
Abstract
Hop powdery mildew [Podosphaera macularis (Wallr.) U. Braun & S. Takam.] is best controlled via the production of resistant varieties. Recent evidence supports selection against plant susceptibility genes to fungal pathogens as a more durable resistance mechanism than selection for resistance genes. The objective of this study was to identify molecular-based QTLs, their genetic effects and epistasis among QTLs associated with susceptibility to powdery mildew. Parents and offspring from the cross, ‘Perle’ × ‘USDA 19058M’, were clonally replicated and inoculated in a greenhouse using a CRD experimental design in Corvallis, OR. DNA was extracted, purified and analyzed via three different marker systems. Analysis of the resulting markers was based upon the “two-way pseudo-testcross” procedure. QTL mapping using multiple interval mapping and Bayesian interval mapping analyses were performed using WinQTL Cartographer 2.5_003. Comparison amongst mapping analyses identified three persistent QTLs on three linkage groups without significant epistatic effect upon expression. The persistent QTL on linkage group C7 had both additive and dominant effects controlling phenotype expression. The presence or absence of the two AFLP markers bordering the QTL on C7 defined susceptibility in offspring. This is the first report in hop identifying molecular markers linked to QTLs associated with disease susceptibility.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A critical problem facing the U.S. hop (Humulus lupulus L.) industry is the need for hop varieties providing superior brewing quality and possessing resistance to powdery mildew. Resistance to powdery mildew [Podosphaera macularis (Wallr.) U. Braun & S. Takam] in hop was described by Mahaffee et al. (2009) as having eight genetic sources of qualitative or “gene-for-gene” resistance as defined by Flor (1971). These R-genes were designated as R1–R7 and Rb gene (blistering gene), and were identified in a range of eight hop genotypes. Neither the function of, nor enzymes encoded for these R-genes have been determined or cloned. Two R-genes, R2 and Rb, have been associated with linked AFLP molecular markers through bulked segregate analysis followed by segregation analysis between markers of interest and resistance in a mapping population (Seigner et al. 2005; Seefelder et al. 2006). Plant resistance to powdery mildew based upon the cultivars ‘Wye Target’ (R2-gene) and ‘Buket’ (Rb gene) are important for disease management in Germany (Seigner et al. 2005). However, R2 and Rb-mediated resistance appear to be easily overcome by most strains of P. macularis in the USA. Specific germplasm and varieties released by the USDA have strong resistance to P. macularis races currently present in the USA (‘Nugget’, Haunold et al. 1984;’Newport’, Henning et al. 2004; ‘19058M’ Haunold 1988). Nonetheless, selection in the field for resistance is hampered by type-II errors with the result that multiple inoculations across environments and years are necessary to accurately ascertain resistance levels. Development and implementation of a marker assisted selection scheme (MAS) using markers identified by QTL mapping strategies would improve both speed of selection as well as accuracy.
Initial studies by Beckmann and Soller (1988) as well as Luo and Kearsey (1989) reported on analytical techniques to associate molecular markers with phenotypic traits. Lander and Botstein’s (1989) refined these techniques by describing “interval mapping” (IM) whereby single QTLs are placed along a linear map of markers previously placed on individual linkage groups. Composite interval mapping (CIM; Zeng 1993, 1994) utilizes interval mapping combined with multiple regression to restrict influences caused by linked or unlinked QTLs. No consideration for epistatic effects between multiple QTLs is included in any of these analyses. Mackay (2001) pointed out that epistatic effects are quite common between QTLs associated with the expression of a specific trait. Estimation of epistatic effects in QTL mapping analyses along with simultaneous mapping of multiple QTLs was first reported by Kao et al. (1999). Multiple interval mapping (MIM; Kao et al. 1999) first locates all possible QTLs, then defines and performs a simultaneous search for epistasis between QTLs. This results in a more refined model than that obtained by any of the previously described analyses. Finally, Satagopan et al. (1996) reported on a Bayesian approach, Bayesian interval mapping (BIM), to identify the best combination of QTLs delineating the best-fit genetic model. BIM utilizes Markov Chain Monte Carlo (MCMC) techniques to simultaneously identify multiple QTLs and the impact of their effect. The benefit of this mapping procedure is that no a priori assumptions are made concerning interactions between loci and the resulting set of QTLs.
A few peer-reviewed studies have reported genetic mapping in hop. Seefelder et al. (2000) were the first to publish hop genomic maps while attempting to identify linkage groups associated with expression of male flowers. This group utilized a “two-way pseudo-testcross” (Grattapaglia and Sederoff 1994) to develop male and female maps. No QTL analyses or statistical association tests were reported but a single linkage group reportedly “co-segregated” with the expression of male flowers. Koie et al. (2005) reported a QTL mapping study on bittering resin components in hop. Genetic linkage maps for female line and male line were constructed. Twelve QTLs were detected for seven of the nine chemical components. Cerenak et al. (2006, 2009a, b) reported QTL analyses for alpha acid levels and yield components. A single major-effect QTL was identified for alpha acid levels while QTLs for yield components were also identified on linkage group one.
The authors are not aware of any peer-reviewed manuscripts reporting on QTL-mapping studies for powdery mildew resistance or susceptibility in hop. A recent review by Pavans et al. (2010) argued the importance of selection against susceptibility to fungal pathogens as a more durable and broad spectrum mechanism for resistance. The authors point out that some pathogen effectors act by suppressing the host plant’s immunity by turning on effector targets whose action lie in functioning as negative regulators of plant immunity. By disrupting the function of this type of effectors’ with negative regulation, a release of the plant’s suppression of disease response activities is triggered—ultimately leading to resistance as suppression of disease response by the pathogen is no longer possible. Pavans et al. (2010) provides an extensive list of documented cases where this observation has been studied. No evidence of such a plant disease resistance mechanism has been documented in hop.
In this study we observed segregation for resistance versus susceptibility to powdery mildew across 124 offspring from the cross between the powdery mildew-susceptible German variety ‘Perle’ and the resistant USDA-ARS male line ‘19058M’. Plant resistance based upon genes from 19058M has not been overcome since powdery mildew was first observed in USA in 1996. This source of resistance appears to be durable and effective against all races of powdery mildew present in the Pacific Northwest USA. To better understand the genetic control over powdery mildew resistance in 19058M, our objectives were to identify molecular-based QTLs, their genetic effects and epistasis among QTLs associated with resistance or susceptibility to powdery mildew.
Materials and methods
Clones (N = 124) from individuals representing the cross between ‘Perle’ and USDA-ARS ‘19058M’ along with parents were established in a greenhouse located near Corvallis, OR. The greenhouse was maintained at 20–25°C with a 14-h photoperiod. Plants were grown in Sunshine Mix #1 (SunGro Horticulture, Bellevue, Washington) in 440 cm3 pots for approximately 14 days and watered daily. Champion 17-17-17 (N-P2O5-K2O) fertilizer with micronutrients (McConkey’s, Portland, Oregon) was applied at each irrigation to promote vigorous growth and greater susceptibility to powdery mildew.
Inoculum of P. macularis representing a wide range of field populations of the pathogen from the Pacific Northwest USA were maintained through successive transfers onto the highly susceptible cultivars ‘Symphony’ and ‘Pacific Gem’ in a growth chamber set at 13°C with a 14 h photoperiod. When inoculum was needed for an experiment, conidia were washed from several heavily infected leaves by rinsing the leaves in a solution of Tween 20 (0.05% vol/vol) and ultra pure water (Nanopure with organic-free cartridge kit [Barnstead, Dubuque, IA]). The inoculum titer was adjusted to 50,000 conidia per ml with the aid of a hemacytometer and promptly sprayed on plants until just before run-off. Plants were air-dried within 30 min of the preparation of inoculum to minimize lysis of conidia before the inoculated plants were placed in a greenhouse maintained at approximately 20°C with a 14 h photoperiod. Plants of cultivar ‘Symphony’ were included in each inoculation as positive controls to verify the infectivity of the inoculum.
After 10 days of incubation (two latent periods on the variety Symphony), plants were rated using a six-step ordinal scale of 0–5, where 0 = no disease symptoms (resistant); 1 = necrotic flecks, non-sporulating blisters, or aborted infection (tolerant); 2 = one or few small lesions on plant with only slight sporulation (moderately resistant); 3 = multiple lesions on a plant, but not on all susceptible leaves (moderately susceptible); 4 = multiple lesions on all susceptible leaves (susceptible); and 5 = coalescing lesions on multiple leaves (highly susceptible). Each genotype was replicated with three or four plants during each inoculation, and inoculations were repeated at least once for all genotypes. Plants scored as a 0 or 1, were re-inoculated to verify that the observed phenotype was not the result of Type II error. Each of these plants was rated again and if the disease reaction varied the greatest observed disease score was used in subsequent analyses.
Young leaves approximately 3 cm in width were chosen for extraction of DNA. Leaf samples were washed with deionized purified water, blot-dried with paper towels, placed in sample vials and immediately stored on ice. Samples were subsequently lyophilized for 24–26 h prior to DNA extraction. DNA extraction and AFLP analysis were performed as described by Townsend et al. (2000) with the exception that AFLP analysis was performed using a 96 well, capillary ABI 3100 (ABI Inc.). AFLP primer combinations were as described by Townsend and Henning (2005). Only 31 out of the original 79 polymorphic loci were included in the integrated map. Microsatellite markers were included in our studies as a co-dominant marker system allowing for future integration with maps from other populations using the same microsatellite markers. Microsatellite markers used in our study were previously described by Jakse et al. (2002), Stajner et al. (2004), and Bassil et al. (2005). Thirty-nine microsatellite primer pairs were pre-screened for polymorphism in parents by 3% agarose gel electrophoresis. Nineteen (HlGT24, HUM-001A, HlGA44, Hl-AGA7, HlGT12, HUM-002B, HlGA58, 11A59, 3A88, Hl-CAG5, HlGA36, HlGA57, Hl-AGA1, Hl-ACA3, Hl-AGA3, HlGT2, HlGA24, HlGA9J and HlGT22) out of these 39 primer pairs proved polymorphic with only four SSR markers (HlGT24, HlGA57, Hl-AGA3 and HlGA9J) showing non-distorted segregation patterns. Of these four SSR markers only one SSR marker segregated with other markers suggesting that SSR markers arise from regions extant to AFLP or DArT markers. Forward primers for these polymorphic simple sequence repeats (SSRs) were fluorescently labeled with WellRed (D2, D3 and D4) tags and the PCR products were separated in the entire population by capillary electrophoresis using the Beckman CEQ 8000 genetic analyzer (Beckman Coulter Inc.). Fragment analysis and visualization were performed with the fragment analysis module of the Beckman CEQ software (microsatellites) or Genographer 2.1 (AFLP; Benham et al. 1999). Diversity Array Technology (DArT) (www.diversityarrays.com/) markers were utilized as previously reported for hop (Whittock et al. 2009) as well as several other plant species (Jaccoud et al. 2001; Xia et al. 2005; Wittenberg et al. 2005). Approximately 550 DArT markers proved to be polymorphic in this population with only 294 of the original 550 DArT markers actually used due to extensive clustering and redundancy. The final number of markers used to construct the genetic map was 326 markers.
Map construction was performed using JoinMap ver 3.0 (Van Ooijen and Voorrips 2001). Map construction followed the 2-way pseudo-testcross approach (Grattapaglia and Sederoff 1994) with specific applicational procedures delineated by Mehlenbacher et al. (2006) using Joinmap 3.0. Mehlenbacher et al. (2006) utilized a reiterative process whereby markers initially placed on male or female linkage groups but not mapped were re-coded as “dummy” markers showing opposite segregation patterns. The idea behind this was to re-code as “trans” to “cis” and vice versa. The analysis in JoinMap 3.0 was subsequently re-run with the dummy variables included. In many cases this resulted in those markers initially not mapped placed onto maps with the original linkage group. Markers re-coded as dummy markers were designated with the addition of a lower case “d” in the name of the marker. AFLP and DArT markers (a = present, 0 = absent) were handled and coded in the same manner as reported by Cerenak et al. (2006, 2009a, b) for dominant markers. The a0 × 00 and 00 × a0 marker groups were analyzed as backcross 1 (BC1) populations with linkage groups initially determined with a log-odds score statistic (LOD) of 4. The a0 × a0 marker group was analyzed as an F2 population. Group delineation for microsatellite markers were designated as ab × cd with all possible variants as determined by presence of alleles in parental lines. After re-analyzing each population type with the newly coded “dummy” variables, the a0 × a0 markers were then added to the data set of the a0 × 00 and 00 × a0 populations. These combined data sets, “a0 × 00 plus a0 × a0” and “00 × a0 plus a0 × a0” were then imported and analyzed separately into JoinMap 3.0 as BC1 populations (as validated by Mehlenbacher et al. 2006) and then re-mapped to produce both male and female maps. Markers from the a0 × a0 group are present in both male and female maps and can subsequently be used to unify both maps into a single integrated map. At this point a decision was made to only include linkage groups having more than five markers covering more than 10 cM for further analysis. Male and female maps were then unified into a single genetic map using the CP (cross pollinated) mapping function in JoinMap 3.0 as described by Mehlenbacher et al. (2006). The CP mapping function represents all possible gene combinations which are then taken into consideration: a0 × a0, a0 × 00, and 00 × a0. Kosambi’s mapping function (Kosambi 1944), an LOD score of five and recombination threshold of 0.45 were used to determine map distance, linkage groups and marker alignment along linkage groups. Again, linkage groups of less than 5 markers and covering less than 10 cM were eliminated for the final genetic map used for QTL analysis.
QTL mapping for resistance to powdery mildew was performed using WinQTL Cartographer Ver. 2.5_006 (Wang et al. 2010). Data were handled as a RF2 population with data recoded according to the specifications of WinQTL. The RF2 population is defined in WinQTL as randomly mated intercross of an F2 population. This population type most clearly defines the hop F1 population (the population used in our study) resulting from a highly heterozygous dioecious plant species. Single marker analysis was subsequently followed by MIM and BIM. Settings for MIM were based upon the default settings of the program. For BIM, one million permutations were used to estimate best fit models.
Results
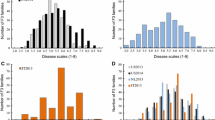
Phenotypic scores for powdery mildew resistance showed significant separation into two groups with the “resistant” group (scores of 0–1) comprising 47.8% of all individuals tested while the “susceptible” group (scores ranging from 3 to 5) comprised 45.8% of all individuals tested (Q1 = 0, median = 1.94, Q3 = 3.33 and max = 4.33) (Fig. 1) with the balance, 6.4%, having phenotypic scores of 1 < × < 3. Segregations into primarily two discrete groups strongly supports single gene control over powdery mildew resistance although distributions within each group would suggest modifier genes associated with penetrance.

Frequency histogram of powdery mildew resistance scores for population 20011 resulting from the cross “Perle × USDA 19058M”
Two maps were initially developed, one based upon segregation for genes from the male resistant parent (19058M) and one for genes from the female susceptible parent (Perle). There were a total of 179 markers covering 238.7 cM used to map the female susceptible parent. Eight linkage groups were identified with 63 markers representing a0 × 00 segregations and 116 markers representing a0 × a0 segregations. For the male resistant parent we identified seven linkage groups covering 529.5 cM with 287 total markers. These markers were distributed as 111 a0 × a0 and 176 00 × a0 markers.
These two maps were then integrated into a single map using JoinMap 3.0’s CP function. Integration of the two maps into a single genetic map resulted in 10 linkage groups identified with 326 DArT, AFLP and microsatellite markers covering 703 cM (Fig. 2). In the development of the male and female maps, significant clustering of DArT markers was observed resulting in a number of polymorphic markers being left out of the analysis. With the exception of four SSR markers (two in the male map and two in the female map) most did not group with DArT or AFLP markers resulting in their non-integration into any linkage group. Linkage group C4 covered the largest region with 19 markers measuring 176.1 cM, while linkage group C5 consisted of 24 markers covering a region of 14.9 cM. When a0 × a0 markers were linked with both a0 × 00 and 00 × a0 markers in their respective parent maps, unification of the linkage groups from both parents was possible. If no a0 × a0 markers were linked to either the a0 × 00 or the 00 × a0 linkage groups in their respective parent maps, no integration between the two maps was possible. Small linkage groups (five or fewer markers covering less than 10 cM) identified in the two BC1 parent maps were not included in the integrated map using the CP function. As a result, a total of 29 markers from both BC1 parent maps were factored out from the development of the resulting integrated map.

Genetic map of cross “Perle × USDA 19058M”. Potential QTL regions identified by multiple interval mapping (MIM) are designated on linkage groups C2, C7 and C8. Total length of map = 703 cM. a Linkage groups C1–C5 with possible QTL located on C2. b Linkage groups C6–C10 with possible QTLs located on C7 and C8
Single marker association tests in WinQTL (Table 1) showed three markers on linkage group C2 (hPb-365378R, hPb-365034R, hPb-365818R), one marker on linkage group C8 (hPd-718377R) and one marker on C9 (hPd-362728S) as significantly associated with powdery mildew resistance (P < 0.05). Several markers on linkage group C7 (200011_488Br, 200011_238.2Ar, 200011_100.6Br, 200011_345Dr, 200011_208Br, 200011_130Dr) as well as linkage group C8 (hPd-618652R, hPd-349787R, hPd-361074R, hPd-366231R, hPd-718377R) exhibited highly significant associations with powdery mildew resistance levels (P < 0.01). Multiple interval mapping analysis revealed one QTL on linkage group C2, one on C7 and one QTL on C8 having highly significant association (P < 0.01) to powdery mildew resistance phenotype (Table 2). The potential QTL on linkage group C2 was located near marker hPd-365818R (at 0.213 cM). The second QTL was located on linkage group C7 between markers 200011_345Dr (at 52.7 cM) and 200011_208Br (at 59.2 cM) while the third QTL was located on linkage group C8 near marker hPd-349787 (at 0.138 cM). No statistically significant epistatic interactions were observed between QTLs.
The “best fit” model using BIM analyses was determined to be a model with two to three markers based upon the number of significant permutations observed in MCMC calculations. Using this statistic as a guide, several models were identified that had significant threshold values of P < 0.05. In almost all cases the previously identified QTLs on linkage group C7 and C8 were included with other plausible QTLs located on either linkage group C2 or C4 as having the best fit of the model controlling powdery mildew resistance. Given the lack of evidence in other analyses for QTLs located on linkage group C4 the most appropriate model for selection using BIM analysis alone would be a model with three QTLs located on linkage groups C2, C7 and C8.
Discussion
This study represents the first attempt to integrate both male and female maps into a single genetic map in hop. As such, our study describes a map with the largest coverage of the hop genome with the greatest number of molecular markers. Mapping QTLs on a single integrated map simplifies the analysis and allows for the identification of epistasis between regions covered in both male and female genomes. Performing both simple and complex QTL analyses—again the first such multi-analyses performed on hop—allowed for a consensus evaluation of QTLs pointing out the single most important QTL for powdery mildew resistance found in 19058M. The use of consensus analyses including more complex QTL analyses demonstrated problems with dependencies upon simple analyses such as single marker analysis or IM.
The results of our single marker analysis highlight the deficiencies of such analyses when multiple QTLs exist within the same linkage group or chromosome. Doerge (2002) points out that single marker analyses (and IM) do not correct for effects caused by regions extant from the marker being evaluated. Thus, type I and type II errors are possible depending upon the effects of the extant region or regions. The likelihood that all markers statistically associated with powdery mildew resistance under single marker analysis are required for successful genetic improvement under MAS remains quite low (Asins 2002) and additional tests were called for.
Interval mapping (data not shown) narrowed the focus for regions containing potential QTLs, while CIM (data not shown) and MIM furthered narrowed down the number of QTLs potentially linked to powdery mildew resistance. Under IM analyses, markers situated on the same linkage group as regions containing true QTLs will often show association with phenotype regardless of relative position to the true QTL and this was observed in our study. Doerge (2002) named such false positive QTL regions “ghost QTLs.” One downfall of IM is the allowance of extant regions to influence identification of QTL locations in other parts of the genome. CIM and MIM analyses tend to minimize this problem by testing individual regions or “windows” of a linkage group while eliminating the effects from neighboring or distant regions associated with expression of phenotype. Analyses performed using MIM help reduce some of the problems associated with CIM analyses—namely reducing the effect of uneven distribution of QTLs across the genome and increasing the statistical power of detecting QTLs by eliminating the use of tightly linked markers as co-factors (Zeng et al. 1999). In our study MIM refined the number of markers observed in single marker analysis by identifying three highly significant QTLs on linkage groups C2, C7 and C8 without showing the presence of significant epistasis between QTLs (Table 2). It is not entirely clear how epistasis, if present, would have affected expression of phenotype in our study other than promoting variability within “resistant” or “susceptible” groups. Further work on epistasis between QTLs is necessary to better define genetic effects.
A set of two markers consistently showed signs of significant association with powdery mildew resistance across almost all analyses: the AFLP markers 200011_345Dr and 200011_208Br. While DArT markers—hPd-365818R and hPd-349787—also showed consistent association with phenotype in several mapping analyses, stepwise Logit regression failed to identify either marker as being consistently present in either diseased or disease-free individuals. As a test for predicting MAS success using these two markers, we analyzed disease scores for each individual genotype from cross 200011 and noted the presence or absence of these two markers. When both markers were present in an offspring, there was a high severity of disease (AVG disease Score = 3.20, STDEV = 0.79, n = 66, RANGE = 1.5–4.33). When only one of the two markers was present (in all cases this was 200011_208Br), mean disease severity was reduced but quite variable with a wide range of expression (AVG Score = 1.80, STDEV = 1.72, n = 11, RANGE = 0–4). When both markers were absent, disease severity was extremely low with genotypes being rated as resistant to powdery mildew infection with little variability among scores (AVG disease score = 0.09, STDEV = 0.16, n = 58, RANGE = 0–0.67). These results suggest that resistance in 19058M could be based at least partially upon lack of susceptibility genes rather than the presence of R-genes. If true, use of these markers in a selection scheme could provide a more durable and broad spectrum form of resistance to powdery mildew.
Our study did not identify a specific R-gene QTL whose presence in an offspring would ensure resistance. We cannot speculate upon genome coverage this study entailed and it is entirely possible that large parts of the genome remain uncharted. Future plans include the analysis of additional SSR and AFLP markers with the intent of incorporating a significant number of these markers onto the current genetic map. Additional markers covering a greater portion of the genome could potentially identify QTL(s) linked to a true R-gene.
Our study focused upon a relatively simple phenotypic trait—powdery mildew resistance—thought to be controlled by one or a few genes (Neve 1991) as proposed by Flor’s “gene-for-gene” hypothesis (Flor 1971). The simplicity of this trait provided a model system for initial QTL mapping in hop using advanced mapping analyses. Consensus results identified a single primary region of association (covering approximately 6.5 cM) on linkage group C7, bordered by two AFLP markers. Results from this study suggest that MAS in populations derived from USDA 19058M for powdery mildew resistance could potentially work by selecting for the absence of markers bordering the susceptibilty QTL, 200011_345Dr and 200011_208Br. Pavan et al. (2010) review on genes controlling susceptibility to plant diseases suggests that selection against susceptibility genes would be a superior method of breeding for resistance to plant diseases as opposed to traditional selection for plant resistance genes. The authors reported that this form of selection resulted in horizontal resistance in the plant as opposed to vertical resistance or the “gene-for-gene” resistance first reported by Flor (1971). Horizontal resistance is thought to be highly durable as compared to vertical resistance. The two AFLP markers, 200011_345Dr and 200011_208Br, border a single major QTL governing susceptibility to powdery mildew in the mapping population used in our study. What is not certain is whether these two markers can be used to select against susceptibility in other populations. Other studies in hop identified essential oils that when present or appear in specific patterns, showed linkage to PM susceptibility (Cerenak et al. 2009a, b). Selection against this pattern resulted in resistance to PM. These results certainly lend credence to the concept that selection against susceptibility genes or markers can lead to durable resistance.
We are in the process of sequencing the two susceptibility markers, 200011_345Dr and 200011_208Br, and will use the resulting sequence information to develop sequence characterized amplified regions (SCAR) markers that can be tested on other hop populations to determine extent of usefulness in MAS schemes. In addition, the authors have combined efforts in the future development of a saturated single genetic map of the hop genome using three different mapping populations and multiple marker systems. This future work will hopefully provide better mapping of the susceptibility QTL as well as other QTLs associated with quantitative or qualitative resistance to powdery mildew and other traits of economic interest.
References
Asins MJ (2002) Present and future of quantitative trait locus analysis in plant breeding. Plant Breed 121:281–291
Bassil NV, Gilmore B, Oliphant J, Henning J, Hummer K (2005) Genbank derived microsatellite markers in hop. Acta Hort 668:47–52
Beckmann JS, Soller M (1988) Detection of linkage between marker loci and loci affecting quantitative traits in crosses between segregating populations. Theor Appl Genet 76:228–236
Benham J, Jeung J-U, Jasieniuk M, Kanazin V, Blake T (1999) Genographer: a graphical tool for automated fluorescent AFLP and microsatellite analysis. http://wheat.pw.usda.gov/jag/papers99/paper399/1999_P3.html. Accessed 29 Sept 2009
Cerenak A, Satovic Z, Javornik B (2006) Genetic mapping of hop (Humulus lupulus L.) applied to the detection of QTLs for alpha-acid content. Genome 49:485–494
Cerenak A, Kralj D, Javornik B (2009) Compounds of essential oils as markers of hop resistance (Humulus lupulus) to powdery mildew (Podosphaera macularis). Acta Agric Slov [Print] 93(3): 267–273. http://dx.doi.org/10.2478/v10014-009-0015-z
Cerenak A, Satovic Z, Jakse J, Luthar Z, Carovic-Stanko K, Javornik B (2009) Identification of QTLs for alpha acid content and yield in hop (Humulus lupulus L.). Euphytica Published Online, March 31, 2009. doi: 10.1007/s10681-009-9920-9
Doerge R (2002) Mapping and analysis of quantitative trait loci in experimental populations. Nat Rev 3:43–52
Flor HH (1971) Current status of the gene-for-gene concept. Ann Rev Phytopathol 9:275–296
Grattapaglia D, Sederoff R (1994) Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: mapping strategy and RAPD markers. Genetics 137:1121–1137
Haunold A (1988) Notice of release of USDA 19058M male hop germplasm. USDA-ARS Publications, Beltsville
Haunold A, Likens ST, Nickerson GB, Hampton RO (1984) Registration of Nugget hop. Crop Sci 24:618
Henning JA, Townsend MS, Mahaffee W, Kenny S, Haunold A (2004) Registration of ‘Newport’ hop. Crop Sci 44:1018–1019
Jaccoud D, Peng K, Feinstein D, Kilian A (2001) Diversity arrays: a solid state technology for sequence information independent genotyping. Nucleic Acids Res 29:e25
Jakse J, Bandelj D, Javornik B (2002) Eleven new microsatellites for hop (Humulus lupulus L.). Mol Ecol Notes 2:544–546
Kao CH, Zeng ZB, Teasdale R (1999) Multiple interval mapping for quantitative trait loci. Genetics 140:1111–1127
Koie K, Inaba A, Okada Y, Kaneko T, Ito K (2005) Construction of the genetic linkage map and QTL analysis on hop (Humulus lupulus L.). Acta Hort (ISHS) 668:59–67
Kosambi DD (1944) The estimation of map distance from recombination values. Ann Eugen 12:172–175
Lander ES, Botstein D (1989) Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121:185–199
Luo ZW, Kearsey MJ (1989) Maximum likelihood estimation of linkage between a marker gene and a quantitative trait locus. Heredity 63:401–408
Mackay TFC (2001) Quantitative trait loci in Drosophila. Nat Rev Genet 2:11–20
Mahaffee WF, Engelhard B, Gent DH, Groves GG (2009) Powdery mildew. In: Mahaffee WF, Pethybridge SJ, Gent DH (eds) Compendium of hop diseases and pests. American Phytopathological Society Press, St. Paul
Mehlenbacher SA, Brown RN, Nouhra ER, Gökirmak T, Bassil NV, Kubisiak TL (2006) A genetic linkage map for hazelnut (Corylus avellana L.) based on RAPD and SSR markers. Genome 49:122–133
Neve RA (1991) Hops. Chapman and Hall Publishers, London 266
Pavan S, Jacobsen E, Visser R, Bai Y (2010) Loss of susceptibility as a novel breeding strategy for durable and broad-spectrum resistance. Mol Breed 25:1–12
Satagopan JM, Yandell BS, Newton MA, Osborn TC (1996) A Bayesian approach to detect quantitative trait loci using Markov Chain Monte Carlo. Genetics 144:805–816
Seefelder S, Ehrmaier H, Schweizer G, Seigner E (2000) Male and female genetic linkage map of hops, Humulus lupulus. Plant Breed 119:249–255
Seefelder S, Lutz A, Seigner E (2006) Development of molecular markers for powdery mildew resistance support breeding for high quality hops. Monatsschrift für Brauwissenschaft 5(6):100–102
Seigner E, Lutz A, Radic-Miehle H, Seefelder S (2005) Breeding for powdery mildew resistance in hop (Humulus L.): strategies at the Hop Research Center, Huell, Germany. Acta Hort 668:19–29
Townsend MS, Henning JA (2005) Potential heterotic groups in hop as determined by AFLP analysis. Crop Sci 45:1907–1910
Townsend MS, Henning JA, Moore DL (2000) AFLP analysis of DNA from dried hop cones. Crop Sci 40:1383–1386
Van Ooijen JW, Voorrips RE (2001) Joinmap® 3.0, software for the calculation of genetic linkage maps. Plant Research International, Wageningen
Wang S, Basten CJ, Zeng Z-B (2010) Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh. http://statgen.ncsu.edu/qtlcart/WQTLCart.htm. Accessed 9 Aug 2010
Whittock S, Leggett G, Jakše J, Javornik B, Carling J, Kilian A, Matthews PD, Probasco G, Henning JA, Darby P, Čerenak A, Koutoulis A (2009) Use of diversity array technology (DArT) for genotyping of Humulus lupulus L. Acta hort (ISHS) 848:59–64
Wittenberg AH, Van Der Lee T, Cayla C, Kilian A, Visser RG, Schouten HJ (2005) Validation of the high-throughput marker technology DArT using the model plant Arabidopsis thaliana. Mol Gen Genet 274:30–39
Xia L, Peng K, Yang S, Wenzl P, Vicente MC, Fregene M, Kilian A (2005) DArT for high-throughput genotyping of cassava (Manihot esculenta) and its wild relatives. Theor Appl Genet 110:1092–1098
Zeng ZB (1993) Theoretical basis of precision mapping of quantitative trait loci. Proc Nat Acad Sci 90:10972–10976
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Zeng ZB, Kao CH, Basteen CJ (1999) Estimating the genetic architecture of quantitative traits. Genet Res 74:279–289
Acknowledgments
The authors wish to acknowledge support from the International Hop DArT Consortium (Members listed alphabetically by first name: Andreja Cerenak; Anthony Koutoulis; Branka Javornik; Claudia Wiedow; Emily Buck; Erin Howard; Gene Probasco; Jernej Jakse; John Henning; Jules Freeman; Paul Matthews; Peter Darby; Rene Vaillancourt; Ron Beatson and Simon Whittock) along with their respective institutions for providing information and support to screen the USDA-ARS mapping population used in this study with diversity array technology (DArT) markers (www.diversityarray.com/). We would also like to thank Andrzej Kilian and Diversity Array Technology Pty Ltd for providing the DArT marker data for QTL mapping analyses.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Henning, J.A., Townsend, M.S., Gent, D.H. et al. QTL mapping of powdery mildew susceptibility in hop (Humulus lupulus L.). Euphytica 180, 411–420 (2011). https://doi.org/10.1007/s10681-011-0403-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-011-0403-4