
Abstract
Sets of polymorphic expressed sequence tag–simple sequence repeat (EST-SSR) markers from the rubber tree (Hevea brasiliensis) have been published by many researchers, but none has been specifically developed to study latex and wood yield traits. In this study, a total 10,321 rubber tree EST sequences, generated from suppression subtractive hybridization-cDNA libraries of bark and latex of high- and low-yielding clones, were used as sources for SSR searching. A total of 432 EST-SSR loci were identified and it was possible to design primer pairs for a subset of 298 EST-SSRs. The highest proportion of EST-SSRs was represented by dinucleotide repeats (46.6 %), followed by trinucleotide repeats (44.3 %). Based on BLASTX analysis, 234 ESTs (80 %) showed similarity to genes in NCBI databases and could be divided into 120 putative proteins with known function and 114 unknown proteins. To enhance the resolution of an existing linkage map from previous work on a rubber tree RRIM600 × PB217 population, 69 EST-SSR markers from the above set were tested to be integrated into the reference genetic map. The enriched map of 18 linkage groups spanned 2054.2 cM in length, showed an average genetic distance of 4.3 cM between adjacent markers, and included 63 new EST-SSR markers. The enhanced map from this study provides a basis for comparative mapping using PCR-based markers and identification of expressed genes possibly affecting important traits of interest.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Natural rubber is an indispensible commodity that is used in manufacturing worldwide. The rubber tree, Hevea brasiliensis (Willd. ex Adr. de Juss) Muell. Arg, a member of the Euphorbiaceae family, the major commercial source of natural rubber, has pivotal roles of production and sustainability in the world economy. Rubber trees start yielding latex after 5–7 years of maturity and have a productive lifespan of 25–30 years (Polhamus 1962). According to the International Rubber Study Group (year 2010), the demand for rubber (natural and synthetic) has steadily risen over the past century and is expected to continue to increase. In addition to natural rubber, rubber trees are used as a source of timber once their latex productivity is no longer economically viable. Rubber wood has become a major timber export of Southeast Asia, as its natural light color and excellent physical properties make it suitable for flooring and household furniture. Owing to the value of this product, several superior latex-timber clones have been developed. The biological characteristics of the rubber tree (i.e. long life-cycle and heterozygous genetic background) and its relatively poorly understood genetics are obstacles to conventional breeding of new varieties. Marker-assisted selection can improve the efficiency of breeding by enabling the direct selection of targeted genotypes. Analysis of genetic linkage among markers and identification of the genetic locations of desirable phenotypes would further improve the selection accuracy. The rubber tree genome includes 18 pairs of chromosomes and is estimated to be ~2.15 Gb in size. About 12.7 % of the 68,955 genes have been identified as associated with rubber biosynthesis, rubber wood formation, disease resistance and allergenicity (Rahman et al. 2013). Recently, we successfully generated Hevea genomic libraries, representing 2.42 haploid genome equivalents (Kanjanawattanawong et al. 2014). The increasing significance of natural rubber has motivated many researchers to unravel the biology and physiology of plant growth as well as the mechanism regulating key product formation.
The aim of rubber tree breeding is to provide a genetically superior planting material with high yield of latex and wood. The development of high-yielding rubber clones is based on conventional breeding which is time-consuming and expensive. Other technologies have become essential tools for increasing the efficiency of rubber breeding. Molecular markers are widely used to track loci and genome regions. Among the various classes of existing markers, microsatellites or simple sequence repeats (SSRs) are the markers of choice because of their properties (Kujur et al. 2013). Microsatellite are short tandem repeat motifs (usually of 1–6 base-pairs) which are distributed throughout the genomes, both in coding and non-coding regions, of prokaryotes and eukaryotes. Due to their co-dominant nature, high polymorphism, reproducibility and transferability across species, SSRs have become effective markers in genetic analysis, population genetic analysis, genetic mapping and marker-assisted selection (MAS) of many plant and animal species. However, the widespread use of these markers is limited by the time and cost involved in their development. For Hevea, microsatellite markers are a very valuable tool for clonal identification and speeding up of the breeding scheme. To date, gene-based markers are more useful in genetic study and one of the most available sources of SSRs is expressed sequence tags (ESTs). ESTs are single-pass reads of around 200–800 base-pairs derived from cDNA clones (Parkinson and Blaxter 2009). The ESTs provide a valuable resource for the identification and development of gene-associated SSR markers linked with agronomically important traits.
Genetic molecular markers are increasingly applied for the identification and authentication of Hevea clones and genotyping of segregating populations (Lekawipat et al. 2003). A large number of rubber tree ESTs have been deposited in various public sequence databases and used for the development of SSR and EST-SSR markers (Bindu et al. 2004; Feng et al. 2009; Triwitayakorn et al. 2011; Li et al. 2012; Cubry et al. 2014). The aim of this study was to develop a new set of functional EST-SSRs from the Hevea EST database, generated from suppression subtractive hybridization (SSH)-cDNA libraries, prepared from bark (Mangpung et al. 2010) and latex (Towaranonte et al. 2010) of high- and low-yielding clones of rubber trees. The potential SSR loci were identified and used to enhance the resolution of an existing linkage map from previous work on a rubber tree RRIM600 × PB217 population. The rubber clone RRIM600 has a high metabolic activity and low level of sucrose in the laticiferous vessels, whereas PB217 has low metabolic activity and a high level of sucrose. The enhanced map from this study provides a basis for comparative mapping using PCR-based markers and identification of expressed genes possibly affecting important traits of interest. The new set of EST-SSR markers developed in this study should facilitate genetic mapping, genetic diversity study and MAS in rubber tree breeding programs.
Materials and methods
Plant material and DNA extraction
A full-sib progeny (F1 progeny) was created at Chachoengsao Rubber Research Center, Thailand in 2000 by crossing highly heterozygous parents (RRIM600 × PB217) using the hand pollination technique. The parents were chosen based on their different characteristics. The rubber clone RRIM600 has a high metabolic activity and low level of sucrose in the laticiferous vessels, whereas PB217 has low metabolic activity and a high level of sucrose. One hundred ninety-six progenies were used in this study. Total genomic DNA of the population RRIM600 × PB217 was isolated from young leaves using the modified CTAB method of Doyle and Doyle (1987). In brief, young leaves from each sample were ground to a fine powder in liquid nitrogen; 200 mg of DNA sample was then mixed with 500 μl CTAB extraction buffer (2 % hexadecyltrimethyl-ammonium bromide (CTAB), 20 mM EDTA, 100 mM Tris HCL pH 8.0, 1.4 M NaCl). Samples were incubated at 65 °C for 30 min followed by extraction with 500 μl chloroform/isoamyl (24:1). The samples were then centrifuged at 10,000 rpm. The upper aqueous phase was transferred to a new tube. DNA was precipitated by adding 1 volume of cold isopropanol with gentle mixing until DNA strands were observed. The precipitate was washed with cold 70 % ethanol at room temperature. The DNA pellet was then dried and suspended in 150 μl TE buffer (10 mM Tris HCl and 1 mM EDTA, pH 8.0). DNA concentration was determined by spectrophotometry and then diluted to a final concentration of 20 ng/μl. DNA samples were kept at −20 °C until used for EST-SSR analysis.
EST-SSR marker development
The Hevea EST database was obtained from various H. brasiliensis bark and latex suppression subtractive hybridization (SSH) libraries generated at Mahidol University, Thailand in collaboration with Institut de Recherche pour le Développement (IRD), Montpellier, France. Without stimulation, rubber tree clones PB260 and PB217, which are high- and low-yielding clones, respectively, were used for SSH. The inner bark and latex samples were collected for RNA extraction and cDNA synthesis and then subjected to SSH. Two inner bark and two latex subtracted-cDNA libraries (high-yielding vs low-yielding) were analyzed and the differentially expressed EST sequences obtained were submitted to the MU-IRD Hevea EST database (Mangpung et al. 2010; Towaranonte et al. 2010).
The 10,321 Hevea ESTs were analyzed with the Expressed Sequence Tag Treatment and Investigation Kit (ESTtik) from the server in CIRAD pipeline (http://esttik.cirad.fr/cgi-bin/SSR_server.cgi) in order to search for SSR regions and then to design primers. ESTtik is a tool dedicated to the analysis and annotation of cDNA raw data. A specific information system of ESTtik has been constructed to process, store and manage this EST collection, allowing the user to query a database. This tool was optimized for the investigation of data related to tropical crops and combines an automatic pipeline, a database and web interfaces to consult and download data. ESTtik is part of the SouthGreen Bioinformatics platform and can be directly accessed using this link: http://esttik.cirad.fr. The search was restricted to perfect dinucleotide motifs with a minimum of six repeat units, and tri-, tetra-, penta- and hexanucleotide motifs with a minimum of five repeat units. Primers were designed based on flanking regions of the SSRs and allowed to generate PCR products of sizes ranging from 100 to 300 bp in length, GC content of 40–60 % and annealing temperature (Ta) of 50–60 °C.
EST-SSR amplification
All EST-SSR primer pairs were evaluated for polymorphism with ten progeny samples and the parental samples. The informative primers were then used to genotype 196 F1 progenies. PCR amplified fragments were evaluated by conventional and automatic methods. The conventional method was performed in 10-µl reaction volumes containing approximately 20 ng of sample DNA, 10× PCR buffer, 1.5 mM MgCl2, 0.2 µM of forward and reverse primers, 200 µM of dNTPs and 1 U of Taq polymerase. All amplification were carried out in a DNA Engine® Thermal Cycler as follows: 5 min at 94 °C, followed by 35 cycles of 30 s at 94 °C, 30 s at 45, 50 or 58 °C according to the optimal temperature for each primer, and 45 s at 72 °C, with a final extension time of 5 min at 72 °C. Amplified products were resolved using 5 % polyacrylamide gel and silver staining. The automatic method was performed in a 10-µl reaction volume containing approximately 5 ng of template DNA, 10× PCR buffer, 50 mM MgCl2, 10 µM of forward-M13 primer, 10 µM of reverse primer, 10 µM of M13 marker, 2 mM of dNTPs and 2 U of Taq polymerase. All amplifications were carried out in Eppendorf Mastercycler® ep384. The conditions for amplification were 4 min at 94 °C, followed by 10 cycles of 45 s at 94 °C, 1 min at 55 °C and 1.15 min at 72 °C, then 25 cycles of 45 s at 94 °C, 1 min at 50 °C and 1 min at 72 °C, with a final extension time of 30 min at 72 °C. The PCR products were analyzed using a capillary gel electrophoresis system (3500xL Genetic Analyzers, Applied Biosystems). GeneMapper® software v4.1 was used to score all SSR alleles.
Gene identity of EST-SSR markers
To predict the functions of ESTs associated with EST-SSR polymorphic markers in this study, EST sequences were searched against protein sequences in NCBI databases using the BLASTX algorithm (http://www.ncbi.nlm.nih.gov/BLAST) software tool (Altschul et al. 1997).
Data analysis and map construction
All EST-SSR markers were screened against the parental samples and ten progeny samples to identify polymorphic markers. All informative primers were genotyped in the mapping population. The genotyping data was scored according to the crossbreeding population type or CP option type code as described in JoinMap® 4.0 manual (Van Ooijen 2006). All markers (AFLP, SSR and EST-SSR) were assigned to linkage groups applying the independence logarithm of odds (LOD) parameter with LOD threshold values ranging from 2.0 to 10.0. Linkage tests were carried out at a LOD threshold of 5.0. The Mendelian segregation ratio of all markers was evaluated using the chi-squared (χ 2) test and distorted markers (P ≤ 0.1) were excluded. The mapping parameters were set with a recombination threshold of 0.5, a jump threshold of 5.0 and a minimum LOD score threshold of 1.0. The Kosambi mapping function was used to determine map distance based on recombination frequencies of 0.5.
Results
Type and frequency of EST-SSRs
The SSR search server in the CIRAD pipeline (http://esttik.cirad.fr/cgi-bin/SSR_server.cgi) was used for the identification of SSRs in EST sequences. The total of 10,321 ESTs from MU-IRD Hevea EST database was assembled into 9721 good quality ESTs, of which 4733 were singletons and 4988 were redundant sequences. Six hundred sequences were excluded from the study due to their poor quality. These available ESTs were clustered in order to reduce the redundancy as well as to develop consensus sequences.
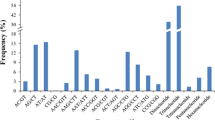
All EST-SSRs were classified into various types depending on the number of repeat units and the arrangement of nucleotides per repeat unit. A total of 298 EST-SSRs were identified, of which 282 (94.63 %) were perfect repeats, 12 (4.03 %) were imperfect repeats with one or more base interruptions in the sequence of repeats, and four (1.34 %) were compound repeats (Supplementary Table S1 and Fig. 1). The perfect repeat types could be classified into five types of repeat motifs: di-, tri-, tetra-, penta- and hexanucleotides. The most abundant microsatellite repeats were di- and trinucleotide repeats (139 and 132 SSRs, respectively), as shown in Fig. 1. The motif AG/TC was the most common motif among dinucleotide repeats (DNRs), accounting for 38.7 %, followed by GA/CT (32.2 %), AT/TA (19.6 %), AC/TG (5 %) and GT/CA (4.5 %). For trinucleotide repeats (TNRs), the three most abundant repeat sequences were AAG/TTC (14.9 %), AGA/TCT (11.9 %) and CTT/GAA (10.3 %).

Frequency distribution of EST-SSR based on motif sequence types
Gene identity of EST-SSR markers
Based on BLASTX analysis of these EST-SSRs, only 234 ESTs (80 %) showed similarity to the information on NCBI databases and could be divided into 120 (51.3 %) putative proteins with known function and 114 (48.7 %) unknown proteins with unknown function. The putative proteins with known function of EST-SSRs could be categorized into eight broad functional types: cell organization/cycle, cellular metabolism/pathway regulation, energy production and conversion, gene expression/chromatin, protein, cellular communication and signaling, stress-defense response and transport (Fig. 2). The largest group was allocated to gene expression/chromatin process (14 %) and the categories of energy production and conversion occupied the smallest proportion (1 %).

Distribution of ESTs by broad functional categories
EST-SSR polymorphism analysis
To identify sufficient number of polymorphic markers between parents is critical for constructing a linkage map. Out of 298 microsatellite markers evaluated, 211 primer pairs successfully produced amplification products and 87 primers failed to amplify any amplicons. Of 211 primer pairs, 69 primers (32.7 %) were polymorphic, 67 primers (31.8 %) were monomorphic, 4 primers (1.9 %) were non-segregated between parents and 71 (33.6 %) showed non-specific amplification or complex amplification patterns. Sixty-nine primers (Supplementary Table S2) were polymorphic within the one or both parent mapping population. These markers were added to a previously produced genetic map (Prapan et al. 2006).
Linkage analysis and map construction
Sixty-nine primer pairs were genotyped with 196 individuals of the F1 progeny. The genotypic data matrix was treated based on the CP model in the JoinMap® 4.0 program. The CP model is specific to a population created from a cross between two heterozygous diploid parents (Van Ooijen 2006). Sixty-nine EST-SSRs showed segregation of 70 loci suitable for mapping. It should be noted that there was one primer pair (Hbe37) in which the amplification of two loci was confirmed. Both of them were segregated, so they were labeled Hbe 37-1 and 37-2. Hbe 37-1 was on linkage group (LG) 5 while Hbe 37-2 was on LG10. The chi-squared test revealed different patterns of segregation among the 70 EST-SSR loci. Forty-three loci exhibited a ratio of 1:1 type for markers segregating in the female or male parent (ab × aa or aa × ab), 29 of them segregating in the female parent and 14 in the male parent. The 1:2:1 type segregation, with the same two alleles present in both parent (ab × ab), was observed in six loci. The remaining 21 loci showed the most informative type of segregation pattern (1:1:1:1), with 20 loci showing segregation in both parents with three alleles (ab × ac or ef × eg) and only one locus segregating with four alleles (ab × cd) (Supplementary Table S3).
The first genetic map of Hevea spp. (2n = 36) was presented in Lespinasse et al. (2000). It was created based on the analysis of a population from an interspecific cross between PB260, a H. brasiliensis cultivated clone, and RO38, a H. brasiliensis × H. benthamiana interspecific hybrid clone. In 2006, Prapan et al. constructed the genetic map of Hevea by using the population RRIM600 × PB217. The map included 18 LGs with 243 genomic SSR and 184 AFLP markers. In this study, 70 EST-SSR loci were produced and added to the above-mentioned Hevea reference genetic map (Prapan et al. 2006). Mapping of the groups using the regression algorithm confirmed the reliability of the grouping and the good assignment of the 496 markers (184 AFLPs, 243 SSRs and 69 EST-SSRs), or so-called loci, to their corresponding groups.
The integration of new markers into the previous map showed that the resulting female map, with 19 LGs, was based on 225 loci (44 AFLP, 144 SSR and 37 EST-SSR loci), the male map, with 19 LGs, was based on 208 loci (46 AFLP, 133 SSR and 29 EST-SSR loci), while the integral map had 18 LGs, with 482 markers included (177 AFLP, 242 SSR and 63 EST-SSR loci). The map was 2054.2 cM in length with an average genetic distance of 4.3 cM between adjacent markers. The length of individual LGs varied from 93.0 cM to 163.8 cM, with the average marker distance per LG ranging from 3.0 cM for the highest to 6.7 cM for the lowest (Supplementary Table S4). Sixty-nine EST-SSRs were distributed across 16 of the 18 chromosomes of previous map (Fig. 3). Only 62 EST-SSR markers out of 69 could fill the gap on the reference map whereas seven markers could not link to any linkage group (Supplementary Table S5).

Genetic linkage map of Hevea brasiliensis with AFLP, SSR and EST-SSR markers
Discussion
For EST-SSR sequence analysis, to date DNRs and TNRs have been mostly reported in plants, with some differences between species. For example, TNRs were the leading repeat motif in Arabidopsis thaliana, wheat, rice, corn, soybean (Cardle et al. 2000), cotton (Qureshi et al. 2004), sugarcane, grape (Cordeiro et al. 2001), barley (Thiel et al. 2003) and citrus (Chen et al. 2006) while DNRs were dominant in kiwi, apricot, peach (Jung et al. 2005), and coffee (Aggarwal et al. 2007). In the rubber tree, based on the results of Feng et al. (2009), DNRs (42.2 %) were the leading repeat motif, followed by TNRs (11.9 %). In addition, AG/TC motifs were the predominant DNRs, consistent with the report of Bindu et al. (2004). Moreover, the observation of AAG/TTC as the leading repeat motif of TNRs in the rubber tree is similar to that of A. thaliana (Cardle et al. 2000), soybean (Gao et al. 2003) and citrus (Chen et al. 2006). In this study, a consistent result similar to other research was obtained. The most abundant microsatellite repeats were di- and trinucleotide repeats (139 and 132 SSRs, respectively). Among the dinucleotide motif sequences in Hevea EST-SSRs, AG/TC was the most common motif (41 %), while AAG/TTC (18 %) was the most abundant trinucleotide motif.
Because the genome sequence of the rubber tree is not yet complete, it is not surprising that around 50 % of the EST-SSR sequences are not related to putative gene function in NCBI databases. ESTs are a potentially rich source of SSRs that reveal polymorphisms not only within the source taxon but also in related taxa (Ellis and Burke 2007). EST-SSR markers can thus reveal putative functional genes and aid in map-based gene cloning. From 69 EST-SSRs, 22 markers related to gene function of various species, 37 markers related to unknown proteins and 10 markers had no hit (Supplementary Table S5).
According to the study of QTL detection for growth and initial latex production in the rubber tree (Rattanawong et al. 2008), Hbg16a131 is a major QTL directly associated with latex production. It was found that this QTL was also associated with other traits that were strongly correlated with production, such as inorganic phosphorus and dry rubber content (latex diagnostic), as well as plugging index. This finding indicates the existence of one major gene (or a cluster of genes) located in LG16 and involved in the genetic determination of latex production. On this map, in LG16, the EST-SSR marker which was located nearest to g16a131 was Hbe 43, and the distance between them was 3.8 cM. Hbe43 was located in LG16 at 2.8 cM and g16a131 at 6.6 cM. However, the function of Hbe43 is still unclear due to the incomplete Hevea genome databases. Moreover, it was also found that the other major QTL, Hbg3a312, was associated with trunk girth (growth). Depending on the age of the tree, Hbg3a312 explained from 16 to 28 % of the genetic variance of the girth. The marker A312 was located on the same linkage group as Hbe154, the nearest EST-SSR marker; the distance between them was 5.9 cM. Hbe154 was located on LG3 at 55.7 cM and Hbg3a312 at 61.6 cM. Hbe154 and Hbe78 are the EST-SSR markers which have functions related to cyclophilins. Cyclophilins have been reported to be involved in the folding of proteins as part of the cellular chaperone machinery, controlling the assembly of protein complexes and membrane translocation (Bozhko et al. 2003). They were shown to be involved in different stress responses and were found to be developmentally regulated in Digitalis lanata (Scholze et al. 1999).
In conclusion, this study presents new rubber tree EST-SSR markers which were developed from ESTs of differentially expressed genes of inner bark and latex samples, from high- and low-yielding tree clones. These markers were mapped into a previously published map, producing a dense framework of AFLP, SSR and EST-SSR marker loci of H. brasiliensis. EST-SSR markers are associated with coding regions, thus making these markers potentially useful for candidate gene mapping at QTLs. This new set of markers provides molecular breeders and researchers with a powerful tool-set which gives an opportunity to map EST-SSRs potentially related to high latex and wood yield in already developed progeny in rubber tree breeding programs.
Abbreviations
- DNRs:
-
Dinucleotide repeats
- EST:
-
Expressed sequence tag
- SSRs:
-
Simple sequence repeats
- TNRs:
-
Trinucleotide repeats
References
Aggarwal RK, Hendre PS, Varshney RK et al (2007) Identification, characterization and utilization of EST-derived genic microsatellite markers for genome analyses of coffee and related species. Theor Appl Genet 114:359–372
Altschul SF, Madden TL, Schäffer AA et al (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Bindu RC, Nazeer MA, Saha T (2004) Identification of simple sequence repeats in rubber (Hevea brasiliensis). Curr Sci 87:807–811
Bozhko M, Riegel R, Schubert R et al (2003) A cyclophilin gene marker confirming geographical differentiation of Norway spruce populations and indicating viability response on excess soil-born salinity. Mol Ecol 2:3147–3155
Cardle L, Ramsay L, Milbourne D et al (2000) Computational and experimental characterization of physically clustered simple sequence repeats in plants. Genetics 156:847–854
Chen CX, Zhou P, Choi YA et al (2006) Mining and characterizing microsatellites from citrus ESTs. Theor Appl Genet 112:1248–1257
Cordeiro GM, Casu R, McIntyre CL et al (2001) Microsatellite markers from sugarcane (Saccharum spp.) ESTs cross transferable to erianthus and sorghum. Plant Sci 160:1115–1123
Cubry P, Pujade-Renaud V, Garcia D et al (2014) Development and characterization of a new set of 164 polymorphic EST-SSR markers for diversity and breeding studies in rubber tree (Hevea brasiliensis Mull. Arg.). Plant Breed 133:419–426
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Ellis JR, Burke JM (2007) EST-SSRs as a resource for population genetic analyses. Heredity 99:125–132
Feng SP, Li WG, Huang HS et al (2009) Development, characterization and cross-species/genera transferability of EST-SSR markers for rubber tree (Hevea brasiliensis). Mol Breed 23:85–97
Gao LF, Tang J, Li H et al (2003) Analysis of microsatellites in major crops assessed by computational and experimental approaches. Mol Breed 113:163–185
Jung S, Albert A, Jesudurai C et al (2005) Frequency, type, distribution and annotation of simple sequence repeats in Rosaceae ESTs. Funct Integr Genomics 5:136–143
Kanjanawattanawong S, Tangphatsornruang S, Triwitayakorn K et al (2014) Characterization of rubber tree microRNA in phytohormone response using large genomic DNA libraries, promoter sequence and gene expression analysis. Mol Genet Genomics 289(5):921–933
Kujur A, Saxena MS, Bajaj D et al (2013) Integrated genomics and molecular breeding approaches for dissecting the complex quantitative traits in crop plants. J Biosci 38:971–987
Lekawipat N, Teerawatanasuk K, Rodier-Goud M et al (2003) Genetic diversity analysis of wild germplasm and cultivated clones of Hevea brasiliensis Muell. Arg. by using microsatellite markers. J Rubber Res 6:36–47
Lespinasse D, Rodier-Goud M, Grivet A et al (2000) S saturated genetic map of rubber tree (Hevea spp.) based on RFLP, AELP, microsatellite and isozyme markers. Theor Appl Genet 100:127–138
Li D, Deng Z, Qin B et al (2012) De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genomics 13:192
Mangpung Y, Kongsawadworakul P, Viboonjun U et al (2010) Identification and characterization of candidate genes related to latex yield in the inner bark tissues of rubber tree (Hevea brasiliensis). Thai J Bot 2(Special Issue):207–214
Parkinson J, Blaxter M (2009) Expressed sequence tags: an overview. In: Parkinson J (ed) Expressed sequence tags (ESTs): generation and analysis. Methods in molecular biology, vol 533. Humana Press, New York, pp 1–12
Polhamus L (1962) Rubber: botany, production, and utilization. World Crops Books, Leonard Hill Limited, London
Prapan K, Lekawipat N, Weber C et al (2006) Molecular genetic markers and rubber breeding in Thailand: 1-Genetic mapping of the family RRIM600 × PB217 by use of microsatellite markers. In: 2006 The 2nd Thai-French seminar rubber cooperation, Bangkok, Thailand, 1 September 2006, pp 1–10
Qureshi SN, Saha S, Kantety RV et al (2004) Molecular biology and physiology: EST-SSR: a new class of genetic markers in cotton. J Cotton Sci 8(2):112–123
Rahman AYA, Usharraj AO, Misra BB et al (2013) Draft genome sequence of the rubber tree Hevea brasiliensis. BMC Genomics 14:75
Rattanawong R, Prapan K, Lekawipat N et al (2008) QTLs detection for growth and initial latex production in rubber (Hevea brasiliensis). In: 2008 IRRDB international natural rubber conference and annual meetings, Kuala Lumpur, 13–14 October 2008. IRRDB, pp 1–28
Scholze C, Peterson A, Diettrich B et al (1999) Cyclophilin isoforms from Digitalis lanata Sequences and expression during embryogenesis and stress. J Plant Physiol 155:212–219
Thiel T, Michalek W, Varshney RK et al (2003) Exploiting EST database for the development and characterization of gen-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106:411–422
Towaranonte B, Chuenwarin P, Kongsawadworakul P et al (2010) Identification and characterization of candidate genes related to latex yield in the laticifers of rubber tree (Hevea brasiliensis). Thai J Bot 2(Special Issue):199–206
Triwitayakorn K, Chatkulkawin P, Kanjanawattanawong S et al (2011) Transcriptome sequencing of Hevea brasiliensis for development of microsatellite markers and construction of a genetic linkage map. DNA Res 18:471–482
Van Ooijen JW (2006) JoinMap® 4, Software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen
Acknowledgments
This research was supported by Rubber Research Institute of Thailand (RRIT), Department of Agriculture, the Thailand Research Fund (TRF) Grant No. MRG5180088 with Faculty of Science, Mahidol University and a grant from Mahidol University, Thailand.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nirapathpongporn, K., Kongsawadworakul, P., Viboonjun, U. et al. Development and mapping of functional expressed sequence tag-derived simple sequence repeat markers in a rubber tree RRIM600 × PB217 population. Mol Breeding 36, 39 (2016). https://doi.org/10.1007/s11032-016-0461-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-016-0461-3