
Abstract
Recent progress in genotyping and resequencing techniques have opened new opportunities for deciphering quantitative trait variation by looking for associations between traits of interest and polymorphisms in panels of diverse inbred lines. Association mapping raises specific issues related to the choice of appropriate (i) panels and marker-densities and (ii) statistical methods to capture associations. In this study, we used a panel of 314 maize inbred lines from the dent pool, composed of inbred material from public institutes (113 inbred lines) and a private company (201 inbred lines). We showed that local LD was higher and genetic diversity lower in the material of private origin than in the public material. We compared the results obtained by different software for identifying population structure and computing relatedness among lines, and ran association tests for earliness related traits. Our results confirmed the importance of the mite polymorphism of Vgt1 on flowering time, but also showed that its effect can be captured by zmRap2.7 polymorphisms located 70 kb apart. We also highlighted associations with polymorphisms within genes putatively involved in lignin biosynthesis pathway, which deserve further investigations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Association mapping, also referred to as linkage disequilibrium (LD) mapping, has been proposed to dissect the genetic basis of quantitative traits in plants. After preliminary attempts in the 1990s (Bar-Hen et al. 1995), it really developed in the early 2000s thanks to new statistical models (Thornsberry et al. 2001). It has been since then increasingly used for dissecting traits of interest, for example in barley (Lorenz et al. 2010), maize (Thornsberry et al. 2001; Flint-Garcia et al. 2003), or Arabidopsis thaliana (Atwell et al. 2010; Aranzana et al. 2005). Compared to linkage based QTL detection, association mapping addresses the relationship between genetic marker polymorphism and phenotypic variation within a population composed of diverse genotypes, ideally non-related by pedigree to each other. It is expected that the recombination events accumulated over the generations leading to such a population have broken the associations (or LD) between loci except for those that are physically close. Hence, compared to conventional QTL detection experiments, where confidence intervals of the estimated QTL positions often exceed 10 centimorgans (cM), association-mapping studies are expected to provide a much higher resolution, depending on the extent of LD (Rafalski 2002). Organizing panels of materials and understanding their properties is of major importance in order to optimize the genotyping strategies and refine the analysis of traits.
Panels assembled from existing material might not be ideal for association mapping. First, population structure within a panel causes LD between distant and physically unlinked loci and may therefore generate false-positive associations, if not controlled properly. The control of population structure was first applied in a human genetics context (Pritchard et al. 2000) and then widely spread to animals and plants studies (Thornsberry et al. 2001). Second, relatedness among lines creates correlations between the performances of the individuals. Yu et al. (2009) showed that the correction for pair wise relatedness in a mixed model significantly decreased false positives as compared to corrections for population structure only. Price et al. (2006) suggested that structure and relatedness could be both taken into account by including principal component analysis (PCA) axes as covariates in the analysis. Several software using different methods and algorithms are available to evaluate population structure with molecular data, (STRUCTURE (Pritchard et al. 2000), Locus Miner (Veyrieras, personal communication, used by Stracke et al. (2009))) or relatedness among lines (Eigensoft (Price et al. 2006), SPAGeDi (Hardy and Vekemans 2002), Emma (Kang et al. 2008)). These aspects are particularly important in maize for which climatic adaptation was accompanied by population differentiation (Camus-Kulandaivelu et al. 2006). This initial trend was further reinforced by hybrid breeding, which led to the definition of heterotic groups. Finally, high intensity selection led to highly related inbred lines with complex pedigree structure (Mikel and Dudley 2006; Van Inghelandt et al. 2010).
In order to find associations, it is necessary to have sufficient LD between QTL and polymorphisms that are analyzed. Depending of the LD extent (Rafalski 2002), two approaches are being used in association studies: (i) candidate gene approach, where the objective is to validate the effect of a specific gene and ideally identify within this gene the polymorphisms underlying trait variation, or (ii) genome-wide association (GWA) approach, using markers whose function is not known a priori to cover the whole genome (Salvi et al. 2007). Linkage disequilibrium patterns vary according to the species, the population which is considered (Flint-Garcia et al. 2003; Tenaillon et al. 2001), and among the chromosomal regions (Thornsberry et al. 2001). In maize, LD decays to less than 20 % (as measured with r² statistics) within 1 kb in maize landraces (Tenaillon et al. 2001). It decays below 10 % after approximately 2 kb in diverse inbred lines (Remington et al. 2001; Wilson et al. 2004). These cases are all appropriate for a candidate gene approach if the causal polymorphism is located within the gene sequence or very close nearby, but necessitate a high marker density if no candidate gene is available. In contrast, LD can extend to 100–500 kb in commercial inbred lines (Ching et al. 2002; Jung et al. 2004; Van Inghelandt et al. 2011) and Belo et al. (2007) suggested that GWA in a maize panel including elite material is possible with 8,590 marker loci. This number is already achievable with high throughput genotyping techniques. Such LD levels can also be interesting in candidate gene approach if causal polymorphisms are distant regulators. This situation is highlighted by Salvi et al. (2007), who demonstrated that the allelic variation responsible for the flowering time QTL Vgt1 was confined to a 2 kb intergenic region located 70 kb upstream from zmRap2.7, an AP2-like flowering time gene, supporting a cis-acting transcription-regulatory role of Vgt1 on this gene (confirmed by expression studies). It is thus of high interest to understand the LD extent in a given panel in order to evaluate the resolution one may expect and the marker density needed to find associations.
In this context, we wanted here to analyze an association panel mainly composed of elite material from a breeding company, in a connected way with a sample of publicly available material representing a broader diversity. This material was a selection of inbred lines from the dent group released during the last 30 years by the public sector and Syngenta Seeds (for the most recent ones), which enabled us to include an historical perspective in the analysis of this panel. Diversity and LD were analyzed for the two different sets of lines and for the whole panel. So far, no association mapping study including both types of germplasms simultaneously has been reported in the literature. We used flowering time related traits, evaluated per se and at the hybrid level, as model traits to run the association tests. We studied polymorphisms located within genes a priori non-candidates for flowering time with the objective of using these genes to estimate the LD extent in our panel. We studied as well sequences within the Vgt1 region, known to be involved in flowering time, to compare our results with those from Ducrocq et al. (2008) and Salvi et al. (2007) obtained with panels of lines of public origin and belonging to different heterotic groups. We tested linear mixed models with different calculations of structure and kinship matrices, showing differences that might appear for a panel mixing different sources of germplasm.
Materials and methods
Plant material
In this study, 314 lines were considered (Supplementary material 1). All were dent lines representing a pool of material complementary to early flint materials to produce hybrids for Northern Europe. To facilitate the trial designs and harvest, they were selected within a restricted earliness window, with sum of temperatures (base 6 °C) at flowering varying from 835 to 1,145 degree-days, based on previous studies. A subset of 113 lines was selected from the panel used by Camus-Kulandaivelu et al. (2006) and subsequent investigations (e.g., Ducrocq et al. (2008)), considering the above mentioned flowering time window and a sum of admixtures of at least 0.572 for Iowa stiff stalk (ISS) and corn belt groups defined in Camus-Kulandaivelu et al. (2006) (“exotic” and “flint” lines were excluded from the panel). Syngenta Seeds provided 201 elite lines, which represented the diversity of the dent germplasm used for Northern European breeding program over the last 15 years. This germplasm included mainly stiff-stalk and iodent lines.
Molecular data
DNA extraction: plants were grown in Klasmann growing medium for seven days in chamber at 24 °C day and 18 °C night. Leaves samples were taken from plants at 1–2 leaves stage. DNA was extracted from leaves using the Macherey–Nagel method (Düren, Germany). Fresh material was sampled from two plants in separate plates (30 mg each) and five repetitions were made. The DNA quantity was amplified using the Genomiphi Kit V2.
To evaluate the relatedness among lines and the population structure, we used a GoldenGate® genotyping assay (1,536 SNP chip, Illumina, San Diego, USA) provided by Syngenta Seeds. These SNPs corresponded to 665 amplicons, all located in coding regions, with one to 12 SNPs per amplicon (with a mean of 3.2 SNPs per amplicon). One thousand two hundred and forty SNPs could be scored with appropriate quality based on laboratory experience following Illumina guidelines (Gunderson et al. 2004). Out of these, 770 were issued from Syngenta Seeds internal projects and 470 SNPs from the HapMap project (SNP originating from Pioneer Hibred) (http://www.panzea.org). When SNPs from the same amplicon were in complete LD (r 2 = 1, 90 pairs), we kept only the SNP with the lower percentage of missing data. Monomorphic markers (47 markers) and markers with minor allele frequency (MAF) less than 3 % (124 markers) were discarded in further analyses, yielding a total of 979 markers representing 422 amplicons. The global rate of missing data for these 979 SNPs was 3.75 %. These SNPs are later referred as SNParray data.
To further investigate diversity, analyze local LD and run association tests, we sequenced 101 amplicons in specific regions. These corresponded to two categories: (i) the Vgt1 region known to have a major impact on flowering time and (ii) 13 regions chosen for their interest in digestibility (Truntzler et al. 2010) but with no a priori effect on flowering time. The 13 regions selected for the digestibility will be analyzed for the variation of digestibility related traits in another study, but we wanted to use them here to assess LD in our panel and take the opportunity to look at the associations for earliness related traits.
For the Vgt1 region, 12 amplicons were sequenced within bins 8.05–8.06. Gene sequences were chosen to be distributed at exponential distance from the targeted QTL Vgt1, considered as a reference due to its major impact on earliness trait (50, 100, 250 then 500 kb from the amplicons highlighted by Ducrocq et al. (2008)). Sequences of these genes were retrieved from MaizeSequence.org (http://maizesequence.org). For the Vgt1 region itself, primers from Ducrocq et al. (2008) were also tested. Two amplicons were sequenced; the first amplicon contained a mite polymorphism, and the other an insertion deletion, further referred to as CGindel587. Both polymorphisms were shown to be associated with flowering time in Ducrocq et al. (2008). PCR reactions were performed as described in supplementary material 2a. Four other amplicons from Ducrocq et al. (2008), called K42, ZmRap2.7, K45 and K46 were also sequenced in the Vgt1 region but using a slightly different protocol (described in supplementary material 2b). The 13 other chromosomal regions were selected from the results of a meta-analysis of QTL involved in digestibility related traits by Truntzler et al. (2010). Five other genes were targeted even though they were not positioned in a meta-QTL region, as they were strong functional candidate genes for cell wall digestibility. Two of the five genes are involved in lignin biosynthesis: cinnamyl alcohol dehydrogenase 2 (CAD2) on bin 2.02; trans-cinnamate 4-hydroxylase (C4H) on bin 8.03 and the remaining three are transcription factors: Geo27 on bin 1.10, Geo16 on bin 5.01 and Lim1 on bin 6.01. We preferentially chose to develop amplicons within candidate gene sequences retrieved in this case from the MAIZEWALL database (Guillaumie et al. 2007), then blasted on MaizeSequence.org in order to design primers. Additional genes were selected with the objective to have at least one amplicon every 250 kb, including the candidate genes in our 13 regions of interest. They were retrieved from MaizeSequence.org. Primers for each amplicon were designed from the transcripts using Primers3 software, and then blasted against BAC sequences to check the specificity of the primers for a single maize gene. We selected only those primers pairs that had a unique amplicon product size less than 2,000 bp and which included intronic regions to increase the probability of detecting SNPs. Several primer pairs for each gene were screened on a mini panel of 16 lines, before choosing the ones used on the whole panel. A total of 101 amplicons were developed over the 13 regions. PCR reactions for these amplicons and the other ten in the Vgt1 region (see above) were performed as described in supplementary material 2b. The primer pairs giving the best sequencing result for each targeted gene were selected to amplify and sequence all 314 lines. Sequencing was performed on an Applied Biosystem 3730XL, using BigDye terminator chemistry, according to manufacturer’s instructions. The amplicons were on average 611 bp long, ranging from 254 to 1,102 bp.
Alignments were performed using Staden package (Staden et al. 1998). For the gene sequences, the alignments were loaded into the software package Tassel in order to extract the polymorphisms (http://www.maizegenetics.net/) (Bradbury et al. 2007). Single nucleotide polymorphisms with minor allele frequency (MAF) of less than 3 % and/or more than 20 % of missing data were removed. When two or more polymorphisms within a same amplicon were in complete LD (r 2 = 1), only one (with the lowest rate of missing data) was considered for further analyses. The extracted polymorphisms are later referred as POLseq.
Phenotypic evaluation
Inbred lines were evaluated for their per se and test-cross (hybrid) values in separate field trials.
Per se trials
Inbred lines were evaluated for their per se values at three locations in 2008. These locations were Le Moulon (MOU), Lusignan (LUS) and Les Pas (LPA), in France. All the lines were replicated twice in every trial in order to form a complete block design. Within a given block, the panel was divided into three maturity sub-blocks based on the sums of temperature required at flowering, in order to limit potential competition between late and early lines. Six lines were duplicated in the adjacent sub-blocks to estimate the sub-block effect. Plant height (PHT) was measured from soil to the first branch, ear height (EARHT) from soil to the node bearing the ear. Male and female flowering time (MFLW and FFLW) were estimated as days since planting, when 50 % of the plants were shedding pollen or extruding silks, respectively.
Hybrids trials
All 314 lines were crossed with a flint inbred line tester and the resulting F1 hybrids were evaluated for two years (2008 and 2009) on one location (Wadersloh, Germany). In each trial, the panel was divided into three blocks, based on the expected performance of the lines (mainly based on the year of release and the expected yield of each hybrid), in order to limit competition effects. Two checks (Syngenta Seeds commercial hybrids) were repeated twice in each block, and two hybrids were repeated 24 times in each field, with eight replicates in each block. The other hybrids (i.e., almost all the hybrids) were not replicated within a given trial. PHT and EARHT were measured in the 2008 trial (WA08) only, whereas FFLW and MFWL were measured in the two years (WA08 and WA09). The traits were measured with the same procedure as for the per se trials.
Molecular data analyses
Gene diversity was calculated using PowerMarker (Liu and Muse 2005) on the SNParray data and on the POLseq. Diversity was estimated for the whole panel and for public and Syngenta lines separately. Hundred bootstrap runs were performed using PowerMarker to estimate the precision of gene diversity (H exp).
Test of neutrality was calculated for the sequences from the Vgt1 region using DNAsp software (Librado and Rozas 2009) with Tajima’s D (Tajima 1989).
Panel structure and kinship matrix
To infer the structure of the population, we used “STRUCTURE” (Pritchard et al. 2000) and “Locus Miner” (Veyrieras et al. 2006) on the SNParray data. Ten independent runs were performed with “STRUCTURE” for two to 15 subpopulations, using the admixture model with the “haploid” option, and a burn-in of 200,000 followed by 100,000 iterations. To infer the most likely number of groups within the population, we used the Evanno transformation method (Evanno et al. 2005) on the STRUCTURE outputs. The algorithm used in “Locus Miner” is described in Stracke et al. (2009). This software implements an Expectation–Maximization (EM) algorithm based on a multivariate model close in spirit to the simple mixture/admixture model of STRUCTURE. The program uses a PCA based approach to evaluate the remaining amount of LD between markers once the estimated population structure has been taken into account (Veyrieras, personal communication). Structure analysis was complemented by the PCA approach implemented in Eigensoft (Price et al. 2006).
Kinship matrices were calculated using SPAGeDi (Hardy and Vekemans 2002) and Emma (Kang et al. 2008). Using the software SPAGeDi, we evaluated two different kinship coefficient estimators, defined by Loiselle (Loiselle et al. 1995) and Ritland (Ritland 1996), respectively. Both estimators give more weight to similarity for rare alleles, proportional to 1/f a for Ritland’s estimator vs. (1−f a)² for Loiselle’s estimator, f a being the allele frequency. Loiselle’s estimator is expected to be unbiased with respect to allelic frequencies (Hardy and Vekemans 2002).
We used the Emma package to estimate identity by state (IBS), which corresponds to the genetic similarity, between lines based on marker data (Kang et al. 2008). To compare the different estimates of kinship, we calculated the correlation coefficients between the different kinship matrices and tested their significance following Mantel (Mantel 1967), using the ade4 R-package in R (Dray and Dufour 2007; R development core Team 2011).
To estimate whether 979 SNPs gave enough information to estimate IBS or not, 20 bootstraps based on the SNParray data were performed using R randomization procedures, yielding 20 kinship matrices with the Emma package.
LD analyses
Linkage disequilibrium between pairwise polymorphisms identified on the sequences was studied using Tassel (Bradbury et al. 2007). We used r 2, which is the squared correlation between alleles at two loci, as a measure of LD. To investigate the effect of allele frequencies we computed LD for polymorphisms belonging to three classes of allelic frequencies: all frequencies, balanced allelic frequencies (both markers of the pair with MAF >0.2) and unbalanced frequencies (at least one marker of the pair with MAF <0.2).
Phenotypic data analyses
Estimation of lsmeans
For per se trials, we used PROC GLM (SAS 1989) to test the block and sub-block effects within each trial, considering the genotypic effect of the lines as a fixed effect. The sub-block effect was not significant; therefore, it was not included in the final models. For each trial, the following model was used:
where Y ikl is the field plot value observed for inbred line i in sub-block l of block k, μ is the general mean, α i the fixed effect of the genotype i, γ k the fixed effect of block k, ε ikl the random residual error. We also analyzed jointly all the trials by adding to the above model a location and a genotype × location interaction effects, considered as fixed.
For hybrid trials, we used PROC GLM in SAS to test the different effects. For each trial, the following model was used:
where Y ik is the value for the trait of interest, μ is the general mean, α i the fixed effect of the genotype i, γ k the block k (fixed effect), ε ik the residual error. For the joint analysis of all trials, the location and the genotype × location interaction effects were added as fixed. When the block effect was not significant for a given trait and a given trial, it was removed from the model. Due to our experimental design, we had a confounding effect between the genotypic value of the lines and the block effect. By considering genotypes as fixed effects in our model, block effects were estimated based on the only hybrids (mainly checks) that were replicated between blocks. Therefore, block effects did not reflect the genotypic differences between hybrids grown in the different blocks but only environmental differences among blocks.
These models were used to compute the adjusted means of the genotypes that were further used to run association tests. Using the different lsmeans, we computed Pearson correlations between traits for a given trial and for the whole trials, and between trials for a given trait. For a given trait, we also computed the correlation between per se and hybrid values for the whole trials.
Analysis of genetic variation and traits heritability
We then used the mixed linear model in ASReml-R to estimate the genetic and residual variances (for a given trial, and jointly for the whole experimental design). In these models, we considered checks as fixed effects and genotype and genotype × location interaction (for the whole trials analysis) as random effects.
Broad sense heritability for each trait on each location (for per se and hybrids trials separately) was computed using the following formula:
with \( \sigma_{\text{g}}^{2} \) being the genetic variance of the lines, and \( \sigma_{\text{e}}^{2} \) the residual variance (within trial) estimated with ASReml-R, K the number of replicates. For each per se trial, K was equal to two; for each hybrid trial, K was equal to one.
Broad sense heritability for each trait on the whole experimental design (for per se and hybrids trials separately) was then computed using the following formula:
with \( \sigma_{\text{g}}^{2} \) being the genetic variance of the lines, \( \sigma_{{{\text{g}} \times {\text{e}}}}^{2} \) the variance of genotype x environment interactions and \( \sigma_{\text{e}}^{2} \) the residual variance (over several trials) estimated with ASReml-R. J is the number of locations and K the number of replicates per location. For the global hybrid trials analysis, J was equal to two for MFLW and FFLW and one for PHT and EARHT. For the global per se trials analysis, J was equal to three, the total number of locations and K was equal to two.
Association tests
To assess the effect of the population structure on the trait variability, we performed linear regression in R (R development core Team 2011) of the lsmeans of the different traits over the covariates issued from STRUCTURE and Eigensoft outputs. We compared the global percentage of variance explained (R²) and p value of the models as well as the Akaike information criterion (AIC) (Sakamato and Kitagawa 1987) to assess which model gave the most parsimonious correction for population structure.
Loiselle and Ritland matrices (K Loiselle and K Ritland) were modified after computation, with diagonal set to two in order to have a semi-definite positive matrix (Kang et al. 2008) and negative values set to zero. Negative values indicate that the two lines are less related than two lines taken at random. Keeping negative values in the kinship matrix would impose a negative correlation between the genetic values of these lines, which is not realistic (Yu and Buckler 2006).
Association tests were performed using Tassel (Bradbury et al. 2007), ASReml-R (Butler et al. 2007) and Emma package (Kang et al. 2008). Association tests were conducted using a linear mixed model, following the approach of Yu et al. (2006). In ASReml-R and Tassel, we used the mixed linear model with Eigenstrat 10 axes and K Loiselle. With Emma package we ran a mixed linear model with K Emma (no Q matrix).
Considering that the 979 SNPs revealed by the Illumina chip were a priori not associated with flowering time related traits, we used these polymorphisms to identify the model best controlling false-positive associations (Yu et al. 2006). Using Tassel, we thus first ran association tests for FFLW with the 979 SNPs revealed by the Illumina chip, with various structures (Q matrices) and K matrices. For each model, we plotted the cumulative proportion of tests considered as significant as a function of the type one error risk considered. The model giving the least false-positive associations was then used to run association tests for flowering time and height related traits, using Tassel and ASReml-R, on the polymorphisms extracted from the sequences. In order to control the false discovery rate (FDR) of multiple association tests, we corrected the p values of each polymorphism with the procedure from Storey and Tibshirani (2003).
Tests were also performed for the Vgt1 region (the four amplicons obtained with the primers from Ducrocq et al. (2008), and the two markers individually genotyped (mite and CGindel857)) for flowering time and height related traits. To compare the association results on the public and Syngenta lines, association tests were compared on the two sub-groups of lines, in addition to the whole panel. In that case, kinship matrices were calculated again within each sub-group.
In order to discriminate the effect of sample size from the origin of inbred lines (Syngenta vs. public), we sampled 50 times 113 lines (number of lines in the public panel) from the 211 Syngenta lines and performed association tests on these 50 samples for the Vgt1 region.
Results
Molecular diversity
Considering the 979 polymorphic SNPs from the Illumina chip, the average gene diversity was 0.30 ± 0.004, 0.33 ± 0.004 and 0.25 ± 0.005 for the whole panel, the public lines and Syngenta lines, respectively. Of the 101 amplicons sequenced, 11 did not show any polymorphisms. A total of 890 non-redundant polymorphisms was identified, including 156 indels and 734 SNPs. On average, there were ten polymorphisms per amplicon, composed of eight SNPs and two indels. Gene diversity over the 890 polymorphisms from the amplicons was 0.29 ± 0.005, 0.33 ± 0.005 and 0.23 ± 0.006 for the whole panel, public lines and Syngenta lines, respectively.
Tajima’s D was calculated in the Vgt1 region and was not significant for any of the amplicons.
Population structure
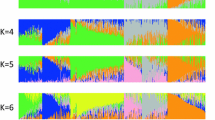
STRUCTURE and Locus Miner provided similar log likelihood values with the 979 Illumina SNPs, the second being twice faster in terms of computation time (48 h for STRUCTURE versus 24 h for Locus Miner with Intel 1.0-GHz mono Core CPU). Evolution of the likelihood according to the number of groups showed that a plateau was reached since two groups for STRUCTURE and Locus Miner (Supplementary material 3). STRUCTURE and Locus Miner outputs were quite similar in terms of distribution of lines within groups from k = 2 to 6 populations. Different runs of STRUCTURE for the same group number showed stable results for the distribution of the lines in each group. Evanno criterion supported the choice of k = 2 for STRUCTURE as the highest level of structure (Evoldir Community 2008). Using Eigenstrat to assess the population structure, we obtained ten axes significant at the 0.05 type I error rate. They accounted for 25.6 % of the genetic variation (data not shown). The first axis accounted for 8.8 % of the variation, whilst the nine others axes accounted from 3 to 1.4 %. This suggested further significant levels of stratification beyond k = 2 (Patterson et al. 2006). Figure 1 showed that the main trend of diversity organization can be interpreted as a differentiation between Syngenta versus public pools, although a few lines of Syngenta remained close (in terms of PCA position or group assignation) to the public ones and vice versa. Beyond k = 2, group assignation was in close agreement with the pedigree of the lines and mostly differentiated sub-groups within Syngenta lines. Based on the log likelihood results and expert knowledge on genetic organization of Syngenta lines, we selected k = 3 for further tests in association mapping.

Plot of the first two axes of principal component analyses for the lines of the panel based on the 979 SNPs from the Illumina chip. a Projection of the Public lines in white and the Syngenta lines in black. b Projection of the two groups of population structure from STRUCTURE output for k = 2 populations (first group in white, second in black)
Relatedness
Mantel tests were performed for the comparison of the 20 K Emma matrices (obtained after 20 bootstraps based on the SNParray data). Pairwise comparisons led to correlations values from 0.94 to 0.97 with an average of 0.95 (significant at a 0.05 level).
Mantel test between K loiselle and K Emma (significant at 0.05 level) gave a correlation of 0.64, whereas it was equal to 0.98 between K Loiselle and K Ritland. The relatedness coefficients varied from −0.29 to 1.24 with a mean of 0.003 for K Loiselle and from 0.59 to 1 with a mean of 0.63 for K Emma. The kinships showed a unimodal distribution for K Loiselle matrix, whereas the distribution appeared more bimodal for K Emma (Fig. 2a and b), indicating that the distribution of kinship was different for the two methods. The correlation graph between Loiselle and Emma coefficients showed three different subsets of points (Fig. 2c). These were clearly separated based on the membership of the lines to the first or second group of structure. For K Loiselle, the relatedness coefficients within groups were reduced to the same variation range, whereas they showed two different ranges of variation for K Emma.

Relatedness coefficients of the kinship matrices. a K Loiselle similarity distribution, b K Emma similarity distribution, and c comparison of K Emma (X axis) and K Loiselle (Y axis). White points correspond to pairwise kinships between lines attributed to the group one (mainly public lines) defined by STRUCTURE, dark gray symbols correspond to pairwise kinships between lines attributed to the group two (mainly Syngenta lines) defined by STRUCTURE, black symbols correspond to pairwise kinships between lines attributed to two different groups defined by STRUCTURE
Local LD investigations
For local LD studies, we removed any polymorphic markers with a MAF inferior to 3 % and more than 20 % of missing data but considered the polymorphisms in complete LD within the same amplicon. According to these criteria, 1,128, 1,067 and 1,155 polymorphisms were kept for the whole panel, the public lines, and for the Syngenta lines respectively. Figure 3a shows the mean r 2 for pairwise comparisons of markers distant from 0 to 1,000 bp. Over this distance, the number of pairwise comparisons was high enough to give reliable means of r 2 and the LD was higher for Syngenta lines (0.61) than for public lines (0.39). From 1 to 5 kb, r 2 was decreasing (Fig. 3b). Only a few pairwise comparisons were available for marker distant between 5 to 100 kb; however, it can be noted that some high r² values were observed. In particular, LD estimated between (i) the mite and CGindel587 located in Vgt1 itself and (ii) 18 polymorphisms within ZmRap2.7, distant from 73 kb, included r² values above 0.72. From 100 to 200 kb, mean r 2 was 0.19, 0.08 and 0.21 for the whole panel, public lines and Syngenta lines respectively, with maximal r 2 values reaching 0.37 for the public lines and 0.66 for Syngenta lines. From 200 to 1,000 kb, mean r 2 was 0.12, 0.06 and 0.19 for the whole panel, public lines and Syngenta lines respectively. For 1,000 kb to 10 Mb, mean r 2 was 0.04, 0.03 and 0.04 for the whole panel, public lines and Syngenta lines, respectively.

Linkage disequilibrium extent (r 2) using the whole polymorphisms, over different distances. a Mean r 2 for each 100 bp. The shapes represent the average r 2, the lines the standard deviation of the r 2 values, b mean r 2 for distances increasing on a logarithmic scale, for markers distant from 0 to 1 Mb. In gray diamond-shaped are the values for the public lines, in gray squares the values for the Syngenta lines and in black triangles the values for the whole panel (right vertical axis). The dark gray area represents the number of pairwise marker comparisons (left vertical axis)
Phenotypic variation
For the per se and hybrids trials, the genetic effect was significant for all the traits in each location at a 0.05 level risk. Genotype x environment interactions were significant for all the traits at 0.05 level (results not shown). Despite this interaction effect, correlations between locations for the per se trials were superior to 0.8 for all traits (data not shown). Correlations between locations were lower for the hybrid trials (from 0.36 to 0.60 depending on trait, results not shown). Heritability was higher for per se trials than for hybrids trials: it ranged from 0.65 to 0.93 for PHT in per se trials, but was 0.63 for hybrids at the single location where this trait was scored (Table 1). Male and female flowering times showed high heritabilities for per se trials, from 0.92 to 0.95. For hybrids trials, heritabilities were very low for these traits at location WA09: 0.26 for FFLW and 0.38 for MFLW but higher at WA08 (0.69).
Correlation between FFLW and MFLW was 0.93 in per se trials and 0.58 in hybrids trials. Positive correlations were also observed for height traits. Correlation between EARTH and PHT was 0.76 for per se trials, but only 0.34 for hybrids trials. Correlations between hybrids and per se performances on the whole experimental design were 0.65 for MFLW and 0.70 for FFLW (Supplementary material 5). For heights, correlations were lower: 0.36 for EARTH and 0.53 for PHT.
Association tests
The effect of the structure obtained from STRUCTURE with k = 3 and Eigenstrat (5 and 10 axes, PCA5 and PCA10, respectively) was significant for the 21 traits (Table 2). Among the three models, PCA10 had the most significant effect on the trait variation, and the lowest AIC for 17 traits out of 21 (data not shown). Using PCA10, the structure accounted for 21 % for FFLW and 32 % for MFLW of the trait variability in the hybrid trials. For k = 3, the structure accounted for 2 (FFLW) to 11 % (MFLW) of the phenotypic variation in the hybrid trials, showing that the three groups were not highly differentiated for earliness.
Association tests with SNPs from the Illumina chip (assumed to have a neutral effect on the traits of interest) were performed in TASSEL with FFLW for hybrids trials to assess which kinship and Q matrices corrected best for false positive associations (Supplementary material 6). The two models that corrected best for false positives were the model with PCA with ten axes and the K Loiselle matrix and the model including only the K Emma matrix.
Over the 18,711 association tests, after corrections with a FDR of 5 %, around 100 associations remained significant for each method (ASReml-R, Tassel and Emma, data not shown). As expected, ASReml-R and Tassel gave similar results. Pearson’s correlations between Emma and ASReml-R p values, and Emma and Tassel ones were 0.94 and 0.95, respectively, for all the associations, and 0.92 and 0.94 for the associations still significant after FDR correction.
Among the 100 associations from ASReml-R still significant after multiple-testing correction, 68 associations were found for polymorphisms located on 5 amplicons on the Vgt1 region (K42, K46, ZmRap2.7 and the two sequences including the mite and CGindel587 polymorphisms). Additional 32 associations corresponded to eight genes located in other regions.
In the Vgt1 region, association tests were performed on the whole panel and also separately on the public and Syngenta lines (Fig. 4). On the whole panel, K42, the two Vgt1 amplicons, ZmRap2.7 and K46 were significantly associated with EARHT, PHT, MFLW and FFLW. Over all the polymorphisms tested, the lowest p value was attributed to the association between the mite and FFLW (p value of 7.74 × 10−9) (Fig. 4; Table 3). Stronger associations were observed for per se trials than for hybrids trials, which can be explained by the higher heritability of the traits in the per se trials. No significant association was found within the public lines panel (Fig. 4). When considering the Syngenta lines only, the CGindel587 polymorphism was no longer significant but the mite and two polymorphisms from ZmRap2.7 (ZmRap2.7-6 and ZmRap2.7-8) remained associated with FFLW. The association tests performed on the 50 samples of 113 Syngenta lines, corresponding to the public lines panel size, were not significant (p values superior to 5 × 10−3).

The p values for association tests run in ASReml-R for female flowering time (FFLW) per se on bin 8.05. Black triangles are p values for the whole panel, gray diamonds for the public lines, and gray squares for the Syngenta lines
Analysis of Vgt1 region was complemented by multilocus analyses. The polymorphisms significantly associated with per se FFLW, jointly explained 6 % of the trait variation.
Considering Vgt1 itself, we noted that the mite effect on flowering time remained significant when tested with CGindel587 as a covariate, whereas the reverse was not true. When considering the haplotype formed by mite/CGindel587 polymorphisms, associations were not significant anymore with EARHT and PHT, and less significant for MFLW and FFLW than for the separate analysis of these polymorphisms. We noted that the CGindel587 was present in more than 80 % of the lines and the mite in 50 % of the lines of the panel. Every line carrying the mite carried as well the CGindel587, except for 2 % of them. Results, therefore, suggested that the main effect at Vgt1 is due to earliness contributed by a haplotype that can be tagged by either the mite only or the mite/CGindel587 combination. We also tested models including the mite in Vgt1 and each of the two ZmRap2 polymorphisms, ZmRap2.7-6 and ZmRap2.7-8, which showed significant associations when tested individually. In both cases, mite was strongly associated with flowering time related traits when tested with either ZmRap2 polymorphisms as covariate. Conversely, ZmRap2 polymorphisms were not associated with any trait anymore when tested with the mite as covariate. This suggested that the effect of ZmRap2.7 polymorphism on flowering time was mainly due to LD with the causal factor in Vgt1, which is more tightly tagged by the mite.
Concerning the 13 selected regions potentially involved in the variation for digestibility (Truntzler et al. 2010), significant associations with an FDR <5 × 10−2 were found for two amplicons from Cinnamoyl-coA reductase (CCR) on bin 1.07 with PHT and FFLW (p values of 6.88 × 10−5 and 2.41 × 10−4, respectively, see Table 3), for a homologous gene with Ricinus root phototropism protein on bin 1.08 with FFLW (4.17 × 10−6), for one copy of Hydroxycinnamoyl-CoA transferase (HCT) on bin 2.04 with PHT (p value of 9.74 × 10−4), for a copy of sucrose synthase with FFLW on bin 3.05 (p value of 1 × 10−4), for a copy of Caffeoyl-CoA 3-O-methyltransferase (CCoAOMT) with PHT on bin 6.01 (p value of 9.12 × 10−4), for a Lim1 transcription factor with FFLW in bin 6.01 (p value of 2.78 × 10−4), for the Cinnamyl alcohol dehydrogenase 2 (CAD2) on bin 7.02 with MFLW (2.94 × 10−5) and for 4-coumarate:coenzyme A ligase (4CL) on bin 9.04 with PHT (3.73 × 10−4).
Discussion
Gene diversity
Gene diversity for the studied polymorphisms was 0.29 on the whole panel, higher for the public lines (0.33) and lower for the Syngenta lines (0.23). These results are comparable to those reported for SNPs in other studies. Hamblin et al. (2007) found a diversity of 0.32 on a set of 259 maize lines from the USA that was representative of global diversity; Lu et al. (2009) found gene diversity from 0.27 to 0.34 on a set of 770 maize lines from six countries; Van Inghelandt et al. (2010) estimated a diversity of 0.32 in a set of 1,537 elite lines representing European and North American diversity. Yang et al. (2010) found a higher gene diversity value (0.39) in a set of 527 lines from very broad origins. This illustrates that gene diversity increases when broadening origins. The lower diversity of the subset of lines from Syngenta is probably due to the fact that elite inbred lines traced to a relatively narrow set of founders lines and have passed through intense selection pressure, as it is frequently the case in private elite breeding programs (Mikel 2008). Furthermore, the private inbred lines used here are restricted to a specific heterotic pool (dent here) and represented only part of the germplasm used by Syngenta in Europe. The gene diversity we observed for this material is similar to the one found by Van Inghelandt et al. (2010) for panels of iodent and stiff-stalk germplasms (0.23 in each case).
Diversity estimation based on POLseq data followed the same trend, although these gene/region sequences were heterogeneous in term of polymorphisms. It was not possible to highlight a pattern based on their physical position or the gene function.
In terms of neutrality test, Tajima’s D values computed in the Vgt1 region did not show any significant evidence of selection or population expansion for this chromosomal region.
Population structure and relatedness
We showed that the first level of structure obtained by STRUCTURE and Locus Miner (k = 2) mostly corresponded to a separation between Syngenta and public lines. This is consistent with the results of PCA analysis. Fifteen lines from the Syngenta lines (most of them are stiff stalk) are grouped with public lines. This isolated group of Syngenta lines has probably not been the principal source of Syngenta germplasm. Reciprocally five public lines are grouped with the Syngenta lines (all of them are iodent) and were probably used as (or were related to) founders of Syngenta lines. This differentiation between a representative pool of public lines and recent corresponding material from a company illustrates that breeding reshapes permanently population structure. Note that this level of stratification, supported by Evanno criterion, can be interpreted as the minimal number of groups that can be considered as independent and does not preclude significant subdivision of these groups (as discussed by Evoldir community (2008)). Indeed, although the evolution of likelihood was limited for further levels of population structure, Eigenstrat analysis suggested that approximately up to ten groups could be defined. Comparison of STRUCTURE runs for a same k value showed stable results until k = 6 and illustrated a subdivision of the Syngenta lines into sub-groups, globally in agreement with pedigree. Such a subdivision according to families of related lines was consistent with observations reported by Camus-Kulandaivelu et al. (2006). In particular, classification of the panel into three sub-groups was highly consistent with expert knowledge from Syngenta germplasm.
By using bootstrap procedures, we showed that 979 SNPs was enough to obtain an accurate estimation for the kinship matrices. It is in agreement with simulation of Yu et al. (2009), which showed that 200 SNPs was enough to estimate Kinship matrix. To date, molecular data used in maize for these purposes have mostly been SSR markers (Lübberstedt et al. 2005; Camus-Kulandaivelu et al. 2006; Yang et al. 2010). Van Inghelandt et al. (2010) compared population structure on 1,537 elite maize inbred lines with 359 SSR and 8,244 SNP markers. They showed that both marker types give same results regarding the population structure and gene diversity, although SSR gave more information. Nowadays with the easy access for SNPs arrays with several thousand of markers, it becomes easier to get as much information as SSR would give.
We showed that kinship matrix K Emma and K Loiselle were partially correlated. This difference for K Emma and K Loiselle coefficients can be interpreted by the fact that K Emma is an IBS allele-sharing matrix, whereas K Loiselle estimates a “relative kinship”, which can be defined as ratios of differences of probabilities of identity in state (Rousset 2002; Vekemans and Hardy 2004) and increases the weight of rare alleles in similarity estimation. We observed that the relationship between the two estimators diverged for the two main groups determined by STRUCTURE software (Fig. 2). Group2, mostly composed of Syngenta lines, displayed higher K Emma similarity levels than group1, which was mostly composed of public material. This is consistent with the lower diversity observed in Syngenta lines. Conversely, K Loiselle displayed the same range of variation for the two groups. This can be interpreted as the effect of the higher weight given to similarity for rare allele in K Loiselle. Indeed, group 2, which is both the most represented and the less diverse group, is expected to yield similarities for alleles that display higher frequencies (in the global population) and, therefore, receive less weight in the computation of K Loiselle.
LD magnitude
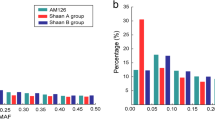
We compared LD in centromeric and telomeric regions but no particular pattern was observed, possibly because we did not have enough segments for comparing these two types of regions, or that the globally high LD magnitude at short distances within our material masked such effects. Patterns of r² appeared dependent on the MAF considered (see supplementary material 4). We observed higher LD for MAF >0.2, suggesting that LD tends to increase when both polymorphisms have balanced frequencies.
Major differences in LD were observed between the whole panel, the public and the Syngenta lines. Mean r 2 over 1 kb was 0.45, 0.40 and 0.60 for the whole panel, public and Syngenta lines respectively. The higher r² for Syngenta lines was consistent with the lower diversity observed for this material, and the higher LD levels for less diverse genetic pools reported by Rafalski (2002). This r 2 values were higher than in some previous studies (Tenaillon et al. 2001, Stich et al. 2005, Remington et al. 2001). For example, Yan et al. (2009) observed a rapid decline of r 2 in their panel since r 2 ranged from 0.16 to 0.24 (depending on gene) for distances reaching 2 kb in their panel of 632 breeding inbred lines. It was expected since these studies have been performed on public lines representing a broad diversity of maize germplasm. On the contrary, Syngenta lines had low diversity due to strong selection of founders and subsequent breeding materials, which caused a higher level of LD (Ching et al. 2002; Jung et al. 2004). This trend towards longer LD extent in groups displaying the highest relatedness and/or lowest diversity was observed as well by Van Inghelandt et al. (2011) and Yan et al. (2009).
Our experiment was not well adapted to global LD investigations in the 100 kb range but high values were nevertheless observed for polymorphisms within this range of distance. In the Vgt1 region, two polymorphisms of ZmRap2.7 were in stronger LD (r 2 > 0.72) with the mite than previously observed (Ducrocq et al. 2008). Mean r 2 for distances between 100 and 200 kb were 0.19. Ersoz et al. (2009) suggested that association mapping could be performed with r 2 values as low as 0.10 so that we can envisage performing GWA with one marker every 100 kb in our panel. This confirms observations from Belo et al. (2007) for similar elite materials.
Association studies
False discovery rate correction was used to correct for multiple tests. As discussed in Müller et al. (2011) the FDR is difficult to interpret when several tests are performed within the same gene. Moreover, this error rate does not control for the genome-wide type I error rate (GWER). In linkage analysis, GWER is often obtained from permutations (Churchill and Doerge 1994). This process is not possible in association mapping studies where one needs to consider both the structure and the pedigree relationships among the different lines. Recently, Müller et al. (2011) proposed a new method to estimate genome-wide error rate that fully takes into account population structure and relatedness through simulations. We hope that this method could be implemented soon in software commonly used for association mapping.
Associations with low p values were more observed with per se values than hybrids, for which no test passed a FDR correction. Previous studies were mainly performed on per se values for maize (Andersen et al. 2008; Chen et al. 2010; Yang et al. 2010). Trait heritabilities were lower in hybrids trials, which hampered the power of association mapping. Part of the explanation for these lower heritabilities may come from the use of a tester line that may have masked part of the genetic variability of our panel. The other explanation may be related to the experimental design itself that was different for the two types of trials. For hybrid data we had two locations only, with one showing very low heritability. Furthermore, we used unreplicated design for the hybrids trials, whereas two replicates of each line were set in the per se trials. This illustrated if needed the importance of high-quality phenotypic data in association genetics studies (Myles et al. 2009).
We compared different models to control for spurious association (false positive) due to population structure and relatedness (Supplementary material 6). We showed that the best model to control false positive rate is PCA10 + K Loiselle. This was in agreement with Zhao et al. (2007) and Zhu and Yu (2009), who showed that PCA performed similarly or better than STRUCTURE to control for spurious associations, even if it does not always reflect population structure, but may reflect family relatedness, long-range LD or assay artifacts (Price et al. 2006). Principal component analysis based approaches are computationally fast and thus a valuable solution to face very large datasets. This might explain why Eigensoft is frequently used for genome-wide association in many species as exemplified by loblolly pine (Eckert et al. 2010), cattle (Porto Neto et al. 2010), etc. New methods have been proposed recently that were not investigated in our study. Zhu and Yu (2009) compared nonmetric multidimensional scaling (nMDS) and PCA to control population structure in genome. They showed that nMDS maintained a lower false-positive rate than using PCA as Q matrix. Jombart et al. (2010) used discriminant analysis of principal components (DAPC), a multivariate method designed to identify clusters of genetically related individuals to account for population structure. Through simulations, they showed that this method performed better than STRUCTURE at characterizing population subdivision. Our dataset was still manageable with the common software but these methods deserve consideration for bigger datasets. Evaluation of population structure remains a central issue, as illustrated by Mezmouk et al. (2011), where different population structures used as cofactors in association tests gave different results in terms of polymorphisms associated with the trait of interest.
Among the most significant polymorphisms associated with flowering time in our study, we highlighted the Vgt1 region localized on bin 8.05. It is probably one of the flowering time QTL that is the more documented in the literature. In their extensive and very complete review on flowering time in maize, including the recent nested association mapping design of Yu et al. (2008), Salvi et al. (2009) showed that bin 8.05 was within the regions with the highest number of QTL detected in different studies. The two polymorphisms mite and CGindel587 as well as the gene Zmrap2.7, found strongly associated with flowering time traits in our study, were previously shown to be involved in flowering time adaptation for maize by Salvi et al. (2007), and their major effects were confirmed by Ducrocq et al. (2008). However, compared to Ducrocq et al. (2008), the mite appeared more significant in our study than CGindel587. Furthermore, Ducrocq et al. (2008) tested the associations with the haplotype mite/CGindel587 and found a higher p value than with each single polymorphism independently, which was not the case in our study. The mite effect still remained significant on flowering time when tested with CGindel587 and ZmRap2.7 polymorphisms separately, suggesting that the mite polymorphism is the causal factor or is in higher LD with the causal factor than CGindel587. This difference in results is probably related to the higher frequency of the CGindel587–mite absence haplotype in the Syngenta pool compared to the public pool, which is consistent with the specificity of this haplotype in the iodent group (Ducrocq et al. 2008), well represented in the Syngenta pool. Compared to Ducrocq et al. (2008), we also had two significant polymorphisms associated in ZmRap2.7, one in K42 and one in K46. The polymorphisms from ZmRap2.7 captured the effect of the mite on flowering time variation, which was not the case in Ducrocq et al. (2008). This shows that for LD mapping, the density of markers needed in this dent panel is probably lower than the density that would be needed in the panel used by Ducrocq et al. (2008).
It can be noted that we observed more significant associations in this region when considering the Syngenta inbred lines only compared to the public inbred lines only. Based on the results of the association tests performed on the 50 samples of 113 Syngenta lines, the difference of results originated mainly from differences in their sample size, showing that 113 lines give limited power for association tests (Yu et al. 2006) compared to the size of our whole panel (316 lines), and not from a difference of effect within each subset.
Association mapping also revealed significant associations for genes that have not been highlighted in previous studies for their role in flowering time variation but in digestibility variation (Barrière et al. 2007). Sibout et al. (2008) suggested that flowering induction is the condition for xylem expansion in hypocotyl and root secondary growth in Arabidopsis thaliana. They showed as well that flowering time and lignin biosynthesis were linked, as major QTL for fiber and xylem expansion were correlated with flowering time QTL.
Further analyses are needed to validate the biological roles of the different genes associated to earliness related traits in our study.
Conclusion and perspectives
Joint analysis of lines from public and private origins within a same genetic group illustrated a major evolution of elite materials, with lower gene diversity accompanied by a higher extent of LD, with average r² values of approximately 0.2 between polymorphisms located 200 kb apart. As a consequence, fewer markers will be needed to conduct GWA for this panel and its elite component than for the broader dent gene pool available to public research. Although this necessitates confirmation, this suggests that the use of 50 kb SNP (approximately one SNP marker every 60 kb) deserves consideration for a first GWA study of this panel. This density seems appropriate to detect QTL explaining at least 10 % of the phenotypic variance (Van Inghelandt et al. 2011). For the identification of QTL with smaller effect, higher density genotyping approaches such as sequencing (Elshire et al. 2011) (genotyping by sequencing) might be necessary to identify polymorphisms associated with trait variation without any prior information.
Even if flowering time can be considered as being a simpler trait than yield, several results highlighted that even if some loci such as Vgt1 seem to play an important role in this trait variation, the number of loci involved certainly exceeds 50 (Chardon et al. 2004; Salvi et al. 2009; Buckler et al. 2009). Genome-wide predictions (Meuwissen et al. 2001; Bernardo and Yu 2007), which use all markers as predictors of performance rather than trying to identify specific loci significantly associated with a trait can be considered as a complementary approach to association mapping to deal with QTL with moderate effects. Our panel, which presents a high level of LD and includes both historical inbred lines and elite material, might be well suited for this kind of approach if the objective is to valorize results in breeding programs.
References
Andersen J, Zein I, Wenzel G, Darnhofer B, Eder J, Ouzunova M, Luebberstedt T (2008) Characterization of phenylpropanoid pathway genes within European Maize (Zea mays L.) inbreds. BMC Plant Biol 8(1):2
Aranzana MJ, Kim S, Zhao K, Bakker E, Horton M, Jakob K (2005) Genome-wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes. PLoS Genet 1(5):e60
Atwell S, Huang YS, Vilhjalmsson BJ, Willems G, Horton M, Li Y, Meng D, Platt A, Tarone AM, Hu TT, Jiang R, Muliyati NW, Zhang X, Amer MA, Baxter I, Brachi B, Chory J, Dean C, Debieu M, de Meaux J, Ecker JR, Faure N, Kniskern JM, Jones JDG, Michael T, Nemri A, Roux F, Salt DE, Tang C, Todesco M, Traw MB, Weigel D, Marjoram P, Borevitz JO, Bergelson J, Nordborg M (2010) Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 465(7298):627–631
Bar-Hen A, Charcosset A, Bourgoin M, Guiard J (1995) Relationship between genetic markers and morphological traits in a Maize inbred lines collection. Euphytica 84(2):145–154
Barrière Y, Riboulet C, Méchin V, Maltese S, Pichon M, Cardinal A, Lapierre C, Martinant JP (2007) Genetics and genomics of lignification in grass cell walls based on Maize as model species. Genes Genom Genomics 1(2):133–156
Beló A, Zheng P, Luck S, Shen B, Meyer DJ, Li B, Tingey S, Rafalski A (2007) Whole genome scan detects an allelic variant of fad2 associated with increased oleic acid levels in Maize. Mol Genet Genomics 279(1):1–10
Bernardo R, Yu J (2007) Prospects for genome-wide selection for quantitative traits in Maize. Crop Sci 47(3):1082–1090
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ram-doss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23(19):2633–2635
Buckler ES, Holland JB, Bradbury PJ, Acharya CB, Brown PJ, Browne C, Ersoz E, Flint-Garcia S, Garcia A, Glaubitz JC, Goodman MM, Harjes C, Guill K, Kroon DE, Larsson S, Lepak NK, Li H, Mitchell SE, Pressoir G, Peiffer JA, Rosas MO, Rocheford TR, Cinta Romay M, Romero S, Salvo S, Sanchez Villeda H, da Silva HS, Sun Q, Tian F, Upadyayula N, Ware D, Yates H, Yu J, Zhang Z, Kresovich S, McMullen MD (2009) The genetic architecture of Maize flowering time. Science 325(5941):714–718
Butler D, Cullis BR, Gilmour AR, Gogel BJ (2007) ASReml-R estimates variance components under a general linear mixed model by residual maximum likelihood (REML). Analysis of mixed models for S language environments. DPI and F Publications, Queensland
Camus-Kulandaivelu L, Veyrieras JB, Madur D, Combes V, Fourmann M, Barraud S, Dubreuil P, Gouesnard B, Manicacci D, Charcosset A (2006) Maize adaptation to temperate climate: relationship between population structure and polymorphism in the dwarf 8 gene. Genetics 172(4):2449–2463
Chardon F, Virlon B, Moreau L, Falque M, Joets J, Decousset L, Murigneux A, Charcosset A (2004) Genetic architecture of flowering time in Maize as inferred from quantitative trait loci meta-analysis and synteny conservation with the rice genome. Genetics 168(4):2169–2185
Chen Y, Zein I, Brenner EA, Andersen JR, Landbeck M, Ouzunova M, Lübberstedt T (2010) Polymorphisms in monolignol biosynthetic genes are associated with biomass yield and agronomic traits in European Maize (Zea mays L.). BMC Plant Biol 10(1):12
Ching A, Caldwell KS, Jung M, Dolan M, Smith OSH, Tingey S, Morgante M, Rafalski A (2002) SNP frequency, haplotype structure and linkage disequilibrium in elite Maize inbred lines. BMC Genet 3(1):19
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138(3):963–971
Dray S, Dufour AB (2007) The ade4 package: implementing the duality diagram for ecologists. J Stat Softw 22(4):1–20
Ducrocq S, Madur D, Veyrieras JB, Camus-Kulandaivelu L, Kloiber-Maitz M, Presterl T, Ouzunova M, Manicacci D, Charcosset A (2008) Key impact of Vgt1 on flowering time adaptation in Maize: evidence from association mapping and ecogeographical information. Genetics 178(4):2433–2437
Eckert AJ, van Heerwaarden J, Wegrzyn JL, Nelson CD, Ross-Ibarra J, Gonzalez-Martinez SC, Neale DB (2010) Patterns of population structure and environmental associations to aridity across the range of loblolly pine (Pinus taeda L., Pinaceae). Genetics 185(3):969–982
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6(5):e19379
Ersoz ES, Yu JM, Buckler ES (2009) Applications of linkage disequilibrium and association mapping in Maize. In: A.L.Kriz BAL (ed) Biotechnology in agriculture and forestry-molecular genetic approaches to Maize improvement, vol 63. Springer-Verlag, Berlin Heidelberg
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620
Evoldir community (2008) Evoldir-month in review. http://evol.mcmaster.ca/evoldir.html
Flint-Garcia SA, Thornsberry JM, Buckler ES (2003) Structure of linkage disequilibrium in plants. Annu Rev Plant Biol 54(1):357–374
Guillaumie S, San-Clemente H, Deswarte C, Martinez Y, Lapierre C, Murigneux A, Barrière Y, Pichon M, Goffner D (2007) MAIZEWALL database and developmental gene expression profiling of cell wall biosynthesis and assembly in Maize. Plant Physiol 143(1):339–363
Gunderson KL, Kruglyak S, Graige MS, Garcia F, Kermani BG, Zhao C, Che D, Dickinson T, Wickham E, Bierle J, Doucet D, Milewski M, Yang R, Siegmund C, Haas J, Zhou L, Oliphant A, Fan J-B, Barnard S, Chee MS (2004) Decoding randomly ordered DNA arrays. Genome Res 14(5):870–877
Hamblin MT, Warburton ML, Buckler ES (2007) Empirical comparison of simple sequence repeats and single nucleotide polymorphisms in assessment of Maize diversity and relatedness. PLoS ONE 2(12):e1367
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2(4):618–620
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC genomics 11(1):94
Jung M, Ching A, Bhattramakki D, Dolan M, Tingey S, Morgante M, Rafalski A (2004) Linkage disequilibrium and sequence diversity in a 500-kbp region around the adh1 locus in elite Maize germplasm. Theor Appl Genet 109(4):681–689
Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, Eskin E (2008) Efficient control of population structure in model organism association mapping. Genetics 178(3):1709–1723
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21(9):2128–2129
Loiselle BA, Sork VL, Nason J, Graham C (1995) Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82(11):1420–1425
Lorenz AJ, Coors JG, Hansey CN, Kaeppler SM, De Leon N (2010) Genetic analysis of cell wall traits relevant to cellulosic ethanol production in Maize (Zea mays L.). Crop Sci 50(3):842–852
Lu Y, Yan J, Guimarães C, Taba S, Hao Z, Gao S, Chen S, Li J, Zhang S, Vivek B, Magorokosho C, Mugo S, Makumbi D, Parentoni S, Shah T, Rong T, Crouch J, Xu Y (2009) Molecular characterization of global Maize breeding germplasm based on genome-wide single nucleotide polymorphisms. Theor Appl Genet 120(1):93–115
Lübberstedt T, Zein I, Andersen J, Wenzel G, Krützfeldt B, Eder J, Ouzunova M, Chun S (2005) Development and application of functional markers in Maize. Euphytica 146(1):101–108
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27(2):209–220
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157(4):1819–1829
Mezmouk S, Dubreuil P, Bosio M, Décousset L, Charcosset A, Praud S, Mangin B (2011) Effect of population structure corrections on the results of association mapping tests in complex Maize diversity panels. Theor Appl Genet 122(6):1149–1160
Mikel MA (2008) Genetic diversity and improvement of contemporary proprietary North American dent corn. Crop Sci 48(5):1686–1695
Mikel MA, Dudley JW (2006) Evolution of North American dent corn from public to proprietary germplasm. Crop Sci 46(3):1193–1205
Müller BU, Stich B, Piepho HP (2011) A general method for controlling the genome-wide type I error rate in linkage and association mapping experiments in plants. Heredity 106(5):825–831
Myles S, Peiffer JA, Brown JB, Ersoz ES, Zhang Z, Costich DE, Buckler ES (2009) Association mapping: critical considerations shift from genotyping to experimental design. Plant Cell 21(8):2194–2202
Patterson N, Price AL, Reich D (2006) Population structure and Eigen analysis. PLoS Genet 2(12):e190
Porto Neto LR, Bunch RJ, Harrison BE, Barendse W (2010) DNA variation in the gene ELTD1 is associated with tick burden in cattle. Anim Genet 42(1):50–55
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38(8):904–909
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959
R Development Core Team (2011) R: a language and environment for statistical computing. 3 R Foundation for Statistical Computing
Rafalski JA (2002) Novel genetic mapping tools in plants: SNPs and LD-based approaches. Plant Sci 162(3):329–333
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, Kresovich S, Goodman MM, Buckler ES (2001) Structure of linkage disequilibrium and phenotypic associations in the Maize genome. PNAS 98(20):11479–11484
Ritland K (1996) Estimators for pairwise relatedness and individual inbreeding coefficients. Genet Res (Camb) 67:175–185
Rousset F (2002) Inbreeding and relatedness coefficients: what do they measure? Heredity 88(5):371–380
Sakamato Y, Kitagawa G (1987) Akaike information criterion statistics. Kluwer Academic Publishers, Norwell
Salvi S, Sponza G, Morgante M, Tomes D, Niu X, Fengler KA, Meeley R, Ananiev EV, Svitashev S, Bruggemann E, Li B, Hainey CF, Radovic S, Zaina G, Rafalski JA, Tingey SV, Miao GH, Phillips RL, Tuberosa R (2007) Conserved noncoding genomic sequences associated with a flowering-time quantitative trait locus in Maize. PNAS 104(27):11376–11381
Salvi S, Castelletti S, Tuberosa R (2009) An updated consensus map for flowering time QTLs in Maize. Maydica 54(4):501–512
SAS (1989) SAS/STAT User’s guide. Version 6, vol 2, 4th edn. SAS Institute Inc., Cary
Sibout R, Plantegenet S, Hardtke CS (2008) Flowering as a condition for xylem expansion in Arabidopsis hypocotyl and root. Curr Biol 18(6):458–463
Staden R, Beal KF, Bonfield JK (1998) The Staden Package. In: Krawetz SMaSA (ed) Computer methods in molecular biology, bioinformatics methods and protocols. vol 132. The Humana Press Inc., Totowa, 115–130
Stich B, Melchinger AE, Frisch M, Maurer HP, Heckenberger M, Reif JC (2005) Linkage disequilibrium in European elite Maize germplasm investigated with SSRs. Theor Appl Genet 111(4):723–730
Storey JD, Tibshirani R (2003) Statistical significance for genome-wide experiments. PNAS 100:9440–9445
Stracke S, Haseneyer G, Veyrieras JB, Geiger HH, Sauer S, Graner A, Piepho HP (2009) Association mapping reveals gene action and interactions in the determination of Flowering time in barley. Theor Appl Genet 118(2):259–273
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tenaillon MI, Sawkins MC, Long AD, Gaut RL, Doebley JF, Gaut BS (2001) Patterns of DNA sequence polymorphism along chromosome 1 of Maize (Zea mays ssp. mays L.). PNAS 98(16):9161–9166
Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler IE (2001) Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet 28:286–289
Truntzler M, Barrière Y, Sawkins M, Lespinasse D, Betran J, Charcosset A, Moreau L (2010) Meta-analysis of QTL involved in silage quality of Maize and comparison with the position of candidate genes. Theor Appl Genet 121(8):1465–1482
Van Inghelandt D, Melchinger AE, Lebreton C, Stich B (2010) Population structure and genetic diversity in a commercial Maize breeding program assessed with SSR and SNP markers. Theor Appl Genet 120(7):1289–1299
Van Inghelandt D, Reif J, Dhillon B, Flament P, Melchinger A (2011) Extent and genome-wide distribution of linkage disequilibrium in commercial Maize germplasm. Theor Appl Genet 123(1):11–20
Vekemans X, Hardy OJ (2004) New insights from fine-scale spatial genetic structure analyses in plant populations. Mol Ecol 13(4):921–935
Veyrieras JB, Camus-Kulandaivelu L, Charcosset A (2006) Etude du déterminisme génétique de caractères quantitatifs chez les végétaux: Méta-analyse de QTL et études d’association. PhD thesis, Agroparistech, Paris
Wilson LM, Whitt SR, Ibanez AM, Rocheford TR, Goodman MM, Buckler IE (2004) Dissection of Maize kernel composition and starch production by candidate gene association. Plant Cell 16(10):2719–2733
Yan J, Shah T, Warburton ML, Buckler ES, McMullen MD, Crouch J (2009) Genetic characterization and linkage disequilibrium estimation of a global Maize collection using SNP markers. PLoS ONE 4(12):e8451
Yang X, Gao S, Xu S, Zhang Z, Prasanna B, Li L, Li J, Yan J (2010) Characterization of a global germplasm collection and its potential utilization for analysis of complex quantitative traits in Maize. Mol Breed. doi:10.1007/s11032-010-9500-7
Yu JM, Buckler ES (2006) Genetic association mapping and genome organization of Maize. Curr Opin Biotechnol 17(2):155–160
Yu JM, Pressoir G, Briggs WH, Bi IV, Yamasaki M, Doebley J, McMullen MD, Gaut BS, Nielsen D, Holland JB, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38(2):203–208
Yu J, Holland JB, McMullen MD, Buckler ES (2008) Genetic design and statistical power of nested association mapping in Maize. Genetics 178(1):539–551
Yu J, Zhang J, Zhu C, Tabanao D, Pressoir G, Tuinstra MR, Kresovich S, Todhunter RJ, Buckler ES (2009) Simulation appraisal of the adequacy of number of background markers for relationship estimation in association mapping. The Plant Genome 2:63–77
Zhao K, Aranzana MJ, Kim S, Lister C, Shindo C, Tang C (2007) An Arabidopsis example of association mapping in structured samples. PLoS Genet 3(1):e4
Zhu C, Yu JM (2009) Nonmetric multidimensional scaling corrects for population structure in association mapping with different sample types. Genetics 182(3):875–888
Acknowledgments
We thank Pascal Delage, Philippe Jamin, Denis Coubriche, Sophie Pin, Dominique Denoué and Christoph Mainka for the set up of the trials, traits measurements and harvest as well as Jean-Paul Muller for insightful discussions on maize germplasm and phenotypic analyses. Part of this work was financed by Syngenta Seeds; we thank them as well for the phenotypic and genetic material. We thank the University of Oslo Bioportal for providing CPU time. Part of this work was carried out by using the resources of the Computational Biology Service Unit from Cornell University, which is partially funded by Microsoft Corporation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Bohn.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Truntzler, M., Ranc, N., Sawkins, M.C. et al. Diversity and linkage disequilibrium features in a composite public/private dent maize panel: consequences for association genetics as evaluated from a case study using flowering time. Theor Appl Genet 125, 731–747 (2012). https://doi.org/10.1007/s00122-012-1866-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-012-1866-y