified more QTL as compared to the MC-QTL mapping approach with most QTL being detected for grain yield (Table 3; Fig. 3). The proportion of genotypic variance explained by a single QTL was highest for a plant height QTL with 13.2%. The total proportion of explained genotypic variance by all detected QTL for one trait was 17.4% for flowering time, 16.2% for plant height, zero for protein content, 2.1% for oil content, 18.3% for glucosinolate content, and 14.7% for grain yield. The full two-dimensional epistasis scan detected two epistatic QTL for flowering time. One between a locus on chromosome C1 at 157 cM and a locus on chromosome C6 at 55 cM and the second between a locus on chromosome C1 at 158 cM and the same locus on chromosome C6 at 55 cM.
Discussion
Properties of the rapeseed population
The present study is based on the experimental data of 391 elite rapeseed genotypes which compared with the previous studies represents a comparably large rapeseed population (e.g. Mahmood et al. 2006; Hasan et al. 2008; Chen et al. 2010). The good quality of the phenotypic data is reflected by the high to very high heritabilities for all traits (Table 1). The nine populations were of varying size, which is typical for applied plant breeding. The principal coordinate analysis revealed that the progenies cluster between their parents and that none of the nine populations is clearly separate from the others (Supplementary Figure S1). In summary, the studied population shows a structure typical for breeding populations and the high heritabilities form an excellent basis for QTL detection in this data set.
Major breeding goals in rapeseed are high yield, high oil content, high protein content, but low glucosinolate content. Both positive and negative correlations have been reported between these traits (e.g. Grami and Stefansson 1977). Thus, an increase in one compound may be achieved only at the expense of another which must be carefully balanced in breeding programs. A possible instrument to break some of the correlations is the identification of QTL and their subsequent implementation in marker-assisted selection programs. As expected, we observed a negative correlation between oil content and glucosinolate content and a similar negative correlation was observed between grain yield and glucosinolate content (Supplementary Figure S3). Potentially, this could be caused by genes having pleiotropic effects on both traits, or by linked genes. In the latter case, the detection of QTL for these traits and their use in breeding programs may accelerate the breeding progress to obtain plants with the desired properties for both traits. It could be speculated that high oil content is correlated with early flowering, as these genotypes have more time to accumulate oil in the seeds. We observed, however, no such correlation between flowering time and oil content. Thus, the time available for oil accumulation does not appear to be a limiting factor in rapeseed and differences between genotypes are rather caused by differences in the efficiency of the underlying metabolic pathways.
Pattern and extent of LD
Linkage disequilibrium between markers and loci associated with the trait is the basis for successful association mapping approaches. In JLAM, the LD structure present in the parental population determines to a large extent the mapping resolution that can be realized in the data set. We found that LD in the population of the ten parents decayed with genetic map distance (Fig. 2). For linked markers with genetic map distances below 5 cM, LD was comparably high with an average r 2 value of around 0.4, whereas for distances greater than 10 cM LD was low and almost indistinguishable from that of unlinked loci. Würschum et al. (2011b) previously suggested as an alternative approach to assess the extent of LD, to examine the highest r 2 value of each marker with any other marker in the genome. In our population, this averaged an r 2 value of 0.94. This represents the highest r 2 value that can be expected on average for a QTL anywhere in the genome.
Our results are in good accordance with those from Ecke et al. (2010) who also observed a decay of LD within a few centimorgan. They concluded that in rapeseed this corresponds to a physical distance of approximately 500–1.000 kb and that for a full genome coverage 1.000–2.500 equally spaced markers would be required. This estimation is based on the LD between closely linked markers. Given the average of the highest r 2 value of each marker observed in this study, this number of required markers may be slightly overestimated. Nevertheless, higher marker densities certainly warrant a better mapping resolution and a higher QTL detection power. With the current advances in sequencing technologies, it can be anticipated that in the near future it will be possible to genotype rapeseed with several thousand markers as is standard already in other crops like maize. As the proportion of variance explained by the marker compared with that explained by the QTL is directly proportional to the r 2 value between the marker and the QTL (van Inghelandt et al. 2011), lower marker densities will only facilitate the detection of QTL with large or medium effects. Taken together, the LD structure present in elite rapeseed germplasm enables association mapping approaches with a good mapping resolution. The marker density applied in this study is sufficient for JLAM with the restriction, that mainly QTL with large or medium effects can be detected.
Comparison of mapping approaches
Two conceptually different biometrical approaches for QTL detection were applied to this data set, MC-QTL mapping and JLAM. MC-QTL mapping is a CIM approach (Jansen and Stam 1994; Zeng 1994) and due to the fact that some of our populations were rather small we used the connected model suggested by Blanc et al. (2006). This approach is an identity-by-descent (IBD) approach based on the parental origin of alleles. QTL effects may vary substantially across populations (Liu et al. 2011; Steinhoff et al. 2011), indicating the interaction of QTL with the genetic background. The connected model is modeled across populations and QTL identified by this approach are thus assumed to be stable across populations, making them very valuable for plant breeders as they can potentially be used in many crosses. The drawback of this approach is the low mapping resolution as indicated by the sometimes rather large support intervals (Table 2).
JLAM on the other hand is an identity-by-state (IBS) approach in which identical marker alleles are treated alike, irrespective from which parent they are derived. The advantage of this approach is the higher mapping resolution, allowing to more precisely pinpoint the detected QTL. It is, however, based on LD and requires much higher marker densities than MC-QTL mapping and disregards the parental origin of alleles.
The data set presented in this study consisted of nine segregating populations of varying size and the plants were genotyped with 253 SNP markers with an average marker distance of 11.6 cM. This situation is typical for applied plant breeding and leaves the question of how to best analyze the data with regard to QTL detection.
In terms of QTL detection, we observed that the JLAM approach detected more QTL which, however, did in general not explain a much higher proportion of the genotypic variance. Of the eight QTL that were detected by MC-QTL mapping, seven were also detected by the JLAM approach. In addition, nine of the JLAM QTL explained <1% of the genotypic variance which likely explains the similar proportion of explained genotypic variance by the detected QTL between the two approaches. This low percentage of explained genotypic variance may be due to colinearity between detected QTL. This holds true even though in all cases these QTL are not genetically linked, as in JLAM the colinearity is caused by LD which we found to be present also between unlinked markers. The plot including both the LOD profile from MC-QTL mapping and the P values from JLAM (Fig. 3), revealed a high consistency between the chromosomal regions where peaks were detected. Differences in the number of detected QTL are thus likely the result of the different threshold levels that are routinely applied in the two approaches. In conclusion, for the presented data set with its rather limited number of markers, both methods MC-QTL mapping and JLAM performed equally well.
Detection of main effects
We performed a literature review for QTL reported in rapeseed in linkage mapping studies or by association approaches (Delourme et al. 2006; Mahmood et al. 2006; Qiu et al. 2006; Hasan et al. 2008, Mei et al. 2009; Chen et al. 2010) and compared the published QTL with those detected in this study by the two mapping approaches (Supplementary Table 2). The drawback that hampers comparisons of QTL between studies is that no common integrated map exists for rapeseed, implying that comparisons must be interpreted cautiously. Nevertheless, the chromosomal positioning of the QTL should be largely comparable across studies and some of the QTL detected in this study collocated with previously described QTL. In addition to probably known QTL, we also detected novel QTL which have not been described so far. The largest effect was of a flowering time QTL on chromosome A2 which amounted an explained genotypic variance of 14.1%.
Interestingly, we also observed the colocalization of QTL from different traits. For MC-QTL mapping and for JLAM the flowering time QTL and the plant height QTL on chromosome A2 and for MC-QTL mapping the plant height and the protein content QTL on chromosome A7 overlap (Table 2; Fig. 3). Both trait combinations showed strong phenotypic correlations (Supplementary Figure S3) and such correlations can be beneficial for a plant breeder as selection for one QTL may improve two traits simultaneously, but can also be disadvantageous when improving one trait will impair the other. In the latter case, future attempts should focus on these regions and saturate them with molecular markers. The higher marker density may enable separation of two linked QTL, allowing to individually select the advantageous allele. A good example is the oil content and glucosinolate content QTL on chromosome A7 detected by JLAM (Table 3). Selection for these QTL alleles may accelerate the breeding process to obtain the desired plants with high oil content, but low glucosinolate content.
Epistatic interactions
Epistatic interactions have recently been shown to affect many important agronomic traits in diverse species (e.g. Reif et al. 2011a, b; Würschum et al. 2011c). We used JLAM and performed a full two-dimensional scan for epistatic interactions. We detected two epistatic QTL for flowering time, which due to the colinearity of the loci on chromosome C1 most likely identify one and the same underlying biological QTL. This epistatic QTL explained 3.2% of the genotypic variance. Neither of the interacting loci was detected as main effect which is in accordance with the results previously reported in sugar beet (Würschum et al. 2011c). We observed no significant LD between the interacting loci suggesting that there is no co-selection on the two loci. Epistasis scans heavily rely on large population sizes to have a sufficient power for QTL detection. Our finding of only one epistatic QTL is most likely due to an insufficient number of lines as several recent results point to the presence of epistasis for many traits, also in elite germplasm (Reif et al. 2011a; Würschum et al. 2011c). Future attempts to detect epistasis in rapeseed should thus build on larger populations to warrant a high QTL detection power.
Conclusions
We have compared two biometrical approaches for QTL detection in multiple segregating populations, JLAM and MC-QTL mapping. Both methods were suited for QTL detection in this population of elite rapeseed breeding lines. Our data underline the power of genomics approaches for applied plant breeding as the detected QTL are of direct relevance for breeding programs. Pyramiding of the detected QTL will now enable the establishment of the next generation of elite rapeseed lines by knowledge-based breeding.
References
Becker HC, Löptien H, Röbbelen G (1999) Breeding: an overview. In: Gómez-Campo C (ed) Biology of Brassica coenospecies. Elsevier, Amsterdam, pp 413–460
Blanc G, Charcosset A, Mangin B, Gallais A, Moreau L (2006) Connected populations for detecting quantitative trait loci and testing for epistasis: an application in maize. Theor Appl Genet 113:206–224
Chen G, Geng J, Rahman M, Liu X, Tu J et al (2010) Identification of QTL for oil content, seed yield, and flowering time in oilseed rape (Brassica napus). Euphytica 175:161–174
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Cochran WG, Cox GM (1957) Experimental designs, 2nd edn. Wiley, New York
Delourme R, Falentin C, Huteau V, Clouet V, Horvais R et al (2006) Genetic control of oil content in oilseed rape (Brassica napus L.). Theor Appl Genet 113:1331–1345
Ecke W, Uzunova M, Weißleder K (1995) Mapping the genome of rapeseed (Brassica napus L.). II. Localization of genes controlling erucic acid synthesis and seed oil content. Theor Appl Genet 91:972–977
Ecke W, Clemens R, Honsdorf N, Becker HC (2010) Extent and structure of linkage disequilibrium in canola quality winter rapeseed (Brassica napus L.). Theor Appl Genet 120:921–931
Flint-Garcia SA, Thornsberry JM, Buckler ES (2003) Structure of linkage disequilibrium in plants. Ann Rev Plant Biol 54:357–374
Gower JC (1966) Some distance properties of latent root and vector methods used in multivariate analysis. Biometrika 53:325–338
Grami B, Stefansson BR (1977) Genetics of protein and oil content in summer rape: heritability, numbers of effective factors, and correlations. Can J Plant Sci 57:937–943
Hasan M, Friedt W, Pons-Kühnemann J, Freitag NM, Link K et al (2008) Association of gene-linked SSR markers to seed glucosinolate content in oilseed rape (Brassica napus ssp. napus). Theor Appl Genet 116:1035–1049
Hill WG, Robertson A (1968) Linkage disequilibrium in finite populations. Theor Appl Genet 38:226–231
Jansen RC, Stam P (1994) High resolution of quantitative traits into multiple loci via interval mapping. Genetics 136:1447–1455
Kimber DS, McGregor DI (1995) The species and their origin, cultivation and world production. In: Kimber D, McGregor DI (eds) Brassica oilseeds: production and utilization. CABI Publishing, Wallingford, pp 1–9
Liu W, Gowda M, Steinhoff J, Maurer HP, Würschum T et al (2011) Joint linkage and association mapping in an elite maize breeding population and implications for genomic selection. Theor Appl Genet 123:847–858. doi:10.1007/s00122-011-1631-7
Lynch M, Walsh B (1998) Genetics and analysis of quantitative traits. Sinauer Associates, Sunderland
Mahmood T, Rahman MH, Stringam GR, Yeh F, Good AG (2006) Identification of quantitative trait loci (QTL) for oil and protein contents and their relationship with other seed quality traits in Brassica juncea. Theor Appl Genet 113:1211–1220
Maurer HP, Melchinger AE, Frisch M (2008) Population genetic simulation and data analysis with Plabsoft. Euphytica 161:133–139
Mei DS, Wang HZ, Hu Q, Li YD, Xu YS et al (2009) QTL analysis on plant height and flowering time in Brassica napus. Plant Breed 128:458–465
Melchinger AE, Utz HF, Schön CC (1998) Quantitative trait locus (QTL) mapping using different testers and independent population samples in maize reveals low power of QTL detection and larger bias in estimates of QTL effects. Genetics 149:383–403
Qiu D, Morgan C, Shi J, Liu R, Zhuang X et al (2006) A comparative linkage map of rapeseed and its use for QTL analysis of seed oil and erucic acid content. Theor Appl Genet 114:67–80
Reif JC, Liu W, Gowda M, Maurer HP, Möhring J et al (2010) Genetic basis of agronomically important traits in sugar beet (Beta vulgaris L.) investigated with joint linkage association mapping. Theor Appl Genet 8:1489–1495
Reif JC, Maurer HP, Korzun V, Ebmeyer E, Miedaner T et al (2011a) Mapping QTLs with main and epistatic effects underlying grain yield and heading time in soft winter wheat. Theor Appl Genet 123:283–292
Reif JC, Gowda M, Maurer HP, Longin CFH, Korzun V et al (2011b) Association mapping for quality traits in soft winter wheat. Theor Appl Genet 122:961–970
SAS Institute Inc (2008) SAS user’s guide, version 9.2. SAS Institute, Cary
Schwarz G (1978) Estimating the dimension of a model. Ann Stat 6:461–464
Smooker AM, Wells R, Morgan C, Beaudoin F, Cho K et al (2011) The identification and mapping of candidate genes and QTL involved in the fatty acid desaturation pathway in Brassica napus. Theor Appl Genet 122:1075–1090
Steinhoff J, Liu W, Maurer HP, Würschum T, Longin FH et al (2011) Multiple-line cross QTL-mapping in European elite maize. Crop Sci (accepted)
Utz HF, Melchinger AE, Schön CC (2000) Bias and sampling error of the estimated proportion of genotypic variance explained by quantitative trait loci determined from experimental data in maize using cross validation and validation with independent samples. Genetics 154:1839–1849
Uzunova M, Ecke W, Weißleder K, Röbbelen G (1995) Mapping the genome of rapeseed (Brassica napus L.). I. Construction of an RFLP linkage map and localization of QTLs for seed glucosinolate content. Theor Appl Genet 90:194–204
Van Inghelandt D et al (2011) Extent and genome-wide distribution of linkage disequilibrium in commercial maize germplasm. Theor Appl Genet 123:11–20
Wright S (1978) Evolution and genetics of populations, variability within and among natural populations, vol 4th. The University of Chicago Press, Chicago, p 91
Würschum T, Liu W, Gowda M, Maurer HP, Fischer S et al (2011a) Comparison of biometrical models for joint linkage association mapping. Heredity. doi:10.1038/hdy.2011.78
Würschum T, Maurer HP, Kraft T, Janssen G, Nilsson C et al (2011b) Genome-wide association mapping of agronomic traits in sugar beet. Theor Appl Genet. doi:10.1007/s00122-011-1653-1
Würschum T, Maurer HP, Schulz B, Möhring J, Reif JC (2011c) Genome-wide association mapping reveals epistasis and genetic interaction networks in sugar beet. Theor Appl Genet 123:109–118
Yu J, Holland JB, McMullen MD, Buckler ES (2008) Genetic design and statistical power of nested association mapping in maize. Genetics 178:539–551
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Acknowledgments
This research was conducted within the Biometric and Bioinformatic Tools for Genomics based Plant Breeding project of the GABI—FUTURE initiative.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by C. Quiros.
T. Würschum and W. Liu contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Würschum, T., Liu, W., Maurer, H.P. et al. Dissecting the genetic architecture of agronomic traits in multiple segregating populations in rapeseed (Brassica napus L.). Theor Appl Genet 124, 153–161 (2012). https://doi.org/10.1007/s00122-011-1694-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-011-1694-5