
Abstract
Verticillium wilt (VW) can cause substantial yield loss in hop particularly with the outbreaks of the lethal strain of Verticillium albo-atrum. To elucidate genetic control of VW resistance in hop, an F1 mapping population derived from a cross of cultivar Wye Target, with the predicted genetic basis of resistance, and susceptible male breeding line BL2/1 was developed to assess wilting symptoms and to perform QTL mapping. The genetic linkage map, constructed with 203 markers of various types using a pseudo-testcross strategy, formed ten major linkage groups (LG) of the maternal and paternal maps, covering 552.98 and 441.1 cM, respectively. A significant QTL for VW resistance was detected at LOD 7 on a single chromosomal region on LG03 of both parental maps, accounting for 24.2–26.0 % of the phenotypic variance. QTL analysis for alpha-acid content and yield parameters was also performed on this map. QTLs for these traits were also detected and confirmed our previously detected QTLs in a different pedigree and environment. The work provides the basis for exploration of QTL flanking markers for possible use in marker-assisted selection.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hop (Humulus lupulus L.) is grown in Europe, USA, China, South Africa, Australia and New Zealand, primarily for its use as an essential flavouring and preservation ingredient in the production of beer and, recently also for pharmaceutical purposes. It is a dioecious perennial climbing plant, the female plants of which are cultivated for the unfertilized inflorescence (cone) carrying lupulin glands rich in resins containing bitter acids and essential oils, components of the bitterness, flavour and aroma of beer, and polyphenols with anti-cancerogenic, anti-inflammatory and phytoestrogenic attributes.
The principal goals of hop breeding programmes have been improvement of yield, the quantity and quality of resins and disease resistance. Hop breeding is a lengthy process, due to the plant’s specific characteristics, such as its perennial nature, late generative phase (2–3 year old plants), dioecism and related high heterozygosity, and time-consuming assessment of production traits in male plants. Additionally, most of the important hop breeding targets are thought to be quantitative traits. The employment of molecular markers in hop genetic studies has opened possibilities of quantitative trait loci (QTL) mapping and the development of trait linked markers for their use in marker-assisted selection (MAS) to support conventional breeding. Markers for simple qualitative traits, such as sex (Jakse et al. 2008b; Patzak et al. 2002; Polley et al. 1997) or markers for powdery mildew resistance locus R 2 (Seefelder et al. 2006) are already available and applied in MAS. However, markers for quantitative traits are not yet available, despite the fact that the construction of hop linkage maps has allowed the identification of QTLs responsible for the chemical structure of hop resins (Koie et al. 2005), QTLs for alpha-acid content and yield parameters (Cerenak et al. 2006, 2009) and, recently, powdery mildew susceptibility (Henning et al. 2011). A number of genes and transcriptional factors involved in the predicted biosynthesis pathway of alpha acids have also been cloned (Matousek et al. 2012; Novak et al. 2003; Okada et al. 2001; Paniego et al. 1999), although they are not yet used in breeding programmes.
The work described here was initiated in an attempt to map Verticillium wilt (VW) resistance, which is poorly understood in hop despite being one of the most devastating diseases in European hop production. Verticillium wilt is a vascular disease of a broad range of plants, caused by the soil borne fungal species Verticillium albo-atrum and V. dahliae. In hop, the disease caused by V. dahliae is comparatively rare and is mild in character, whereas V. albo-atrum causes the majority of outbreaks in both mild and lethal forms (Neve 1991). The appearance of disease forms is attributed to pathogen virulence, the sensitivity of hop cultivars and ecological factors (Isaac and Keyworth 1948; Sewell and Wilson 1984). In general, the mild form varies in intensity from year to year and rarely causes plant death, whereas lethal wilt is less influenced by seasonal climatic variations and causes very severe symptoms, with rapid plant withering and dieback. The V. albo-atrum strain with increased virulence in hop (pathotype PV1) was first discovered in the UK in 1933 (Keyworth 1942), followed by outbreaks in Slovenia in 1997 (Radisek et al. 2003) and in Germany in 2005 (Seefelder et al. 2009). Host resistance accompanied by strict phytosanitary measures is the most effective disease control, so wilt resistant breeding is very important. Hop VW resistance traits were introduced in UK breeding programmes in the 1930s from two distinct sources of an American wild hops (H. lupulus var. neomexicanus), designated Y90 and AA 7 (Darby 2001). The mean values of assessed symptoms for resistance in progeny derived from crosses with the Y90 source indicated an additive action of genes, without evidence of the discontinuity of individual values, thus suggesting polygenic control. On the other hand, control by two dominant complementary genes was predicted according to the segregation ratios observed in the progeny of crosses made with parents having an AA 7 source (Darby 2001), which became the main source of Verticillium resistance in hop breeding programmes.
The best studied example of VW resistance is in tomato, in which resistance against V. dahliae race 1 is conferred by a single dominant gene Ve, which was mapped on the short arm of chromosome 9 (Diwan et al. 1999). Two genes were isolated from the mapped Ve locus by positional cloning, of which only Ve1 showed functional resistance against V. dahliae race 1 (Fradin et al. 2009; Kawchuk et al. 2001). The Verticillium resistance Ve1 gene codes for extracellular leucine rich repeat (LRR) receptor-like protein, which recognizes the recently discovered effector Ave1 and induces a defence response (de Jonge et al. 2012).
Several mapping efforts have been made in cotton, in which VW is one of the most serious constraints to production. QTL analysis on interspecific mapping populations identified several QTLs explaining the variation in resistance to V. dahliae (Bolek et al. 2005). In Gossypium barbadense, QTL analysis of F2 and BC1 populations revealed two stable QTLs in both populations and four QTLs at both seedling and mature stages (Yang et al. 2008). However, mapping in upland cotton (G. hirsutum) of F2 progeny showed clusters of several QTLs on two chromosomal regions, suggesting a genetic model of two major genes conferring resistance to V. dahliae (Jiang et al. 2009). In contrast, single and two dominant cotton genes conferring resistance to V. dahliae strains were reported by Mert et al. (2005).
Linkage maps for tracing VW resistance have also been constructed in several other crops. In oilseed rape, in which V. longisporum causes major yield losses, such an approach identified two major and two minor QTL regions in multiple environments for resistance against VW, using a doubled haploid mapping population (Rygulla et al. 2008). In lettuce, the production of which is also severely affected by VW, the F6 RIL population was genotyped using SNP assay for map construction. QTL analysis detected a single gene of major effect, conferring a high level of resistance to V. dahliae race 1, which was supported by Mendelian analysis of phenotypic data of several additional lettuce populations (Hayes et al. 2011).
In the present study, a hop genetic linkage map was constructed with a high number of SSR markers on an F1 population segregating for VW resistance using a pseudo-testcross strategy. The known hop VW resistant source is described and hop QTL for VW resistance was identified for the first time. In addition, QTLs for yield and hop alpha acids were also located on the constructed map, corresponding to QTLs from our previous study employing a different mapping family.
Materials and methods
Plant material
The F1 mapping population was derived from a cross between the VW highly resistant English variety Wye Target and the susceptible Slovene male breeding line BL2/1. The cross was done in 2004. Stratified seeds were sown in a commercial soil mixture in a tray and placed in a greenhouse. Seedlings with 2–3 leaves were transplanted to pots and subsequently multiplied by cuttings. Those plants were either used for VW resistance scoring or were transplanted to the hop field for evaluation of yield traits.
The analysis included 144 F1 full-sib genotypes, consisting of 99 female and 45 male plants, as determined by the sex-linked microsatellite marker HlAGA7 (Jakse et al. 2008b). In addition to VW resistance variability, the progeny also displayed phenotypic variations in traits, such as growth rate, vigour and alpha-acid content.
The hop varieties, susceptible Celeia and Fuggle and moderately resistant cv. Wye Challenger, together with parents, resistant cv. Wye Target and susceptible BL2/1, were used as reference varieties for VW scoring.
VW resistance scoring
One hundred and seventeen F1 generation genotypes were randomly chosen, together with reference varieties, for resistance testing to the highly virulent V. albo-atrum hop isolate (Radisek et al. 2006). Twelve clonally replicated plants per genotype of 2-month-old cuttings were used for inoculation. We were able to test 20 genotypes (12 clones per genotype, three standard varieties and non-infected controls, in total 384 plants) per experiment in the growing chamber and only twice per year because of hop dormancy. Fungal inoculum was prepared by growing isolate cultures in liquid general fungal medium (Radisek et al. 2003) on a rotary shaker for 5 days at 50 rpm and room temperature in the dark. Conidia were removed from the mycelium by 100 μm cell strainer (Falcon, BD Biosciences, USA) and adjusted by Thoma counting chamber to a concentration of 2 × 106 conidia/ml. Plants were inoculated by dipping the roots in the conidial suspension for 10 min (Clarkson and Heale 1985), after which each plant was transplanted into an 8 cm pot containing commercial sterilized potting substrate (S01-Presstopf Gramoflor, Germany). Control plants were mock inoculated using sterile distilled water. Plants were grown in a growing chamber (RK-13300, Kambic) under a 12 h photoperiod of fluorescent light (L 58 W/77; Fluora, Osram) at 22 °C temperature and 65 % relative humidity during the light period and 18 °C and 70 % during the dark period, in five independent tests. After the first symptoms appeared, plants were assessed for the severity of foliar symptoms at weekly intervals for 3 weeks, using a scale of 0–5. A score of 0 indicates no leaf symptoms, 1 = 1–20 % leaf area wilted, 2 = 21–40 % leaf area wilted, 3 = 41–60 % leaf area wilted, 4 = 61–80 % leaf area wilted and 5 = 81–100 % leaf area wilted. Plant infection was confirmed by re-isolation of the fungus from roots and stems. Cut pieces of the vascular tissue were placed on potato dextrose agar and the presence of mycelium development was examined by light microscopy after 3–5 days. Within 12 clonally replicated plants per genotype some were not successfully infected, as shown by the re-infection test and were subsequently omitted from the scoring. Final genotype assessment was expressed as a disease severity index (DSI), calculated as mean wilt score of the infected plants on the last foliage assessment. Based on DSI, genotypes were classified into six numeric resistance categories corresponding to a 0–5 scale of foliar wilt assessment.
Alpha-acid content
Determination of the alpha-acid content (lead conductance value—LCV) was performed by the standard method (Analytica EBC/European brewery convention 2000) on the cone harvest in 2008 and 2009 (Cerenak et al. 2009).
Dry cone weight and harvest index
The parameters of yield were determined as reported previously (Cerenak et al. 2009). The hop plants were grown in the hop field in the main hop growing area of Slovenia (Savinja valley), maintained by standard agricultural practice in terms of maintenance, fertilizing and disease and pest protection. The cones were picked in 2008 and 2009. They were dried at 103–104 °C for 5 h, weighed and the percentage of moisture calculated and the dry cone weight (DCW) was determined. The harvest index (HI) was calculated as the ratio of fresh cone weight to the total plant weight.
Molecular markers analysis
Total genomic DNA was extracted from young hop leaves using a common CTAB method (Kump and Javornik 1996). DNA concentrations were measured by means of fluorimetry (Hoefer DyNA Quant 200). DNA samples were stored in TE buffer at −20 °C until further use.
Various sets of molecular markers were used for genotyping the 144 F1 progeny and two parents: anonymous genomic SSRs, EST-derived SSRs, AFLPs, SCARs, SNPs and RAPDs.
Anonymous genomic SSRs were either (a) developed in our laboratory from microsatellite enriched genomic libraries (Jakse et al. 2002; Jakse et al. 2008a; Stajner et al. 2005), (b) taken from the literature (Brady et al. 1996; Hadonou et al. 2004; Kanai et al. 2006) or (c) acquired through collaboration (Seefelder, unpublished). EST-derived SSRs were developed from hop EST/cDNA sequences (Jakse et al. 2011). Microsatellite loci were first tested on both parents and six randomly chosen F1 plants for polymorphism and segregation and those exhibiting segregation were used for genotyping the mapping population. PCR reactions were performed in 15 μl, using 20 ng of hop DNA, 1× supplied PCR buffer, 2 mM MgCl2 and 0.5 μM of locus-specific primer pair. Forward primers were either labelled with CY5 dye at the 5′ ends or a three primer protocol allowing economic labelling of amplified fragments was used according to the published procedure (Schuelke 2000). Fluorescently labelled microsatellite alleles were separated on an ALFExpressII automatic DNA sequencer. Formamide loading dye (formamide, 5 mg/ml dextran blue) was added to the PCR reactions (1:1) and 5 μl of heat-denaturated sample was loaded onto 7 % denaturing polyacrylamide gel (19:1 acrylamide:bisacrylamide, 7 M urea) prepared in a short cassette. Samples were subjected to electrophoresis at 55 °C for the amount of time required to separate the longest allele of the locus at a constant power of 15 W. The same gel was reloaded two or three times. Data were acquired with the ALFWin analysis module and chromatograms were analysed with Allele Locator 1.03 software.
AFLP analysis (Vos et al. 1995) was performed using 12 primer combinations with three or two selective nucleotides employing EcoRI (E) or PstI (P) restriction sites in combination with a frequent 4-base cutter MseI (M): E-ACG + M-CTA, E-ACG + M-CTT, E-ACG + M-CAT, E-AGC + M-CAG, E-AGC + M-CTT, E-AGC + M-CAC, E-AGC + M-CTC, E-AGC + M-CTA, P-AAC + M-CTT, P-AA + M-CAC, P-AAC + M-CT and P-AAC + M-CAA. The amplification protocol was performed using 5′ CY5 labelled E or P primers (Gril et al. 2008). Amplification reactions were electrophoresed using the same equipment, chemicals and conditions as described in the case of microsatellite markers, with an electrophoresis time of 300 min.
Three SCAR markers, sex-linked RAPD-Y (Polley et al. 1997), LRR sequence WT3B12 (Kozjak et al. 2009) and HOP-B (Araki et al. 1998), 13 SNP markers based on LRR hop sequences (designated as SNP followed by a number; unpublished) and ten RAPD decamer primers (Sustar-Vozlic and Javornik 1999) were also genotyped on the mapping population.
All marker data were stored according to the required JoinMap codes in an Excel spreadsheet.
Data analysis
Marker and phenotype segregation were tested for goodness-of-fit to Mendelian inheritance ratios using the Chi-square test. All markers were used for mapping, including those demonstrating a significant departure from the expected Mendelian ratios. The molecular-marker information was used to construct separate linkage maps for the female (Wye Target) and male (BL2/1) parents, using the two-way pseudo-testcross strategy (Grattapaglia and Sederoff 1994) by the JoinMap® 4.0 program (Van Ooijen 2006). Kosambi’s mapping function was applied and a LOD 6.0 threshold with a recombination fraction of 0.35 was used to determine linkage groups. A LOD score of 2.0 was used to determine the optimal marker order by applying a ripple value of 1.0 and a jump threshold of 5.0.
MapQTL® 5 (Van Ooijen 2004) was used for identification and location of QTLs associated with VW resistance, alpha-acid content, dry cone weight and harvest index in each year. Single trait analysis was performed using the Kruskal–Wallis non-parametric test, as well as an interval mapping approach (Lander and Botstein 1989) followed by multiple-QTL mapping (MQM) (Jansen and Stam 1994). The linkage groups were scanned at 1 cM intervals. The most likely QTL position corresponded to the locus of the marker exhibiting the strongest association with the trait. By performing a backward elimination procedure, co-factors significantly associated with each trait at P < 0.02 were selected and used in the MQM. Genome-wide threshold values (P < 0.05) for declaring the presence of QTLs were estimated from 1,000 permutations of each phenotypic trait (Churchill and Doerge 1994). The interaction effects were estimated for each QTL using the results of QTL analysis based on the corresponding parental map. In a pseudo-testcross progeny, a QTL can segregate for four different alleles (Q1Q2 × Q3Q4). The means of the distribution of the quantitative trait associated with four different QTL genotypes (Q1Q3, Q1Q4, Q2Q3 and Q2Q4) can be estimated. Following the method proposed by Knott et al. (1997), three allelic effects can be calculated: effects for each parent individually (difference in the effect of the alleles inherited from each parent) and interaction effects (i.e., deviation from additivity, whereby a value of zero indicates complete additivity).
One- and two-LOD support intervals for the position of each QTL were calculated as described by Conneally et al. (1985). Maps were drawn using MapChart version 2.2 software (Voorrips 2002).
Results
The F1 population of 144 plants was developed from the VW-resistant mother plant Wye Target and susceptible male breeding line BL2/1, to obtain a mapping population segregating for VW. The tested progeny exhibited a range of variability of resistance to VW, from highly susceptible to completely resistant genotypes. We scored 117 F1 generation genotypes randomly chosen out of 144 genotypes used for map construction, due to time and labour limitations. We believed that the chosen number of genotypes was sufficient for mapping the disease-related QTL. At harvest, when the plants reached technological ripeness, the alpha-acid content, yield (dry cone weight) and harvest index (ratio of fresh cone weight to total plant weight) were also measured in two subsequent years on female plants, in order to map QTLs for these traits (Table 1). The correlations between the 2 years were significant but moderate in case of alpha-acid content (r = 0.643; P < 0.001) and dry cone weight (r = 0.695; P < 0.001), while in case of harvest index the correlation was low (r = 0.190; P = 0.032).
VW resistance assessment
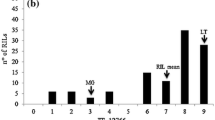
In total, 117 hop plants were scored for resistance to VW, in a growing chamber in five independent tests after infection by the highly virulent pathotype of V. albo-atrum, which induces severe and lethal disease symptoms. In all tests, the first symptoms were observed as leaf chlorosis approximately 3 weeks after inoculation on the susceptible reference varieties Celeia and Fuggle and on susceptible plants of genotypes classified four and five from the mapping family. Re-infection analysis of inoculated plants revealed 100 % successful infection in genotypes assessed as class four and five, whereas in genotypes 0–3 it varied between 67 and 92 % and thus only infected plants were included in the expression of DSI. Based on DSI, genotypes were classified into six numerical resistant categories (0–5): 39 plants with score 0, 23 plants with score 1, 11 plants with score 2, 17 plants with score 3, 14 plants with score 4 and 13 plants with score 5 (Fig. 1).

Distribution of 117 analysed plants according to 6 VW resistance scores
Map construction
One hundred and forty-four genotypes were included in the marker analysis for linkage map construction. We were able to produce 296 scorable polymorphic markers (111 anonymous genomic SSRs, 9 EST-derived SSRs, 153 AFLPs, 3 SCARs, 7 RAPDs and 13 SNPs). Marker segregation analysis revealed a high percentage (116 markers, 39.19 % at P < 0.01) of them to be distorted. Nevertheless, out of 296 markers, 203 markers (106 anonymous genomic SSRs, 7 EST-derived SSRs, 68 AFLPs, 3 SCARs, 6 RAPDs and 13 SNPs) were mapped (Table 2). Of these 203 mapped markers, 67 still displayed distorted segregation (33.00 %). The majority of the ninety-three markers that did not map were AFLPs (85), with five anonymous genomic SSRs, two EST-derived SSRs and one RAPD, since they did not show any linkage.
A total of 177 markers (93 anonymous genomic SSRs, 4 EST-derived SSRs, 64 AFLPs, 2 SCARs, 5 RAPDs and 9 SNPs) were placed on the maternal Wye Target map, forming 10 major linkage groups and three doublets, covering 552.98 cM of the genome. The average distance between adjacent markers was 5.88 cM. Ten major linkage groups consisted of 4–31 markers, and ranged in size from 35.64 to 77.04 cM (Table 2). The percentage of markers exhibiting distorted segregation in the ten major linkage groups was below 50 %, ranging from 6.45 % (LG01) to 42.86 % (LG05), with the exception of LG02, which had 84.62 % of distorted markers.
A total of 108 markers (53 anonymous genomic SSRs, 3 EST-derived SSRs, 45 AFLPs, 1 SCARs, 1 RAPDs and 5 SNPs) were placed on the paternal BL2/1 map, defining 441.1 cM of the total map distance, with an average inter-marker distance of 6.42 cM. Ten major linkage groups and three doublets were detected. Ten major linkage groups consisted of 4–22 markers, and ranged in size from 12.05 to 72.34 cM (Table 2). The distribution of markers exhibiting distorted segregation was uneven among the ten major linkage groups of the BL2/1 map. In six out of ten major groups, the percentage of distorted markers was below 50 %, ranging from 0.00 % (LG01 and LG14) to 38.89 % (LG04). Two relatively large groups (LG02 and LG05) had 77.27 and 77.78 % of distorted markers, respectively, while two smaller groups (LG03 and LG04) consisted solely of markers exhibiting segregation distortion.
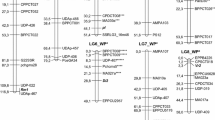
Out of 203 mapped markers, 82 (40 anonymous genomic SSRs, 41 AFLPs and 1 SNP) were mapped on both female and male maps. Due to the presence of these common biparental markers, homology of the eleven linkage groups was inferred (Fig. 2), leaving two linkage groups mapped solely on the Wye Target map and two on the BL2/1 map.



Linkage maps of female (Wye Target) and male (BL2/1) parent developed using their F1 progeny. Map positions are given in cM using the Kosambi mapping function. Linkage groups 1–10 are homologous in the two maps. Common biparental markers are shown in bold. Bar positions indicate the locations of quantitative trait loci (QTLs) for Verticillium resistance (vh) alpha-acid content (alpha), dry cone weight (dcw) and harvest index (hi) as detected in each year (2008 and 2009). Outer and inner intervals corresponding to 1-LOD and 2-LOD support interval are indicated as a full box and a line, respectively
The marker distribution in the obtained maps was generally fairly even, with larger gaps of around 20 cM between two markers being evident in 2 LGs on the paternal map. Marker clustering occurred only on the larger LG01 and LG02 of the maternal and paternal maps.
QTL analyses of Verticillium wilt resistance
Genetic effects related to VW resistance were identified on one chromosomal region on LG03 of both parental maps (Fig. 2). By using the Kruskal–Wallis non-parametric test, as well as both interval mapping and MQM, QTLs for VW resistance were identified at LOD 7, explaining from 24.2 to 26.0 % of the phenotypic variance. The flanking markers of the QTL determined on the female map were anonymous microsatellite EMHL052 and AFLP marker P-AA_M-CAC_91, both segregating only in the female parent. The peak of the putative QTL determined on the male map was found exactly at the location of the monoparental anonymous microsatellite marker Ho0084 (Table 3).
QTL analysis of the alpha-acid content
In 2009, the QTL for alpha-acid content was identified on LG01 of both maps (Fig. 2). On the female map, the flanking marker was monoparental AFLP marker P-AAC_M-CT_287, while on the male map, the flanking marker was biparental anonymous microsatellite HlGT4. Their LOD values ranged from 5.33 to 6.77, explaining from 31.1 to 37.6 % of the phenotypic variance. The maternal effects of both QTLs were larger than both paternal and interaction effects (Table 3). In 2008, no QTL were found with LOD >4.0, although the correlation of alpha content between years was significant (0.64, P < 0.001), indicating the presence of QTLs with a lower LOD score.
QTL analysis of yield parameters DCW and HI
QTLs associated with quantity of yield (DCW, HI) were obtained in 2008, while no significant QTLs were detected in the following year, 2009 (Fig. 2) with LOD >4.0 although a high correlation (0.69, P < 0.001) was found for DCW between the years. Genetic effects related to quantity of yield were identified in the chromosomal region on LG03 of both parents. The LOD values for these putative QTLs for HI varied from 4.46 to 5.92, explaining from 25.4 to 28.8 % of phenotypic variance. The position of the QTL on the male map was found at exactly the same position as the QTL for VW resistance, at the location of marker Ho0084. The third putative QTL, relating to quantity of yield (DCW), was positioned on the female map, with two AFLP flanking markers, explaining 23.5 % of the phenotypic variance (Table 3).
Discussion
Verticillium wilt is becoming a serious threat to hop production in Europe due to outbreaks of lethal wilt caused by a highly virulent strain of V. albo-atrum. To enhance resistance breeding, since host resistance is the most effective control of the disease, marker-assisted selection could greatly improve the lengthy hop breeding process. The main source of VW resistance in hop breeding is the cultivar Wye Target, bred in the UK, where they first encountered the lethal form of hop VW (Neve 1991), although data from our recent hop breeding programme suggest that other VW unknown resistant sources have also been effective. Wye Target carries resistance from AA 7, which is based on two dominant genes with complementary action, as proposed by Darby (2001).
In our attempt to establish the gene model for VW resistance and to map the region in the hop genome carrying resistance genes, in order to find markers for MAS, we constructed a mapping family using Wye Target as a source of VW resistance. Several segregation patterns of resistance scores for mono- and dihybrid crosses were tested for goodness-of-fit to Mendelian inheritance ratios. When we put our phenotypic data into two categories, resistant and susceptible, ignoring the variation of symptoms among susceptible plants, the segregation pattern of a two-gene model was obtained, consistent with the study of Darby (2001). However, this segregation ratio was not supported by the segregation of markers linked to the identified QTL, nor did we find any marker linked to either gene from the anticipated two-gene model.
The evaluation of VW symptoms in hop is as demanding as in other crops susceptible to these soil-born pathogenic fungi. We have established a pathogenicity test for testing the virulence of field isolates of V. albo-atrum using a root dipping method, growing plants in a growth chamber and evaluating the symptoms at time intervals (Radisek et al. 2006). The same method was applied for evaluating symptoms after inoculation of the mapping progeny with a highly virulent strain of V. albo-atrum. A continuous distribution of symptoms was observed from symptomless plants, through moderately affected plants and completely diseased plants, correlating with resistant, moderately resistant/susceptible and highly susceptible plants. Such an expression of phenotypes suggests a dosage effect of alleles of major gene(s) or the additional action of minor genes, as has already been discussed for other crops (Bae et al. 2008; Fradin et al. 2009; Hayes et al. 2011; Jansky et al. 2004). Additionally, a continuous distribution of symptoms can also be ascribed to the aetiology of VW disease. Plants get infected through the roots after resting mycelia of V. albo-atrum are stimulated to germinate by root exudates. Hyphae invade the roots, enter the xylem vessels and form conidia, which are then carried upwards by the transpiration stream and subsequently stopped at vessel trapping sites, where they germinate and penetrate adjacent vessel elements and begin a new infection cycle. Colonization of plants is thus not even, plants can be infected at different times and colonized to a different extent, due to the pathogen infection strategy, as well as the influence of environmental factors and all these are reflected in symptom development, which is scored at fixed time intervals.
The linkage map using the F1 population from the cross between VW-resistant cultivar Wye Target and susceptible BL2/1 is the first map constructed for mapping VW resistance in hop. It is also the hop map that includes the highest number (119) of microsatellite markers developed primarily for mapping purposes (Jakse et al. 2008a; Jakse et al. 2011). To date, five linkage maps have been published in peer reviewed journals for hop families segregating for different traits (Cerenak et al. 2009; Cerenak et al. 2006; Henning et al. 2011; Koie et al. 2005; Seefelder et al. 2000), which were mainly constructed with AFLP markers, including a small number of microsatellites, while the most recent map used a larger number of DaRT markers. Microsatellites, as co-dominant markers, are highly applicable in a pseudo-test cross mapping strategy of dioecious hop, as has been shown in our previous mapping studies (Cerenak et al. 2009; Cerenak et al. 2006), since they readily allow comparison of male and female linkage groups and the construction of an integral map and are transferable between maps. On the other hand, it is also important to include various types of molecular markers in linkage studies, since specific marker types can be differentially distributed at different genomic positions (Semagn et al. 2006). Our map is based on 203 polymorphic markers including anonymous and EST developed microsatellites, AFLPs, SNPs, RAPDs and SCARs. The constructed map consists of eight common biparental LGs with more than two markers in the group, while both maps have two parental specific LGs. The total number of LGs is close to the haploid chromosomal number 10 of the hop genome. Although we report a shorter total length of the present male and female maps compared to our previous two maps used for QTL analysis of alpha-acid content and yield parameters (Cerenak et al. 2006; Cerenak et al. 2009), the average marker distance of the new map is greatly improved; in the female map by 5.63 and 7.17 cM and by 5.84 and 5.22 cM in the male map, respectively.
Although segregation analysis revealed a high percentage of distorted markers, we decided to include them in the map construction. Segregation distortion of markers in mapping efforts has been confirmed in numerous plant species. The phenomenon of distortion of nuclear genes is well documented in plants (Zamir and Tadmor 1986). The deviations are explained by the many different selection forces, from the period of formation of the gametes until their fusion into zygotes (Gillet and Gregorius 1992). However, mapping studies have shown that distortion does not affect the estimate of map distance (Lu et al. 2002).
QTL analysis carried out on the constructed linkage map to detect the QTL for VW resistance gave a positive result. We located the QTL on a single chromosomal region on LG03 in both parents (Fig. 2), with a significant QTL for VW tolerance at LOD 7 explaining 24.2–26.0 % of the phenotypic variance and confirming the action of more than one gene (Table 3). How many genes might be positioned at this single, well-supported QTL is not known. However, Mendelian segregation analysis of the phenotypic data of VW in the F1 mapping population was consistent with the action of two dominant, complementary genes, suggesting that at least one gene is within the detected QTL. The maternal effect of the detected QTL is much larger than both paternal and interaction effects, which is to be expected, since the maternal parent carried the resistance. The flanking AFLP and microsatellite markers of the maternal QTL are not very tightly linked, so the region would require fine mapping to find candidate markers linked to resistant gene(s). A more promising candidate marker for MAS is microsatellite marker Ho0084, which is co-located (0 cM) with the QTL on the paternal map.
The main aim of our research was determination of QTL(s) for wilt resistance, but we extended the mapping efforts to traits related to hop quality and yield in order to verify our previously detected QTLs (Cerenak et al. 2009). We measured the content of alpha acids, the main hop quality parameter, and harvest index (hi) and dry cone weight (dcw), the two yield parameters, over 2 years in field conditions.
In 2009, we were able to detect QTL for alpha-acid content on LG01 of both parental maps, describing a reasonably high proportion of phenotypic variation despite the fact that the maternal parent (Wye Target) displayed low variation in alpha-acid content in comparison with the maternal parent cultivar Magnum used in our previous QTL mapping of alpha-acid content (Cerenak et al. 2009). The male parent was the same in both mapping populations and a number of common microsatellite markers were found on the corresponding LGs. The number of common microsatellite markers used in both studies enables comparison of the QTL location for alpha-acid content on the two maps. LG Wye-1, with the detected QTL for alpha-acid content (alpha-09), corresponds to LG Magnum-3 with located alpha-acid QTLs (α1-03, α3-04, α1-05, α1-06). Microsatellite marker HlGT4 is particularly interesting. It was detected as a flanking marker of a QTL (alpha-09) on LG BL2/1-1, as well as of two QTLs (α2-02 and α4-04) located on the corresponding LG from the Magnum × BL2/1 population. The related QTLs for yield were also detected on LG Wye-3 and BL2/1-3, which correspond to LG Magnum-1 and BL2/1-1, on which several QTLs for harvest index and dry cone weight have been previously reported. The present male LG-3 is very short. However, it carries the HlAGA6 marker, which is also the flanking marker for several maternal and paternal QTLs for harvest index on LG-1 of Magnum and BL2/1. The comparison of QTLs on the Wye Target × BL2/1 and Magnum × BL2/1 maps showed co-location of QTLs for specific traits on both maps and thus confirmation of the same QTLs in different genetic backgrounds and environments. The common flanking markers (HlGT4 and HlAGA6) are also good candidates to be explored as markers for alpha-acid content and yield in hop. The male markers deserve special attention, since paternal inheritance of the above traits is very poorly studied because male plants usually participate in the breeding process as unknown pollinators.
To the best of our knowledge, this is the first report of the QTL for hop resistance against the lethal strain of V. albo-atrum, which has become the major limiting factor in European hop production. These results are an important first step in the investigation of the genetic basis of a currently available hop VW resistance source, as well as opening possibilities of marker development and exploitation in MAS resistant breeding. A marker linked to VW resistance would allow selection of resistant genotypes at the seedling stage from large numbers of progeny, without the time-consuming screening of artificially infected plants. Together with successful marker development for alpha-acid content and yield parameters, this work will significantly contribute to the implementation of MAS for VW resistance and complex traits in breeding strategies.
References
Analytica EBC/European brewery convention (2000) Nuernberg, EBC Analysis Committe–Nuernberg, Carl, Hans, Getränke–Fadverl. Grundwerk: Section 7
Araki S, Tsuchiya Y, Takashio M, Tamaki T, Shinotsuka K (1998) Identification of hop cultivars by DNA marker analysis. J Am Soc Brew Chem 56:93–98
Bae J, Halterman D, Jansky S (2008) Development of a molecular marker associated with Verticillium wilt resistance in diploid interspecific potato hybrids. Mol Breed 22:61–69
Bolek Y, El-Zik KM, Pepper AE, Bell AA, Magill CW, Thaxton PM, Reddy OUK (2005) Mapping of verticillium wilt resistance genes in cotton. Plant Sci 168:1581–1590
Brady JL, Scott NS, Thomas MR (1996) DNA typing of hops (Humulus lupulus) through application of RAPD and microsatellite marker sequences converted to sequence tagged sites (STS). Euphytica 91:277–284
Cerenak A, Satovic Z, Javornik B (2006) Genetic mapping of hop (Humulus lupulus L.) applied to the detection of QTLs for alpha-acid content. Genome 49:485–494
Cerenak A, Satovic Z, Jakse J, Luthar Z, Carovic-Stanko K, Javornik B (2009) Identification of QTLs for alpha acid content and yield in hop (Humulus lupulus L.). Euphytica 170:141–154
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Clarkson JM, Heale JB (1985) Pathogenicity and colonization studies on wild-type and auxotrophic isolates of Verticillium-albo-atrum from hop. Plant Pathol 34:119–128
Conneally PM, Edwards JH, Kidd KK, Lalouel JM, Morton NE, Ott J, White R (1985) Report of the committee on methods of linkage analysis and reporting. Cytogenet Cell Genet 40:356–359
Darby P (2001) Single gene traits in hop breeding. In: Seigner E (ed) Scientific Commission of the International Hop Growers Convention IHGC. Canterbury, UK, pp 76–80
de Jonge R, van Esse HP, Maruthachalam K, Bolton MD, Santhanam P, Saber MK, Zhang Z, Usami T, Lievens B, Subbarao KV, Thomma B (2012) Tomato immune receptor Ve1 recognizes effector of multiple fungal pathogens uncovered by genome and RNA sequencing. Proc Natl Acad Sci USA 109:5110–5115
Diwan N, Fluhr R, Eshed Y, Zamir D, Tanksley SD (1999) Mapping of Ve in tomato: a gene conferring resistance to the broad-spectrum pathogen, Verticillium dahliae race 1. Theor Appl Genet 98:315–319
Fradin EF, Zhang Z, Ayala JCJ, Castroverde CDM, Nazar RN, Robb J, Liu CM, Thomma B (2009) Genetic dissection of Verticillium wilt resistance mediated by tomato Ve1. Plant Physiol 150:320–332
Gillet E, Gregorius HR (1992) What can be inferred from open-pollination progenies about the source of observed segregation distortion? A case-study in Castanea sativa Mill. Silvae Genet 41:82–87
Grattapaglia D, Sederoff R (1994) Genetic-linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: mapping strategy and RAPD markers. Genetics 137:1121–1137
Gril T, Celar F, Munda A, Javornik B, Jakse J (2008) AFLP analysis of intraspecific variation between Monilinia laxa isolates from different hosts. Plant Dis 92:1616–1624
Hadonou AM, Walden R, Darby P, Darby W (2004) Isolation and characterization of polymorphic microsatellites for assessment of genetic variation of hops (Humulus lupulus L.). Mol Ecol Notes 4:280–282
Hayes RJ, McHale LK, Vallad GE, Truco MJ, Michelmore RW, Klosterman SJ, Maruthachalam K, Subbarao KV (2011) The inheritance of resistance to Verticillium wilt caused by race 1 isolates of Verticillium dahliae in the lettuce cultivar La Brillante. Theor Appl Genet 123:509–517
Henning JA, Townsend MS, Gent DH, Bassil N, Matthews P, Buck E, Beatson R (2011) QTL mapping of powdery mildew susceptibility in hop (Humulus lupulus L.). Euphytica 180:411–420
Isaac I, Keyworth WG (1948) Verticillium wilt of the hop (Humulus lupulus). Ann Appl Biol 35:243–249
Jakse J, Bandelj D, Javornik B (2002) Eleven new microsatellites for hop (Humulus lupulus L.). Mol Ecol Notes 2:544–546
Jakse J, Luthar Z, Javornik B (2008a) New polymorphic dinucleotide and trinucleotide microsatellite loci for hop Humulus lupulus L. Mol Ecol Resour 8:769–772
Jakse J, Stajner N, Kozjak P, Cerenak A, Javornik B (2008b) Trinucleotide microsatellite repeat is tightly linked to male sex in hop (Humulus lupulus L.). Mol Breed 21:139–148
Jakse J, Stajner N, Luthar Z, Jeltsch JM, Javornik B (2011) Development of transcript-associated microsatellite markers for diversity and linkage mapping studies in hop (Humulus lupulus L.). Mol Breed 28:227–239
Jansen RC, Stam P (1994) High-resolution of quantitative traits into multiple loci via interval mapping. Genetics 136:1447–1455
Jansky S, Rouse DI, Kauth PJ (2004) Inheritance of resistance to Verticillium dahliae in diploid interspecific potato hybrids. Plant Dis 88:1075–1078
Jiang F, Zhao J, Zhou L, Guo WZ, Zhang TZ (2009) Molecular mapping of Verticillium wilt resistance QTL clustered on chromosomes D7 and D9 in upland cotton. Sci China Life Sci 52:872–884
Kanai D, Kirita M, Sakamoto K (2006) Method for judging kind of hop using microsatellite DNA. Japan Patent 2006-034142. Asahi Breweries LTD, Japan
Kawchuk LM, Hachey J, Lynch DR, Kulcsar F, van Rooijen G, Waterer DR, Robertson A, Kokko E, Byers R, Howard RJ, Fischer R, Prufer D (2001) Tomato Ve disease resistance genes encode cell surface-like receptors. Proc Natl Acad Sci USA 98:6511–6515
Keyworth WG (1942) Verticillium wilt of the hop (Humulus lupulus). Ann Appl Biol 29:346–357
Knott SA, Neale DB, Sewell MM, Haley CS (1997) Multiple marker mapping of quantitative trait loci in an outbred pedigree of loblolly pine. Theor Appl Genet 94:810–820
Koie K, Inaba A, Okada Y, Kaneko T, Ito K (2005) Construction of the genetic linkage map and QTL analysis on hop (Humulus lupulus L.) In: Hummer KE, Henning JA (eds) ISHS Acta Horticulturae 668: I International Humulus Symposium. ISHS, Corvallis (Oregon), USA, pp 59–66
Kozjak P, Jakse J, Javornik B (2009) Isolation and sequence analysis of NBS-LRR disease resistance gene analogues from hop Humulus lupulus L. Plant Sci 176:775–782
Kump B, Javornik B (1996) Evaluation of genetic variability among common buckwheat (Fagopyrum esculentum Moench) populations by RAPD markers. Plant Sci 114:149–158
Lander ES, Botstein D (1989) Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121:185–199
Lu H, Romero-Severson J, Bernardo R (2002) Chromosomal regions associated with segregation distortion in maize. Theor Appl Genet 105:622–628
Matousek J, Kocabek T, Patzak J, Fussy Z, Prochazkova J, Heyerick A (2012) Combinatorial analysis of lupulin gland transcription factors from R2R3Myb, bHLH and WDR families indicates a complex regulation of chs_H1 genes essential for prenylflavonoid biosynthesis in hop (Humulus Lupulus L.). BMC Plant Biol 12:27
Mert M, Kurt S, Gencer O, Akiscan Y, Boyaci K, Tok FM (2005) Inheritance of resistance to Verticillium wilt (Verticillium dahliae) in cotton (Gossypium hirsutum L.). Plant Breed 124:102–104
Neve RA (1991) Hops. Chapman and Hall, London
Novak P, Matousek J, Briza J (2003) Valerophenone synthase-like chalcone synthase homologues in Humulus lupulus. Biol Plant 46:375–381
Okada Y, Sugimoto M, Ito K (2001) Molecular cloning and expression of farnesyl pyrophosphate synthase gene responsible for essential oil biosynthesis in hop (Humulus lupulus). J Plant Physiol 158:1183–1188
Paniego NB, Zuurbier KWM, Fung SY, van der Heijden R, Scheffer JJC, Verpoorte R (1999) Phlorisovalerophenone synthase, a novel polyketide synthase from hop (Humulus lupulus L.) cones. Eur J Biochem 262:612–616
Patzak J, Vejl P, Skupinova S, Newadba V (2002) Identification of sex in F1 progenies of hop (Humulus lupulus L.) by molecular marker. Rost Vyroba 48:318–321
Polley A, Seigner E, Ganal MW (1997) Identification of sex in hop (Humulus lupulus) using molecular markers. Genome 40:357–361
Radisek S, Jakse J, Simoncic A, Javornik B (2003) Characterization of Verticillium albo-atrum field isolates using pathogenicity data and AFLP analysis. Plant Dis 87:633–638
Radisek S, Jakse J, Javornik B (2006) Genetic variability and virulence among Verticillium albo-atrum isolates from hop. Eur J Plant Pathol 116:301–314
Rygulla W, Snowdon RJ, Friedt W, Happstadius I, Cheung WY, Chen D (2008) Identification of quantitative trait loci for resistance against Verticillium longisporum in oilseed rape (Brassica napus). Phytopathology 98:215–221
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234
Seefelder S, Ehrmaier H, Schweizer G, Seigner E (2000) Male and female genetic linkage map of hops, Humulus lupulus. Plant Breed 119:249–255
Seefelder S, Lutz A, Seigner E (2006) Development of molecular markers for powdery mildew resistance to support breeding for high quality hops. Monatsschrift Brauwiss 59:100–104
Seefelder S, Seigner E, Niedermeier E, Radišek S, Javornik B (2009) Genotyping of Verticillium pathotypes in the Hallertau: Basic findings to assess the risk of Verticillium infections. In: Seigner E (ed) CICH—IHB—IHGC International Hop Growers` Convention. Leon, Spain, pp 67–69
Semagn K, Bjornstad A, Skinnes H, Maroy AG, Tarkegne Y, William M (2006) Distribution of DArT, AFLP, and SSR markers in a genetic linkage map of a doubled-haploid hexaploid wheat population. Genome 49:545–555
Sewell GWF, Wilson JF (1984) The nature and distribution of Verticillium albo-atrum strains highly pathogenic to the hop. Plant Pathol 33:39–51
Stajner N, Jakse J, Kozjak P, Javornik B (2005) The isolation and characterisation of microsatellites in hop (Humulus lupulus L.). Plant Sci 168:213–221
Sustar-Vozlic J, Javornik B (1999) Genetic relationships in cultivars of hop, Humulus lupulus L., determined by RAPD analysis. Plant Breed 118:175–181
Van Ooijen J (2004) MapQTL® 5, Software for the mapping of quantitative trait loci in experimental populations, 5th edn. Kyazma B.V, Wageningen
Van Ooijen J (2006) JoinMap® 4, Software for the calculation of genetic linkage maps in experimental populations, 4th edn. Kyazma B.V, Wageningen
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Vos P, Hogers R, Bleeker M, Reijans M, Vandelee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zabeau M (1995) AFLP—a new technique for DNA-fingerprinting. Nucleic Acids Res 23:4407–4414
Yang C, Guo WZ, Li GY, Gao F, Lin SS, Zhang TZ (2008) QTLs mapping for Verticillium wilt resistance at seedling and maturity stages in Gossypium barbadense L. Plant Sci 174:290–298
Zamir D, Tadmor Y (1986) Unequal segregation of nuclear genes in plants. Bot Gazette 147:355–358
Acknowledgments
The authors acknowledge funding by the Slovenian Research Agency, research program Grant no. P4-0077. We thank Dr. Stefan Seefelder from Bayerische Landesanstalt für Landwirtschaft, Institut für Pflanzenbau und Pflanzenzüchtung, Freising, for kindly providing primer sequences for microsatellite markers of the Ho series.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by C. Gebhardt.
Rights and permissions
About this article
Cite this article
Jakse, J., Cerenak, A., Radisek, S. et al. Identification of quantitative trait loci for resistance to Verticillium wilt and yield parameters in hop (Humulus lupulus L.). Theor Appl Genet 126, 1431–1443 (2013). https://doi.org/10.1007/s00122-013-2062-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-013-2062-4