
Abstract
Linkage mapping based on multiple-line crosses is a promising strategy for mapping quantitative trait loci (QTL) underlying important agronomic traits. The main goal of this survey was to study the advantages of QTL mapping across versus within biparental populations using experimental data from three connected sugar beet (Beta vulgaris L.) populations evaluated for beet yield and potassium and sodium content. For the combined analysis across populations, we used two approaches for cofactor selection. In Model A, we assumed identical cofactors for every segregating population. In contrast, in Model B we selected cofactors specific for every segregating population. Model A performed better than Model B with respect to the number of QTL detected and the total proportion of phenotypic variance explained. The QTL analyses across populations revealed a substantially higher number of QTL compared to the analyses of single biparental populations. This clearly emphasizes the potential to increase QTL detection power with a joint analysis across biparental populations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Linkage mapping based on multiple-line crosses (MC) has been suggested as a promising strategy for identifying quantitative trait loci (QTL) underlying important agronomic traits (Blanc et al. 2006; Verhoeven et al. 2006; Coles et al. 2010). In contrast to linkage mapping based on a single biparental population, MC-QTL mapping shows an enhanced statistical power of QTL detection and an improved precision for estimating QTL locations (Rebaï and Goffinet 1993; Muranty 1996; Blanc et al. 2006; Buckler et al. 2009, Coles et al. 2010; Negeri et al. 2011). Moreover, MC-QTL mapping allows the investigation of variation in allele substitution effects across different genetic backgrounds (Steinhoff et al. 2011), which is of paramount importance in evaluating the stability of diagnostic markers.
MC-QTL mapping relies on a high colinearity among genetic maps of different biparental populations. For sugar beet, contrasting results ranging from high (Hallden et al. 1996; Schneider et al. 2007) to low colinearity (Schumacher et al. 1997; Weber et al. 1999) among genetic maps have previously been reported. It is therefore important to study the colinearity among genetic maps before conducting MC-QTL mapping studies in sugar beet.
Different biometric models have been suggested for MC-QTL mapping assuming either random (e.g., Xu 1998) or fixed QTL effects (e.g., Rebaï and Goffinet 1993). Random allele effect models are powerful in situations with a large number of segregating families, because the number of QTL parameters is independent of the number of families. Therefore, the number of parameters in the model does not increase with the number of parental lines as in a fixed model. When the number of segregating families is low, fixed allele effect models possess a higher QTL detection power compared to random allele models (Blanc et al. 2006). Rebaï and Goffinet (2000) proposed two fixed allele models, termed the disconnected and connected models.
We used experimental data from three connected elite testcross populations of sugar beet evaluated for beet yield as well as potassium and sodium content and studied the QTL detection power and mapping resolution of fixed allele effect MC-QTL mapping approaches. In particular, the objectives of our work were to (1) study the colinearity of genetic maps among three biparental populations sharing one parent, (2) examine the consistency of QTL results across three biparental populations, (3) compare the QTL detection power by applying two different MC-QTL mapping models based on the selection of cofactors across segregating populations (Model A) and on a cofactor selection specific for every segregating population (Model B), and (4) evaluate the importance of epistatic interactions for agronomic traits in sugar beet.
Materials and methods
Plant materials and field experiments
Four sugar beet (Beta vulgaris L.) inbred lines (A, B, C, and D) from the pollen parent pool were crossed using an incomplete diallel mating design and three populations (Pop-A×B with 74 individuals, Pop-A×C with 82 individuals, Pop-A×D with 83 individuals) with a total of 239 F2 families were produced (Table 1). The 239 F2 families were crossed with an F1 CMS (cytoplasmic male sterility) tester from the seed parent pool, i.e., a tester representing the opposite heterotic pool. The test crosses were carried out with cloned F2 plants. The test-cross progenies were evaluated in an alpha-lattice design at six locations with two replications in Germany, France, and the Netherlands. Data recorded were the agronomically important traits beet yield (BY, Mg ha−1), potassium content (K, decamol Mg−1), and sodium content (Na, hectomol Mg−1). The latter two traits are important characteristics for the quality of sugar beet for sugar production. All plant materials used in this study are proprietary to KWS SAAT AG (Einbeck, Germany). Beet yield was determined by weighing the washed harvested beets. Sodium and potassium content were measured with a photometer based on the fine pulp of the harvested beets, which was cleaned with aluminium sulphate (Al2(SO4)3).
Phenotypic data analysis
Ordinary lattice analyses of variance were performed for each location separately (Cochran and Cox 1957). Adjusted means were used for a combined analysis across locations, applying the following statistical model: Y im = μ + g i + l m + ε im, where Y im was the adjusted mean of the ith test-cross family in the mth environment, μ was an intercept term, g i was the genetic effect of the ith test-cross family, l m was the effect of the mth location, and ε im was the residual. Variance components were determined by the restricted maximum likelihood (REML) method assuming a random model. Heritability (H²) on an entry-mean basis was calculated as the ratio of genotypic to phenotypic variance. In addition, best linear unbiased estimates (BLUEs) across environments were estimated assuming fixed effects for test-cross families (Supplementary File S1). Analyses were performed using SAS statistical software (SAS Institute 2008). Simple correlation coefficients (r) were calculated between all traits based on BLUEs of the test-cross progenies of each population.
Genotyping and linkage map construction
Single nucleotide polymorphism (SNP) markers and simple sequence repeat (SSR) markers were provided by KWS Saat AG, where genotyping was conducted. The markers have not been used in other studies previously. The genotyping was done by 384er muliplex GoldenGate assay with VeraCode Technology (Illumina). Observed genotypic frequencies at each marker locus were checked for deviations from Mendelian segregation ratios and allele frequencies of 0.5 using a χ2 test. High-quality molecular data, i.e., markers adhering to the expected Mendelian segregation ratio and with less than 20 % missing values, were produced for 481 SNP and 40 SSR markers. The software JoinMap Version 3.0 (van Ooijen and Voorrips 2001) was used to construct maps of the three populations, applying the Kosambi mapping function (Kosambi 1944). A LOD threshold of 2.0–4.0 was used to construct nine linkage groups. Single maps were used to create an integrated genetic map. Colinear markers were removed, resulting in an integrated map of 394 markers, which was used for further analyses.
QTL mapping in individual populations
For each trait and each population, QTL mapping was conducted based on the integrated genetic map applying composite interval mapping (CIM) by the regression approach (Haley and Knott 1992) in combination with the use of cofactors (Jansen and Stam 1994). An additive genetic model was chosen for the analysis of test-cross progenies, as described by Utz et al. (2000). Testing for the presence of a putative QTL in an interval was performed using a likelihood-ratio test. The experiment-wise type I error was determined to be P < 0.10 based on 2,000 permutation runs (Doerge and Churchill 1996). The LOD threshold of 3.8 for Pop-A×B, between 3.3 and 3.4 for population Pop-A×C, and 3.6 for Pop-A×D was used for all three traits. Based on simulation studies on the size of confidence intervals for different scenarios (Visscher et al. 1996), we defined two QTL to be distinct if their estimated map position had a genetic map distance larger than 20 cM, due to the fact that we used a 1-LOD support interval.
QTL mapping in combined populations
For the joint QTL analyses across the three populations, two different fixed allele models were proposed: (1) in the disconnected model specific QTL effects are assumed for every population (Model A), and (2) in the connected model specific QTL effects for parental lines are assumed (Model B) (Blanc et al. 2006). For our data set, the degrees of freedom do not vary between the two models and they therefore yield the same results.
We implemented the models outlined above as described by Steinhoff et al. (2011, 2012a, b). We applied a two-step procedure for QTL detection. In the first step, we identified cofactors using PROC GLMSELECT implemented in the statistical software SAS. Here, we varied the way cofactor selection was implemented: in Model A, we assumed that cofactors are the same for every segregating population. In contrast, in Model B, we selected cofactors specific for every segregating population. For both models, all markers were used for cofactor selection. Cofactor selection was based on stepwise selection using the Schwarz Bayesian Information Criterion (BIC, Schwarz 1978). In the second step, testing for presence of a putative QTL in an interval was performed by a likelihood-ratio test using the statistical software R (R Development Core Team 2010). A LOD threshold of 3.5 was used in Model A, and a LOD score between 5.1 and 5.2 was used in Model B, which correspond to an experiment-wise type I error of P < 0.10, based on 2,000 permutations (Doerge and Churchill 1996). Permutation was performed within the segregating populations and cofactors were estimated for each permutation run.
The proportion of the genotypic variance explained by all detected QTL was estimated as p G = R² adj /H². For both single population QTL analyses and combined analyses, the support of a QTL was defined as a LOD fall-off of 1.0 expressed as position on the chromosome (Lander and Botstein 1989) and cofactors were excluded where the genetic map distance to the marker interval under consideration was smaller than 10 cM.
In addition, we performed a full two-dimensional scan for pairwise interactions using Model A. The two-dimensional scan was based on a comparison of a full versus a reduced model (Steinhoff et al. 2012a). The full model included all selected cofactors plus the main effects of the two loci under consideration and the interaction effect between the loci. The reduced model included all factors of the full model except the interaction term between the two loci under consideration. We used an α-level of 0.05 and followed the suggestion of Holland et al. (2002) dividing the α-level by the number of possible independent pairwise interactions between chromosome regions, assuming two separate regions per chromosome (P < 3.3e-4).
Results
The genotypic variances were significantly larger than zero (P < 0.01) for all three agronomic traits and in all three populations (Table 1). Heritability (H 2) for each trait was high in all three populations with values above 0.83. Consequently, the quality of the phenotypic data represents a solid base for QTL analyses.
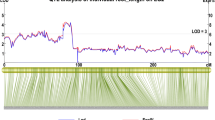
The integrated map of sugar beet had a total length of 543 cM with 395 polymorphic markers distributed across nine chromosomes with an average interval of 1.4 cM between adjacent markers (Supplementary Fig. S1). Average marker densities for the genetic maps of individual populations were between 2.4 cM (Pop-A×B, Pop-A×C) and 2.9 cM (Pop-A×D) and the maximum genetic map distances between adjacent polymorphic markers were smaller than 10 cM with only a few exceptions (Supplementary Fig. S2). The comparison between the genetic maps of the individual populations revealed a high overall colinearity in marker order (Supplementary Fig. S3). We observed significant (P < 0.01) deviations from the expected segregation ratio on chromosome 3 for Pop-A×B and on chromosome 8 for Pop-A×C (Fig. 1).

Segregation distortion in the three populations studied. The significance threshold (P < 0.01) is indicated as a dashed line
QTL analyses of individual populations detected between two and seven QTL for potassium content and ten QTL for beet yield (Table 2). The proportion of phenotypic variance explained by all QTL in single populations ranged from 24.0 % for beet yield in Pop-A×B to 79.5 % also for beet yield in Pop-A×C. The length of the support intervals averaged across all three populations and traits was 7.1 cM.
The analyses across populations revealed as many as 27 QTL for all three traits for Model A compared to 20 QTL for Model B (Table 2, Fig. 2). The total proportion of phenotypic variance was also higher for Model A compared to Model B for all three traits. The length of the support intervals averaged 2.9 cM for Model B and 3.9 cM for Model A. The overlap of QTL detected with single populations as well as Model A and B ranged from 40 % for sodium content to 56 % for potassium content (Fig. 3). The allele substitution effects estimated in each population for the QTL detected with Model A revealed large variation among populations (Supplementary Fig. S4).

LOD curves of three agronomic traits based on single-population analysis and joint QTL analyses applying Model A and Model B

Venn diagrams for QTL detected based on Model A (A) as well as combined population analyses based on Model B (B) and on single population analyses (C) for beet yield, potassium content, and sodium content
Two-dimensional genome scans for all three traits revealed a total of four significant epistatic interactions (Supplementary Fig. S4). Three of these interactions involved chromosome 7, which interacted with regions on chromosomes 5 and 6 for sodium content and with chromosome 3 for beet yield. For potassium content, we observed an epistatic interaction between chromosomes 4 and 5. In addition, main effect QTL were detected for some of the regions involved in the epistatic interactions detected.
Discussion
Resequencing projects revealed large variation in the presence or absence of specific genomic regions within species (Medini et al. 2005), which can ultimately lead to a strong variation in the locus order of genetic maps. MC-QTL mapping relies on a high colinearity among genetic maps of single populations. In contrast to some previous findings in sugar beet (Weber et al. 1999, 2000), we observed a good congruency of locus order among genetic maps of the three populations (Supplementary Fig. S3). This underlines the robustness of the constructed integrated map used for the QTL mapping across populations (Supplementary Fig. S1). Results of a simulation study suggested that for linkage mapping a marker density of 10 cM is necessary to detect QTL with optimal power (Piepho 2000). We observed only a few adjacent marker pairs with a genetic map distance larger than 10 cM and never exceeding 20 cM (Supplementary Fig. S2). Therefore, in this study the QTL detection power is not limited by the marker density.
Single population based QTL analysis
The reliability of the QTL detected and their usefulness in marker-assisted selection (MAS) is largely influenced by congruency in QTL position estimates and their effects across different populations. Previously in sugar beet, no common QTL were observed between two unrelated biparental populations (Weber et al. 1999). In contrast to this, we observed at least two to three congruent QTL for each trait across two populations. The average degree of overlap was 26.7 %, which is in the range observed in previous studies for other crops like maize (Beavis et al. 1991; Mihaljevic et al. 2004; Blanc et al. 2006; Steinhoff et al. 2011). The observed moderate congruency of QTL can be partly explained by the absence of QTL with large effects and low population size with, on average, 82 individuals per population, which led to reduced power of QTL detection.
Multiple-line cross QTL mapping
Joint analyses across populations have been proposed to increase the power of detecting QTL in multiple-line crosses (Blanc et al. 2006; Negeri et al. 2011; Steinhoff et al. 2011). In accordance with this expectation, we observed a higher number of QTL for Models A and B than in the single population analyses, resulting in a substantially larger proportion of explained phenotypic variance (Table 2). This is in agreement with previous studies in maize for grain yield, grain moisture, and flowering time (Steinhoff et al. 2011, 2012a, b) and clearly underlines the potential of increasing QTL detection power with a joint analysis across biparental populations.
Besides the higher QTL detection power, the support intervals of each QTL detected with Models A and B were smaller compared to the QTL detected in single populations (Table 2). This finding is in agreement with previous studies in maize (Coles et al. 2010; Negeri et al. 2011; Steinhoff et al. 2011). Consequently, the joint analyses also allow estimating QTL positions with higher precision compared to a single population analysis.
We observed a higher QTL detection power for Model A compared to Model B (Table 2, Fig. 2). This finding can be explained by differences in the selection of cofactors: in Model A, cofactor selection was implemented across the three segregating populations. In contrast, in Model B cofactors were selected specific for every segregating population. This resulted in a higher number of cofactors for Model A than Model B (Supplementary Tables S1, S2, S3). A lower number of cofactors can lead to an increase in the genetic background noise and consequently also in a higher significance threshold determined by permutation analysis. Consequently, our findings suggest that cofactor selection should be implemented across populations as this can result in a higher power of detecting QTL of interest compared to cofactor selection specific within populations.
Variation in allele substitution effects and presence of epistasis
We observed a large variation in allele substitution effects across the three segregating populations (Supplementary Fig. S4). Explanations that have been suggested for the observed variation in allele substitution effects include multiple alleles (Buckler et al. 2009, Steinhoff et al. 2012a), variation in allele frequencies (Steinhoff et al. 2012b), and presence of epistasis (Jannink and Jansen 2001).
Previously, significant epistatic interactions were reported for agronomic traits in sugar beet (Reif et al. 2010; Würschum et al. 2011a, b, 2012). In accordance with this, we also observed a total of four significant pairwise epistatic interactions in the full two-dimensional genome scan for beet yield, potassium content, and sodium content (Supplementary Fig. S5). It must be noted here that for test-cross populations only a limited proportion of the variance can be exploited for the detection of epistatic QTL as compared to QTL detection based on per-se performance. The fact that, despite this, epistatic QTL were detected confirms the contribution of epistasis to the expression of complex agronomic traits in sugar beet. Consequently, epistasis may be one of the potential causes for the observed variation in allele substitution effects (Supplementary Fig. S6). The analysis of epistatic interactions involving the main effect QTL detected, however, revealed a distribution of P values similar to that expected in the absence of epistasis (Supplementary Figs. S7, S8, S9). This suggests that the variation in allele substitution effects observed in this study may primarily be caused by multiple alleles at the QTL.
Conclusions
We observed a higher power for detecting QTL and resolving QTL position for the joint versus the single population QTL analyses. In addition, our study suggests that cofactor selection in multi-population QTL analyses should be implemented across populations rather than fitting cofactors specific for the single populations. This cofactor selection contributes substantially to a higher QTL detection power.
We applied a QTL mapping approach exploiting identical by-descent probabilities, assuming that the four parental lines are unrelated by pedigree (for review, see Würschum 2012). Switching approaches, which in addition take the relatedness among parental lines into account, promises to further increase QTL detection power and mapping resolution in multi-cross designs. The basic requirement for such approaches is a higher marker density (Liu et al. 2012). With the first draft of the sugar beet genome (Weißhaar et al. 2011) dense marker information is no longer a limiting factor and opens new avenues for unraveling the genetic architecture of relevant agronomic traits.
References
Beavis WD, Grant D, Albertsen M, Fincher R (1991) Quantitative trait loci for plant height in four maize populations and their associations with qualitative genetic loci. Theor Appl Genet 83:141–145
Blanc G, Charcosset A, Mangin B, Gallais A, Moreau L (2006) Connected populations for detecting quantitative trait loci and testing for epistasis: an application in maize. Theor Appl Genet 113:206–224
Buckler ES, Holland JB, Bradbury PJ, Acharya CB, Brown PJ et al (2009) The genetic architecture of maize flowering time. Science 325:714–718
Cochran WG, Cox GM (1957) Experimental designs, 2nd edn. Wiley, New York
Coles ND, McMullen MD, Balint-Kurti PJ, Pratt RC, Holland JB (2010) Genetic control of photoperiod sensitivity in maize revealed by joint multiple population analysis. Genetics 184:799–812
Doerge RW, Churchill GA (1996) Permutation tests for multiple loci affecting a quantitative character. Genetics 142:285–294
Haley CS, Knott SA (1992) A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Theor Appl Genet 103:601–606
Hallden C, Hjerdin A, Rading IM, Sall T, Fridlundh B, Johannisdottir G, Tuvesson S, Akesson C, Nilsson N (1996) A high density RFLP linkage map of sugar beet. Genome 39:634–645
Holland JB, Portyanko VA, Hoffman DL, Lee M (2002) Genomic regions controlling vernalization and photoperiod responses in oat. Theor Appl Genet 105:113–126
Jannink J, Jansen R (2001) Mapping epistatic quantitative trait loci with one-dimensional genome searches. Genetics 157:445–454
Jansen RC, Stam P (1994) High resolution of quantitative traits into multiple loci via interval mapping. Genetics 148:1203–1213
Kosambi D (1944) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Lander ES, Botstein S (1989) Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121:185–199
Liu W, Reif JC, Cossic F, Würschum T (2012) Comparison of biometrical approaches for QTL detection in multiple segregating populations. Theor Appl Genet 125:987–998
Medini M, Hamza S, Rebaï A, Baum M (2005) Analysis of genetic diversity in Tunisian durum wheat cultivars and related wild species by SSR and AFLP markers. Genet Resour Crop Evol 52:21–31
Mihaljevic R, Utz HF, Melchinger AE (2004) Congruency of quantitative trait loci detected for agronomic traits in testcrosses of five populations of European maize. Crop Sci 44:114–124
Muranty H (1996) Power of tests for quantitative trait loci detection using full-sib familiars in different schemes. Heredity 76:56–165
Negeri AT, Coles ND, Holland JB, Balint-Kurti PJ (2011) Mapping QTL controlling southern leaf blight resistance by joint analysis of three related recombinant inbred line populations. Crop Sci 51:1571–1579
Piepho HP (2000) Optimal marker density for interval mapping in a backcross population. Heredity 84:437–440
R Development Core Team (2010) R: a language and environment for statistical computing. Available at http://www.R-project
Rebaï A, Goffinet B (1993) Power of tests for QTL detection using replicated progenies derived from a diallel cross. Theor Appl Genet 86:1014–1022
Rebaï A, Goffinet B (2000) More about quantitative trait locus mapping with diallel designs. Genet Res 75:243–247
Reif JC, Liu W, Gowda M, Maurer HP, Möhring J, Fischer S, Schechert A, Würschum T (2010) Genetic basis of agronomically important traits in sugar beet (Beta vulgaris L.) investigated with joint linkage association mapping. Theor Appl Genet 121:1489–1499
SAS Institute (2008) SAS/STAT 9.2 user’s guide. SAS Institute, Cary
Schneider K, Kulosa D, Soerensen TR, Möhring S, Heine M, Durstewitz G, Polley A, Weber E, Jamsari, Lein J, Hohmann U, Tahiro E, Weisshaar B, Schulz B, Koch G, Jung C, Ganal M (2007) Analysis of DNA polymorphisms in sugar beet (Beta vulgaris L.) and development of an SNP-based map of expressed genes. Theor Appl Genet 115:601–615
Schumacher K, Schondelmaier J, Barzen E, Steinrücken G, Borchardt D, Weber WE, Jung C, Salamini F (1997) Combining different linkage maps in sugar beet (Beta vulgaris L.) to make one map. Plant Breed 116:23–38
Schwarz G (1978) Estimating the dimension of a model. Ann Stat 6:461–464
Steinhoff J, Liu W, Maurer HP, Würschum T, Longin CFH (2011) Multiple-line cross QTL-mapping in European elite maize. Crop Sci 51:2505–2516
Steinhoff J, Liu W, Maurer HP, Würschum T, Longin CFH, Ranc N, Reif JC (2012a) Exploitation of elite maize (Zea mays L.) germplasm across maturity zones. Crop Sci 52:1534–1542. doi:10.2135/cropsci2011.10.0533
Steinhoff J, Liu W, Reif JC, Porta GD, Ranc N, Würschum T (2012b) Detection of QTL for flowering time in multiple families of elite maize. Theor Appl Genet. doi:10.1007/s00122-012-1933-4
Utz HF, Melchinger AE, Schön CC (2000) Bias and sampling error of the estimated proportion of genotypic variance explained by quantitative trait loci determined from experimental data in maize using cross validation and validation with independent samples. Genetics 154:1839–1849
van Ooijen JW, Voorrips RE (2001) JoinMap 3.0: Software for the calculation of genetic linkage maps. Plant Research International BV, Wageningen
Verhoeven KJF, Jannink J, McIntyre LM (2006) Using mating designs to uncover QTL and the genetic architecture of complex traits. Heredity 96:139–149
Visscher PM, Thompson R, Haley CS (1996) Confidence intervals in QTL mapping by bootstrapping. Genetics 143:1013–1020
Weber WE, Borchardt DC, Koch G (1999) Combined linkage maps and QTLs in sugar beet (Beta vulgaris L.) from different populations. Plant Breed 118:193–204
Weber WE, Borchardt DC, Koch G (2000) Marker analysis for quantitative traits in sugar beet. Plant Breed 119:97–106
Weißhaar B, Dohm JC, Minoche A, Schulz B, Kraft T, Wolf M, Holtgraewe D, Himmelbauer H (2011) The draft genome sequence of sugar beet (Beta vulgaris). Plant & Animal Genomes XIX Conference W563: Sugar Beet
Würschum T (2012) Mapping QTL for agronomic traits in breeding populations. Theor Appl Genet 125:201–210
Würschum T, Maurer HP, Schulz B, Möhring J, Reif JC (2011a) Genome-wide association mapping reveals epistasis and genetic interaction networks in sugar beet. Theor Appl Genet 123:109–118
Würschum T, Maurer HP, Kraft T, Janssen G, Nilsson C, Reif JC (2011b) Genome-wide association mapping of agronomic traits in sugar beet. Theor Appl Genet 123:1121–1131
Würschum T, Liu W, Gowda M, Maurer HP, Fischer S, Schechert A, Reif JC (2012) Comparison of biometrical models for joint linkage association mapping. Heredity 108:332–340
Xu S (1998) Mapping quantitative trait loci using multiple families of line crosses. Genetics 148:517–524
Acknowledgments
This research was conducted within the “Biometric and Bioinformatic Tools for Genomic based Plant Breeding” project of the GABI-FUTURE initiative. D.D. Schwegler was supported by DFG within the project “Genetische Architektur der Eigen- versus Testkreuzungsleistung für wichtige agronomische Merkmale beim Roggen” (Grant ID:MI/519/1-1). M. Gowda was supported by BMBF within the HYWHEAT project (Grant ID: FKZ0315945D).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Schwegler, D.D., Liu, W., Gowda, M. et al. Multiple-line cross quantitative trait locus mapping in sugar beet (Beta vulgaris L.). Mol Breeding 31, 279–287 (2013). https://doi.org/10.1007/s11032-012-9788-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-012-9788-6