
Abstract
Autoinflammatory disorders are sterile inflammatory conditions characterized by episodes of early-onset fever and disease-specific patterns of organ inflammation. Recently, the discoveries of monogenic disorders with strong type I interferon (IFN) signatures caused by mutations in proteasome degradation and cytoplasmic RNA and DNA sensing pathways suggest a pathogenic role of IFNs in causing autoinflammatory phenotypes. The IFN response gene signature (IGS) has been associated with systemic lupus erythematosus (SLE) and other autoimmune diseases. In this review, we compare the clinical presentations and pathogenesis of two IFN-mediated autoinflammatory diseases, CANDLE and SAVI, with Aicardi Goutières syndrome (AGS) and monogenic forms of SLE (monoSLE) caused by loss-of-function mutations in complement 1 (C1q) or the DNA nucleases, DNASE1 and DNASE1L3. We outline differences in intracellular signaling pathways that fuel a pathologic type I IFN amplification cycle. While IFN amplification is caused by predominantly innate immune cell dysfunction in SAVI, CANDLE, and AGS, autoantibodies to modified RNA and DNA antigens interact with tissues and immune cells including neutrophils and contribute to IFN upregulation in some SLE patients including monoSLE, thus justifying a grouping of “autoinflammatory” and “autoimmune” interferonopathies. Understanding of the differences in the cellular sources and signaling pathways will guide new drug development and the use of emerging targeted therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Systemic autoinflammatory diseases were introduced as a concept by Dan Kastner in 1999 to distinguish periodic fever syndromes that lack autoantibodies or antigen-specific T cells from autoimmune diseases such as systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) [1]. The characterization of patients with “autoinflammatory phenotypes” led to identification of pathogenic genetic defects in innate immune sensors and pattern recognition receptors (PRRs) pathways, pioneered by mutations in the IL-1 activating inflammasome genes MEFV encoding pyrin (familial Mediterranean fever), NLRP3 (cyropyrinopathies) and, recently NLRC4 (NLRC4-associated macrophage activation syndrome), linking increased cytokine and chemokine production and recruitment of innate immune cells to the development of tissue-specific sterile organ inflammation and damage [2]. A novel group of monogenic disorders presenting with early-onset systemic and organ-specific inflammation and a prominent IFN response gene signature (IGS) including Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperatures (CANDLE) [3] and STING-Associated Vasculopathy with onset in Infancy (SAVI) [4] and Aicardi Goutières syndrome (AGS) [5] suggested a key role for type I IFN in disease pathogenesis. In 2003, the IGS was described in SLE [6] followed by other autoimmune diseases, such as juvenile dermatomyositis (JDM) [7]. In 2011, Yanick Crow suggested the term “interferonopathies” to indicate that IFN drives shared immunopathology [8]. The discovery of genetic causes of interferonopathies has propelled research and provided insights into innate (autoinflammatory) and autoantibody or antigen-specific T cell-mediated (autoimmune) mediated pathology in diseases with IGS. This review compares the clinical disease manifestations and pathogenesis of CANDLE and SAVI with AGS and monogenic forms of SLE caused by mutations in the complement system (C1Q) [9] and DNA nucleases (DNASE1 and DNASE1L3) [10, 11]. Based on differences in immunopathogenesis, we suggest grouping these conditions as “autoinflammatory” and “autoimmune” interferonopathies.
Clinical and pathogenic description of the Mendelian (monogenic) autoinflammatory interferonopathies CANDLE and SAVI
CANDLE/PRAAS (MIM#256040)
In 2010 and 2011, several studies found that autosomal recessive (AR) loss-of-function (LOF) mutations in the proteasome subunit beta type 8 (PSMB8) cause conditions referred to as JMP (joint contractures, muscular atrophy, microcytic anemia, and panniculitis-induced lipodystrophy) [12], Nakajo-Nishimura syndrome (NNS) with nodular erythema, elongated and thickened fingers and emaciation [13], Japanese autoinflammatory syndrome with lipodystrophy (JASL) [14], and CANDLE [3], demonstrating a phenotypic disease spectrum with variable disease severity [3, 15], also referred to as proteasome-associated autoinflammatory syndromes (PRAAS). In addition to mutations in PSMB8 [3, 12–14, 16] in CANDLE/PRAAS, we described loss-of-function mutations in other proteasome subunits (PSMB9, PSMB4, PSMA3) and the proteasome assembly protein, POMP, with expansion of the inheritance to compound heterozygous (mutations in PSMB4), digenic recessive (combinations of PSMA3, PSMB4 and/or PSMB9), and autosomal dominant (AD) (POMP) [17]. CANDLE is a rare disease with less than 100 cases described worldwide.
Clinical manifestations
Clinical manifestations of PRAAS/CANDLE are summarized in Tables 1 and 2. Long-standing inflammation causes organ damage (progressive lipodystrophy, joint contractures, and basal ganglia calcifications in the CNS). While inflammatory features are more prominent early in life, damage features manifest later in life. Clinical manifestations include the following:
Systemic inflammation: This includes fevers and elevated acute phase reactants [3, 13–21].
Skin manifestations: Nodular or plaque-like violaceous skin rashes and violaceous periorbital rash with or without swelling are thought to be manifestations of panniculitis that evolve to lipodystrophy [3, 13–22]. Skin biopsies show immature neutrophils (Leder stain positive) and myeloid precursors (myeloperoxidase positive) as well as atypical mononuclear cells, which are likely activated macrophages (positive for CD68 and CD163, negative for Leder stain) [3].
Musculoskeletal manifestations: Musculoskeletal findings include myositis and arthritis [3, 13–22]. Joint contractures are common and develop even early in childhood [3, 13–15, 17, 18, 21, 22].
Metabolic manifestations: Prominent metabolic features include truncal obesity, dyslipidemia, insulin resistance, and acanthosis nigricans consistent with metabolic syndrome [3, 13, 15–18, 20–22], which are exaggerated by steroids [3].
Cardiovascular manifestations: Recently, primary pulmonary hypertension has been observed in 2 young patients with CANDLE/PRAAS [23].
Mortality: Early mortality due to sudden death in the context of infections, cardiomyopathy, and cardiac arrhythmias has been reported [3, 15, 18]. See Table 1 for details.
Treatment: Methotrexate, azathioprine, cyclosporine, tacrolimus, rituximab, IL-1 blockade, and tumor necrosis factor antagonist (infliximab, etanercept, adalimumab) are ineffective; partial response to anti IL-6 therapy and/or high doses of steroids is seen [3, 13–15, 18]. Janus kinase or JAK inhibitors are being used with promising results (see below).
Pathogenesis
The proteasome is a tubular protein degradation system for misfolded or damaged intracellular proteins, particularly those marked for degradation by polyubiquitination, which are proteolytically cleaved by the caspase, chymotrypsin, and trypsin-like activities [24]. The proteasome mutations decrease the proteolytic activities [12–14] and might impair adipocyte differentiation based on PSMB8 knockdown experiments [14]. Clues to pathogenesis came from observations of high serum levels of IFN γ inducible protein 10 (IP-10) or CXCL10 and a prominent IGS [3, 12–14]. Knockdown experiments in healthy fibroblasts identified a critical threshold of proteasome dysfunction that leads to induction of type I IFN gene transcription. Proteasome dysfunction induced in wild-type cells by proteasome inhibitors results in dose-dependent increases in IFNA and IFNB transcription [17]. Although the exact mechanism leading to IFN transcription in CANDLE/PRAAS remains unknown, the type I IFN induction is independent of STING (stimulator of IFN genes protein) and MAVS (mitochondrial antiviral signaling protein), adaptors of RNA and DNA viral sensors, respectively [17]. Patient cells are hyper-responsive to IFN γ [3] and IFNα (unpublished) stimulation and respond with increased STAT-1 phosphorylation; environmental stressors such as infections can trigger clinical flares [25].
SAVI (MIM #615934)
We recently described dominant gain-of-function mutations in TMEM173, encoding stimulator of interferon genes (STING), as the cause for the autoinflammatory syndrome STING-associated vasculopathy with onset in infancy (SAVI) with disease-causing mutations thus far limited to exon 5 in TMEM173 (residues V147, N154, V155) [4, 26–29], with clinical features summarized in Tables 1 and 2. A report of a family with 4 mutation positive (V155 M) members in 3 generations with variable clinical phenotype and onset (ranging from onset at 1 yo with febrile attack and rash with lung disease and failure to thrive in one individual to adult-onset arthralgia and systemic inflammation), all with overexpression of interferon-regulated genes [26], suggests modifiers may influence the severity of SAVI.
Clinical manifestations
Clinical manifestations of SAVI are summarized in Tables 1 and 2.
Systemic manifestations Rash with systemic inflammation (fever, elevated acute phase reactants) is typically seen in the first months of life [4, 26–29].
Skin manifestations Vasculopathic lesions are most prominent in cold-sensitive acral areas and present as violaceous plaques and/or nodules on the face, nose, and ears, and distal ulcerations. Features of vascular and/or tissue damage include nail dystrophy, gangrene/infarcts of fingers/toes with tissue loss, and nasal septal perforation [4, 26–28]. In lesional skin biopsy endothelial cells, there is upregulation of endothelial inflammation (inducible nitric oxide synthase), coagulation (tissue factor), and endothelial cell adhesion and activation (E-selectin). Although immune complexes (ICs) are seen in partially destroyed vessels, many vessels did not have immune complex (IC) deposition and affected cutaneous small-vessels were surrounded by neutrophils and leukocytoclasia [4].
Lung manifestations Paratracheal adenopathy, abnormal pulmonary function tests, and variable interstitial lung disease on CT are seen with or without symptoms; the extent of lung fibrosis varies between patients [4, 26–29]. No vasculitis is observed in the lung of SAVI patients. STING is expressed in alveolar macrophages, type 2 pneumocytes, and bronchial epithelium. SAVI patients present with increased alveolar macrophages, activation of type 2 pneumocytes, follicular hyperplasia, and prominent fibrosis [4].
Musculoskeletal manifestations Myositis, arthritis, and arthralgia are variable [4, 26, 29].
Mortality: Early mortality due to respiratory failure likely related to underlying SAVI lung disease often in the context of infections is seen [4, 26, 27]. See Table 1 for details.
Treatment: Immunosuppressive agents including high-dose steroids, azathioprine, colchicine, methotrexate, mycopheonlate mofetil, cyclophosphamide, tumor necrosis factor antagonist, IL-1 blockade, IL-6 blockade, and anti-CD 20 therapy have not resulted in sustained responses [4, 26–29].
Pathogenesis
STING is a key dimeric adaptor protein at the endoplasmic reticulum that is essential for interferon beta induction. Viral or self double-stranded DNAs (dsDNAs) are sensed in the cytosol by cyclic GMP-AMP synthase or cGAS ligand [30, 31]. Upon binding cGAS, cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) is released as a second messenger, which binds STING, leading to phosphorylation of TANK-binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF-3); IRF3 then translocates into the nucleus leading to IFNB1 (interferon β) transcription. The disease-causing STING mutations constitutively activate the pathway resulting in IFNB1 transcription, prominent IGS, elevated serum levels of interferon-inducible protein 10 (IP-10) and constitutively elevated STAT-1 phosphorylation in patient T and B lymphocytes [4]. Activation of the STING pathways with cGAMP in endothelial cells in vitro leads to transcription of activation markers and clotting factors as well as cell death, suggesting a role for STING in tissue homeostasis [4].
Treatment considerations for CANDLE and SAVI
Inhibition with Janus kinase (JAK) inhibitor tofacitinib decreased the STAT-1 phosphorylation and IP-10 in mononuclear cells (PBMCs) from CANDLE patients stimulated with IFNγ [3]. In vitro experiments in PBMCs from SAVI patients showed that constitutive phosphorylation of STAT1 in CD4 T and CD19 B cells was reduced in cells co-cultured with JAK inhibitors (tofacitinib, ruxolitinib, and baricitinib). This phenomenon was also observed in fibroblasts. cGAMP stimulated interferon-regulated genes (CXCL10, MX1, and OAS3) were suppressed when cells were co-cultured with tofacitinib [4]. These observations and the absence of other suitable treatment options led to the use of JAK inhibition as therapy in CANDLE and SAVI (NCT01724580). Preliminary data show clinical improvement with significant decrease in symptom scores and daily steroid requirement in CANDLE patients [32].
Comparison of Aicardi-Goutières syndrome (AGS) with CANDLE and SAVI
Mutations in 7 genes cause the AGS phenotype: AGS1 (TREX1) (MIM#225750), AGS2 (RNASEH2B) (MIM#610181), AGS3 (RNASEH2C) (MIM#610329), AGS4 (RNASEH2A) (MIM#610333), AGS5 (SAMHD1) (MIM#612952), AGS6 (ADAR1) (MIM#615010), AGS7 (IFIH1) (MIM#615846).
Clinical presentation in AGS compared to CANDLE and SAVI
AGS is characterized by an early-onset, often monophasic, congenital infection-like syndrome resulting in encephalopathy with microcephaly, dystonia, basal ganglia calcifications, white matter abnormalities, and cerebral atrophy [33]. Cerebrospinal fluid analysis shows chronic lymphocytosis and elevated IFNα levels [34]. Expression of interferon-regulated genes (IGS) in peripheral blood is also upregulated, which is sustained over time. [5].
Systemic manifestations: Unexplained fevers and hepatosplenomegaly are noted with AGS and are present in the context of the early/initial disease. Systemic inflammation is typically not persistent; acute phase reactants are not consistently reported [33, 35].
CNS manifestations The encephalopathy and white matter disease of AGS are not seen in CANDLE or SAVI; however, CANDLE patients present with headaches and mild lymphocytosis in CSF suggestive of intermittent lymphocytic meningitis. Basal ganglia calcifications are most prominent in AGS (AGS > CANDLE > SAVI), are present in most CANDLE patients and less common in SAVI. In AGS, the onset and severity of encephalopathy correlates with CNS damage (i.e., severe brain atrophy, white matter disease, and resulting clinical sequelae), but AGS may present later in life (i.e., patients with SAMHD1 mutations can present with later-onset vasculopathy and moyamoya disease later in childhood) [33]. Autoantibodies that trigger the CNS disease have not been described and the early-onset of AGS makes an innate immune dysregulation resulting in organ (CNS) damage most likely.
Skin manifestations “Chilblains” or cold-induced acral dermatosis is present in up to 40 % of patients with AGS, but the face is typically spared. Out of 7 chilblain cases assessed in AGS, 4 were noted to have IgM deposition in the basement membrane (positive in 1 with mutated TREX1 and 1 with mutated RNASEH2B, and 2 not genetically analyzed, one of whom was noted to have anti-C1q antibodies which raises the possibility of cofactors [36]; negative in 2 with mutated SAMHD1 (at least 1 AR) and 1 with AR TREX1 mutations) [37]. Chilblain-like vasculopathy is also seen in SAVI, but not CANDLE. Lobar neutrophilic panniculitis leading to lipodystrophy is seen in CANDLE, but is not present in SAVI and is rare in AGS (<1 %) [33].
Hematologic manifestations Hematologic manifestations including thrombocytopenia, lymphopenia, and leukopenia may be autoantibody-mediated or the result of IFN-mediated bone marrow suppression. Cytopenias with IFN treatment are reported [38, 39] and IFN-mediated transient bone marrow suppression (mostly lymphopenia) is the likely mechanism of cytopenias in patients with SAVI and CANDLE. Thrombocytosis is common in SAVI. In AGS, thrombocytopenia and leukopenia have been reported, but their association with antibodies or flare-induced bone marrow suppression remains unclear [33, 40].
Cardiovascular manifestations Systemic hypertension and pulmonary hypertension are recently observed morbidities in CANDLE and SAVI, which suggest a shared role of interferon in vasculopathy, but have not been reported in AGS.
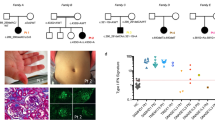
Autoantibodies Features of complex SLE were systematically assessed in 18 mutation positive AGS patients such as oral ulcers (3/18, 16.7 %) and detectable autoantibody titers including ANA (3/18, 16.7 %), anti-dsDNA (1/18, 5.6 %), and anti-cardiolipin (ACA) (2/18, 11.1 %) antibodies [40]. Some SLE clinical criteria [41] thought to be triggered by respective immune complex deposition include classic photosensitive butterfly rash (Fig. 1) [42], pleuritis [43], SLE nephritis and presence of antibodies to chromatin (anti-dsDNA) and to spliceosome components (anti-Sm) and these criteria are rarely positive in AGS patients (Table 4). Autoantibodies are also detected with mostly low titers in CANDLE (12 ANA, 3 anti-dsDNA, 2 antiphospholipid (anti-cardiolipin, beta 2 glycoprotein) antibodies, 2 anti-SSA, 1 anti-smRNP, 1 anti-SmB, 5 ANCA), and SAVI (9 ANA, 2 anti-dsDNA, 7 antiphospholipid antibodies, 2 anti-RNP, 1 anti-Ro, 6 ANCA). Variable lupus anticoagulant positivity was found in 7/15 (46.7 %) CANDLE and in 4/9 (44.4 %) SAVI. Data are based on patients who had at least one positive result over time. Autoantibody titers fluctuate and do not correlate with disease activity in CANDLE and SAVI and are thus likely secondary to persistent IFN upregulation rather than driving pathology. Metabolic and pulmonary manifestations seen in CANDLE or SAVI have not been described in AGS.

Different patterns in facial rash of SAVI and SLE. In SAVI patients (upper panels), the rashes are aggravated by cold exposure and are prominent in cold-sensitive areas including the tip of the nose and the lower part of the cheeks and the ears (not shown). The characteristic malar (facial) rash with a butterfly distribution on pictures from genetically undefined SLE patients (lower panels) show a photosensitive rash that is often induced by exposure to sunlight and associated with immune complex deposition at the dermal-epidermal junction (LE band) on biopsy. References: Lower left photo: © 2016 American College of Rheumatology. Used with permission. Lower right photo: www.mollysfund.org. Used with permission.
Genetics and pathogenesis of AGS compared to SAVI and CANDLE
The pathogenesis of most forms of AGS is thought to result from accumulation of nucleic acids that cannot be sufficiently metabolized, thus raising the pool of cytoplasmic nucleic acids that signal through the DNA sensor cGAS which activates adaptor STING and/or RNA sensors MDA-5 and RIG-I which activate adaptor MAVS [45]. In 2006, AR LOF mutations in 3′ repair exonuclease TREX1 became the first monogenic cause of AGS [46]. Since then, AR LOF mutations in components of the RNASEH2 endonuclease complex (RNASEH2A, RNASEH2B, and RNASEH2C) [47], a deoxynucleotide triphosphohydrolase SAMHD1 [48], and an adenosine deaminase, ADAR1 or ADAR [49] were found to cause AGS. Most recently, GOF AD mutations in IFIH1 (interferon-induced with helicase C domain 1 or AGS7), also known as MDA5 or melanoma-differentiation-associated protein 5 [50, 51] have been reported to cause an AGS phenotype with more variable disease severity.
As the major cytosolic 3′-to-5′ exonuclease, TREX1 deficiency can lead to the accumulation of interferogenic DNA-RNA triggers. TREX1 can act in concert with a granzyme A-activated endonuclease, NM23-H1, to degrade DNA during granzyme A-dependent apoptosis. A defect in TREX1 would lead to decreased degradation of damaged DNA that is generated with granzyme A-dependent apoptosis [52]. Furthermore, the absence of TREX1 in Trex1−/− mouse embryo fibroblasts upon gamma radiation destabilizes Chk2, a central DNA damage checkpoint [53], and results in the accumulation of aberrant extranuclear single-stranded DNA (ssDNA) that result from uncleared DNA replication intermediates and leads to chronic ATM–mediated Chk2 activation [54]. Lastly, the absence of TREX1 has also been associated with the accumulation of reverse-transcribed DNA derived from transcription of endogenous retroeelements [55]. The accumulation of cytoplasmic DNA leads to STING-dependent but mostly endosomal TLR-independent upregulation of type I IFN transcription in the cell types examined [55, 56]. LOF mutations in RNASEH2 endonuclease complex are associated with increased formation of RNA:DNA hybrids [57] that are involved in the regulation of a wide range of functions including immunoglobulin class switching, regulation of gene expression and replication of mitochondrial DNA [58]. Furthermore, mutations in the RNASEH2 complex result in defective removal of misincorporated ribonucleotides from DNA due to deficient ribonucleotide excision repair which causes DNA damage and triggers a cGAS and STING-dependent type I IFN response [59, 60]. LOF mutations in SAMHD1 reduce DNA damage repair and increase the dNTP pools [61], similarly resulting in STING-dependent type I IFN signaling [60, 62, 63]. Adenosine deaminase ADAR has dsRNA-editing function through deaminating adenosine bases to inosines [49, 64]. When mutated, there is accumulation of dsRNA stimulating MAVS-dependent type I IFN transcription [65]. Finally, GOF mutations in IFIH1 lead to MAVS-dependent IFN production without nucleic acid accumulation with incomplete penetrance and variable clinical phenotypes suggesting a possible role for modifier genes [50, 51].
The actual pathomechanisms that lead to CNS damage in AGS patients are unknown. In a mouse model transgenic for a glial fibrillary acidic protein-interferon-alpha fusion protein where IFNα is expressed in an astrocyte-restricted fashion, transgenic animals develop a progressive inflammatory encephalopathy with neuropathologic features similar to AGS [66] suggesting a neurotoxic role of IFNα in the developing brain. In contrast, the disease-causing mutations in CANDLE and SAVI are not known nor expected to alter nucleic acid metabolism. The type I IFN production in CANDLE is MAVS and STING independent [17]. In SAVI, GOF STING mutations directly increase IFNB1 transcription.
Comparison of clinical features in IFN and IL-1 mediated autoinflammatory diseases
Clinical features that are overlapping in at least 2 of the 3 disease groups (AGS, CANDLE, SAVI) are summarized in Table 3 and contrasted with clinical features that are seen in patients with IL-1 mediated diseases [44]. The features more commonly seen in patients with known IFN dysregulation may suggest a role for IFN in perpetuating organ inflammation and damage and may help with defining a pathogenic role of IFN in yet uncharacterized autoinflammatory diseases.
Familial chilblain lupus (FCL), retinal vasculopathy with cerebral leukodystrophy (RVCL), and Singleton Merten syndrome (SMS)
FCL phenotype has been associated with AD mutations in TREX1 [67, 68], SAMHD1 [69] and, more recently, TMEM173/STING [70]. FCL is characterized by painful bluish-red papular or nodular lesions of skin in acral locations precipitated by cold or wet exposure, some with ulceration and generally without photosensitivity, consistent with cutaneous chilblain lupus. Four different AD TREX1 mutations have been reported in 4 kindreds with FCL. The most common FCL mutation (D18N) is also associated with heterozygous AGS, usually de novo, but has also been seen with both phenotypes (AGS, FCL) in the same family; this same mutation has been seen in an asymptomatic mother who has a child with a FCL phenotype [71, 72]. An AD SAMHD1 mutation, which has also been observed with AGS when inherited as recessive mutation, has been reported in 1 family with chilblain lupus and photosensitivity but a negative autoantibody panel [69]. In most patients with FCL the positive ANA antibody titers fluctuate over time, and anti-dsDNA Abs or immune-complex mediated kidney disease have not been described [67, 71]. Upregulation of IFN-stimulated genes in peripheral blood from FCL patients and increased expression of IFN-inducible IFN myxovirus resistance protein A (MxA) and CXCL10 in lesional skin biopsies from FCL patients have been seen [71, 73]. Cold-induced skin features of FCL resemble the cutaneous features in SAVI in some [74] and a distinct TMEM173 mutation identified in a single kindred has the phenotype of FCL [70], but no lung disease in FCL has been reported as seen in SAVI. The pathophysiology of FCL remains unclear. The fact that mutations in 3 different genes cause a FCL phenotype suggests additional modifiers that lead to the disease. Whether the vasculitis in cold-exposed areas is mediated by direct endothelial cell damage or by cold-induced autoantibodies needs further investigation.
RVCL is an adult-onset autosomal dominant neurological disease caused by frameshift mutations in the C-terminus of TREX1 that are distinct from the AGS causing mutations. Prominent features include vascular retinopathy leading to blindness, neuropsychiatric symptoms, and progressive neurologic disease with characteristic neuroimaging abnormalities [72, 75]. Though there is a shared gene, relationship to IFN signaling in RVCL is not yet established [72, 76].
SMS is associated with AD presumed gain-of-function mutations in IFIH1 and DDX58 [77, 78], characterized by dental dysplasia, early-onset aortic and valvular calcification, and acro-osteolysis. One IFIH1 mutation, which had not previously been associated with AGS phenotype, was identified in 3 families with SMS phenotype, with upregulation of nterferon-regulated genes [78]. Interestingly, a distinct mutation in IFIH1 in a family characterized by acral lentigines, scarring and ulceration of distal ears, erythematous cheeks, loss of secondary dentition, contractures, variable neurologic involvement, skeletal abnormalities, and glaucoma was associated with overlapping features of AGS, FCL, and SMS and upregulated interferon-stimulated genes [79]. Two DDX58 mutations in 2 respective families were associated with atypical SMS presenting with glaucoma, aortic calcifications, and skeletal abnormalities and notably normal dentition [77]. The more variable clinical features in the context of vascular calcifications suggest that other genetic and/or environmental factors may influence the disease phenotype.
Comparison of the “autoimmune” interferonopathies (complement deficient SLE, and DNAse deficient SLE) with the “autoinflammatory” interferonopathies SAVI and CANDLE
The IGS was initially described in SLE in 2003 [6] and in other polygenic autoimmune conditions. The discovery of “monogenic interferonopathies” raises the question of similarities and differences of monogenic versus complex autoimmune diseases. The finding of monogenic mutations in a complement factor (C1Q) and the DNA nucleases (DNASE1 and DNASE1L3), which encode proteins involved in the rapid clearance of apoptotic cells, provide monogenic disease models of SLE (monoSLE) that are invaluable in comparison with CANDLE, SAVI, and AGS.
MonoSLE
AR LOF mutations in complement component 1 (C1q) that lead to C1q deficiency (C1Q MIM#613652), and AD LOF mutations in DNASE1 (MIM# 152700), and AR LOF mutations in DNASE1L3 (MIM#614420) predispose to development of SLE with characteristic clinical features, including positive anti-dsDNA antibodies, positive anti-spliceosome antibodies (i.e., anti-Sm), immune-complex-mediated glomerulonephritis (GN), malar photosensitivity rashes, and LE (lupus erythematosus) band seen on skin biopsy indicating complement or IC deposition in the dermis-epidermis interface [41, 42]. More than 90 % of individuals with C1q deficiency develop SLE features with equal incidence among males and females with childhood-onset of disease. The high incidence and defined clinical presentation of SLE associated with mutations in these genes led to their designation as a “monogenic form of SLE.” LOF mutations in other complement components (i.e., C1r/C1s, C2, C3, C4) are associated with a lower risk of developing SLE and other autoimmune conditions, with a SLE risk of about 65 % with C1r/C1s deficiency, 10–20 % for patients with C2 and C3 deficiencies, and about 80 % for patients with C4 deficiencies respectively [9, 80]. C1r and/or C1s deficiency is associated with a prominent immunodeficiency phenotype with recurrent bacterial, viral, or fungal infections in 85 % and many dying at a young age due to severe infection [80]. C4 deficiencies have also been associated with increased infections, autoimmune hepatitis, and type I diabetes [81, 82]. This suggests that these mutations are strong modifiers of autoimmunity but may not cause a “monogenic form of SLE.”
Clinical features in monoSLE compared to CANDLE, SAVI, and AGS
C1q deficiency
Kidney disease reported is primarily mesangial proliferative glomerulonephritis, but IgA nephropathy has been reported and IC deposition has been demonstrated [83, 84]. Variable rashes have been reported including a photosensitive rash with a positive LE band on skin biopsy reported in one case [84, 86, 87]. Antibodies are not uniformly assessed or reported. In one family, ANA and anti-Ro antibodies are reported in 2/2 [86] and in another kindred, 6/6 were ANA positive, 1/7 had positive anti-Sm antibody, and 2/6 had positive anti-Ro antibody titers [87]. Elevated IP-10 and IFNα in peripheral serum have been reported [85].
DNASE1 deficiency
AD mutation in DNASE1 resulting in low serum DNASE1 activity was identified in 2 unrelated patients with juvenile-onset SLE (jSLE) (13yo, 17yo) who presented with fever and generalized erythematoid rash; 1 patient also had parotitis and renal disease with mesangial IgG deposition. Both patients had high-titer ANA, and positive anti-dsDNA, anti-RNP, and anti-SSA antibody titers [10].
DNASE1L3 deficiency
AR LOF mutations in DNASE1L3 resulting in absent DNAse activity in vitro, cause jSLE as reported in 17 individuals (11 male) from 6 Arab families. In 11 individuals (from 4 families) positive ANCA antibodies were measured. 15/17 patients had positive anti-dsDNA antibodies and all 17 had a positive ANA with low C3 and C4. Positive anti-cardiolipin antibody (ACA) titers were seen in 5 patients from 2 families and 11 patients from 5 families had nephritis [11].
SLE-specific diagnostic criteria are listed in Table 4 and were compared in monoSLE, CANDLE, SAVI, and AGS. Photosensitive rashes, positive LE band on skin biopsy, glomerulonephritis, and positive anti-dsDNA and anti-Sm antibodies are seen in patients with monoSLE, but rarely in patients with CANDLE (n = 3 with anti-dsDNA, n = 1 with anti-SmB antibody), SAVI (n = 2 with anti-dsDNA, none with anti-Sm antibody reported), and AGS (n = 1 with anti-dsDNA and none with anti-Sm antibody reported). Interestingly, IgM deposition at the basement membrane or dermal-epidermal junction consistent with a LE band has been described with chilblain lesions in AGS patients [37, 46] as noted above. Other disease manifestations are as follows:
Systemic manifestations: Systemic inflammation with fever similar to CANDLE and SAVI is seen in early-onset of monoSLE [10].
Skin manifestations: The differences in the distribution of the facial rashes in SAVI and genetically undefined SLE are depicted in Fig. 1 with representative images [88, 89]. In SAVI, the erythematous rash is localized to acral areas such as the tip of the nose, cheeks and the ears and is aggravated by cold. In contrast, the characteristic malar rash in SLE is distributed in a butterfly pattern and photosensitive [42]. Skin biopsies from SAVI show small vessel vasculopathy and a predominantly neutrophilic infiltrate but absence of an LE band; however, a LE band at the dermal-epidermal junction has been described in monoSLE (C1q deficiency). In SAVI, ICs are seen in some vessels but typically later in the course when significant destruction has occurred, thus suggesting IC deposition in the context of clearance of tissue damage rather than as early triggers of the inflammatory response. In CANDLE, neutrophilic lobar panniculitis on biopsy is thought to lead to adipocyte destruction and lipoatrophy [14]. Panniculitis has not been reported in SAVI with rare reports in AGS [33] and so far no reports in monoSLE. In contrast to CANDLE, histologic features of SLE panniculitis (SLE profundus) include lobular lymphocytic infiltrates with increased interstitial mucin and hyaline necrosis in subcutaneous tissue [90], suggesting a distinct innate pathogenesis in CANDLE.
Lung manifestations: Interstitial lung disease in SAVI often with significant fibrosis at the time of biopsy is very rare in CANDLE, not reported in AGS, and not characteristic of SLE. Although not reported in monoSLE, pleuritis is a common lung manifestation of not yet genetically defined SLE. The presence of ICs in pleural fluid has been reported but their contribution to pathology is unclear [91].
Autoantibodies: Although autoantibodies are more frequently seen in CANDLE and SAVI than in IL-1 mediated autoinflammatory disease, the titers are typically low and the presence or absence of an autoantibody does not correlate with disease severity. Importantly, IC-mediated glomerulonephritis is a feature not yet described with CANDLE, SAVI, or AGS in contrast to monoSLE.
Hematologic and CNS manifestations: Hematologic features are part of the diagnostic clinical criteria of SLE [41] (Table 4) but have not systematically been assessed or described in reports of monoSLE. CNS disease has not yet been reported in monoSLE.
Pathogenesis of “monogenic SLE” compared to SAVI, CANDLE, and AGS
The development of pathogenic ICs against nucleosomes including chromatin, ribonucleoproteins, and histones that are generated in the context of delayed clearance of apoptotic debris are thought to be the hallmark in SLE pathogenesis [92, 93]. The characterization of patients and their respective murine models with C1q-deficiency [94] after DNASE1 [95, 96] and DNASE1L3 [97] deficiency have provided insights into the pathogenic role of immune complexes and pathomechanisms that lead to the autoimmune-mediated organ damage that has significant overlap with the pathogenesis of respective clinical features in not yet genetically defined SLE.
-
1.
Clearance of apoptotic debris: C1q is a strong opsonin for phagocytosis of late apoptotic and necrotic cells; it is essential for effective uptake of degraded chromatin by monocyte-derived phagocytes [94, 98]. A role of the two major serum DNAses (Deoxyribonuclease I (DNASE1) and Deoxyribonuclease I-like 3 (DNASE1L3)) in degrading chromatin has been described [96, 97]. DNASE1L3 has a specific role in removing chromatin from the surface of microparticles that are released from apoptotic cells thus indicating a direct mechanism in controlling circulating levels of immunogenic chromatin [97]. Impaired clearance of apoptotic tissue and hematopoietic cells including neutrophil extracellular traps (NETs) induces secondary necrosis, which leads to presentation of dsDNA-protein including chromatin and RNA-protein complexes (ribonuclear proteins) on the cell surface (i.e., in blebs on apoptotic keratinocytes [99], on glomerular basement membrane (GBM) in kidneys [96], or released during neutrophil death by NETosis [100]). Secondary necrotic cell-derived material (SNECs) is more immunogenic and associated with exposure of autoantigens that are potent stimulators of autoantibodies [101].
-
2.
Development of autoantibodies and break of tolerance: The cellular/tissue locations of the break in tolerance remain unclear and are likely variable and specific to autoantibodies present. Dendritic cells containing apoptotic bodies migrate from peripheral sites to draining lymph nodes (LN). Chromatin taken up in apoptotic blebs can induce myeloid dendritic cell (myDC) maturation [122], upregulation of proinflammatory cytokines, expression of MHCs and co-stimulatory molecules [102–104], and effective stimulation of autoreactive T-helper cells and autoantibody production by autoreactive B cells. Chromatin-Ig complexes are found in non-genetically defined SLE patients and can activate B cells by dual engagement of IgM through the B-cell receptor, and through the endosomal Toll-like receptors (TLRs) [106, 107]. Furthermore, DNASE-1 and DNASE1L3 deficiency affects tissue susceptibility to organ injury and damage. In a Dnase1 deficient murine model, anti-chromatin antibodies selectively bind epitopes in renal tissue where Dnase-1 deficiency leads to necrotic death of kidney cells and the generation of chromatin fragments. Evidence of in situ immune complex deposition by binding to uncleared cellular debris at tissue sites or blood have been supported in murine models of GN [108] and reflect a bidirectional interaction between the immune response and the target tissues (recently reviewed in detail in [92]).
-
3.
Complement activation in tissues and tissue injury: Circulating autoantibodies/ICs can bind to tissue antigens exposed during cell stress, such as injury [92]. The clearance of antinuclear immune-complexes via Fc-gamma receptors by blood phagocytes, macrophages, and dendritic cells that get recruited to tissues may lead to the secretion of proinflammatory cytokines (IL-8, IL-1β, TNFβ, IFNα, IL-18) and tissue inflammation [105]. In the kidney, an organ sensitive to IC-mediated injury, anti-chromatin containing immunoglobulin (Ig) binds to the cell basal membranes, stimulating a local inflammatory response [109] and activating the complement cascade; The attack complex causes damage to the GBM for example and perpetuates an abnormal immune response [110].
-
4.
Amplification of a vicious cycle of dysregulated immune responses and the role of IFN: The clearance of NETs is facilitated by DNASE1 and C1q-assisted extracellular preprocessing [98, 111]. After phagocytosis, NETs are normally degraded in an immunologically “silent” process [111]. Delay of NET clearance (i.e., in the context of anti-HMGB1, anti-LL37 and/or anti-C1q antibody and possibly anti-chromatin autoantibody interference that prevents access of DNASE1) can extend the survival of the NETs that are immunogenic and interferogenic [109, 112]. Type I interferon aggravates the abnormal tissue responses by accelerating autoreactive B cell maturation, precipitating cell death and organ damage [113], and by increased NETosis induction [114]. Uncleared NETs serve as a source of interferogenic autoantigens by exposure of chromatin, myeloperoxidase, proteinase 3, and release of oxidized mitochondrial DNA, thus fueling a type I IFN amplification loop that perpetuates the abnormal immune responses in SLE [100, 115]. IFN upregulation has been reported in C1q deficiency; pathogenic mechanisms in DNASE1 and DNASE1L3 deficiency suggest similar upregulation of IFN.
Distinction between autoinflammatory and autoimmune interferonopathies
SAVI and CANDLE patients do not have genetic defects that affect clearance of apoptotic debris. In SAVI, constitutive transcription of IFNβ and the tissue-specific effects of STING signaling on cell homeostasis can lead to immune-complex and/or autoantibody independent organ inflammation and damage [4]. In SAVI, the lungs and small vessels in the skin are affected, but not the kidneys. The specific interferogenic pathomechanism in CANDLE remains incompletely defined, but the absence of significant autoimmunity despite significant tissue damage (high LDH, and development of lipodystrophy, myositis and other organ manifestations) and our data on independence of MAVS and STING signaling suggest “independence” from the endosomal and RNA/DNA sensing pathways as well [17]. Although the persistent abnormal and chronic IFN response can and might stimulate autoimmunity [116] and may lead to “secondary autoimmune manifestations,” the CANDLE and SAVI-specific disease manifestations seen early in life are independent of autoantibody production and immune complex deposition. In that context, the presence of positive anti-citrullinated peptide antibody (ACPA) erosive RA in a 7 year old SAVI patient [4] may have been facilitated by the abnormal type I IFN production on the background of other genetically predisposing variants or modifiers (i.e., presence of shared epitope in the patient).
The overlap with autoimmunity, however, may occur more frequently in patients with AGS and SPENCDI (Fig. 2). Although the accumulation of cytoplasmic nucleic acids in AGS seems to mainly trigger the disease manifestations through stimulation of STING and MAVS dependent nucleic acid receptors, it cannot be excluded that stimulation through endosomal TLRs in B cells also occurs and may trigger autoimmune manifestations. Similarly, SPENCDI is caused by recessive mutations in ACP5, which encodes tartrate-resistant phosphatase (TRAP) and presents with some features similar to lupus including anemia, nephritis, autoantibody positivity, thrombocytopenia, and myositis [117, 118] with 6/10 cases with dsDNA antibodies and 3/10 with lupus nephritis in one report [117]. Interestingly, increased levels of intracellularly phosphorylated osteopontin (OPN) are seen in the context of impaired TRAP function, which mediates signaling through TLR9 in predominantly plasmacytoid dendritic cells (pDCs), resulting in increased IFNα production, and thus activating a signaling pathway more likely to induce autoimmunity [66, 117, 118].

Comparative pathogenic considerations in autoinflammatory and autoimmune interfernopathies. On the left end of the spectrum, in monogenic conditions CANDLE/PRAAS and SAVI, the IFN signature is derived from proteasome suppression leading to IFNA and IFNB1 transcription and constitutively increased IFNB1 transcription, respectively, and are not triggered by immune complex deposition and can thus be called autoinflammatory. In AGS 1–6, sterile pyrexia and CNS damage are likely driven STING- or MAVS-dependent type I IFN production, suggesting innate (autoinflammatory) immune activation triggered by intracellular nucleic acid accumulation. IFIH1 a (AGS7) mutations directly activate MAVS-dependent upregulation of IFN production without nucleic acid accumulation. The incomplete penetrance and variable clinical phenotype with IFIH1 and TREX1 mutations suggests a possible role for modifier genes. FCL and SMS have clinical features not seen in other interfernopathies. Though upregulated interferon-regulated genes are reported in SMS, little information regarding pathogenesis is available. Both autoimmune and autoinflammatory features are seen in the middle box. On the right end of the spectrum, clinical features of monoSLE (C1q deficiency, DNA clearance defects due to deficiency in DNASE1 or DNASE1L3) are likely driven by immune complex deposition (see text). The immune complexes can drive IFN production through activation of interferogenic dendritic and B cell responses or interferogenic release of oxidized mitochondrial DNA, suggesting involvement of adaptive “autoimmune” pathways in disease pathogenesis. Pathogenic antibodies and immune complexes are seen in SLE, and likely in JDM and SSc
Scleroderma (SSc) [119, 120] and juvenile dermatomyositis (JDM) [7, 121] are genetically unsolved autoimmune diseases with prominent blood and tissue IGS that fluctuate with disease severity [7, 119, 120] and have overlapping clinical features with CANDLE and SAVI (JDM: myositis, lipodystrophy, vasculopathy, and lung disease; SSc: vasculopathy and lung fibrosis). A comparison of these conditions with CANDLE, SAVI, and the monogenic forms of SLE will likely provide clues to the pathogenesis of these diseases and to pathways leading to the IFN signature.
Conclusion
Recent studies on a number of monogenic conditions that present with an IFN signatures and overlapping clinical features have provided insights into the different molecular pathways that drive type I IFN production. The comparisons of SAVI and CANDLE with AGS and monogenic forms of SLE (C1q deficiency, DNAse1 deficiency, DNAse1L3 deficiency) point to differences in innate and adaptive immune dysregulation that lead to IFN production. The type I IFN production is induced by constitutive IFNB1 transcription in SAVI, and in most forms of AGS (AGS1-6), the accumulation of nucleic acids triggers innate immune nucleic acid (RNA and DNA) sensors in a STING or MAVS-dependent fashion causing innate immune activaiton. In contrast, when modified, chromatin fragments are exposed in the context of clearance or apoptosis defects that underlie the pathogenesis of monoSLE and polygenic forms of SLE. These modified autoantigens can trigger adaptive immune dysregulation by stimulating autoreactive B cell differentiation and production of interferogenic ICs (i.e., against chromatin and spliceosome antigens) which can trigger IFN responses. In the autoinflammatory (CANDLE and SAVI) and autoimmune (monoSLE, some forms of SLE) interferonopathies, type I IFN leads to an amplification loop that reinforces the underlying abnormal pathomechanisms seen in these conditions and may provide a target for treatment. Insights gained from the study of these monogenic conditions can guide our understanding of other as yet genetically undefined autoinflammatory and autoimmune interferonopathies, guide new drug development, and provide rational approach to the use of emerging therapies targeting the type I IFN pathway.
References
Kastner DL, Aksentijevich I, Goldbach-Mansky R (2010) Autoinflammatory disease reloaded: a clinical perspective. Cell 140:784–790
de Jesus AA, Canna SW, Liu Y, Goldbach-Mansky R (2015) Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu Rev Immunol 33:823–874
Liu Y, Ramot Y, Torrelo A, Paller AS, Si N, Babay S, Kim PW, Sheikh A, Lee CC, Chen Y, Vera A, et al. (2012) Mutations in proteasome subunit beta type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheumatism 64:895–907
Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, et al. (2014) Activated STING in a vascular and pulmonary syndrome. N Engl J Med 371:507–518
Rice GI, Forte GM, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, Ackroyd S, Allcock R, Bailey KM, Balottin U, et al. (2013) Assessment of interferon-related biomarkers in Aicardi-Goutieres syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol 12:1159–1169
Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V (2003) Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 197:711–723
Baechler EC, Bauer JW, Slattery CA, Ortmann WA, Espe KJ, Novitzke J, Ytterberg SR, Gregersen PK, Behrens TW, Reed AM (2007) An interferon signature in the peripheral blood of dermatomyositis patients is associated with disease activity. Mol Med 13:59–68
Crow YJ (2011) Type I interferonopathies: a novel set of inborn errors of immunity. Ann N Y Acad Sci 1238:91–98
Sturfelt G, Truedsson L (2012) Complement in the immunopathogenesis of rheumatic disease. Nat Rev Rheumatol 8:458–468
Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, Kuroda Y (2001) Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet 28:313–314
Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, Al Sonbul A, Sewairi W, Qari A, Abdallah E, et al. (2011) Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet 43:1186–1188
Agarwal AK, Xing C, DeMartino GN, Mizrachi D, Hernandez MD, Sousa AB, Martinez de Villarreal L, dos Santos HG, Garg A (2010) PSMB8 encoding the beta5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Gen 87:866–872
Arima K, Kinoshita A, Mishima H, Kanazawa N, Kaneko T, Mizushima T, Ichinose K, Nakamura H, Tsujino A, Kawakami A, et al. (2011) Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc Natl Acad Sci U S A 108:14914–14919
Kitamura A, Maekawa Y, Uehara H, Izumi K, Kawachi I, Nishizawa M, Toyoshima Y, Takahashi H, Standley DM, Tanaka K, et al. (2011) A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J Clin Invest 121:4150–4160
Torrelo A, Patel S, Colmenero I, Gurbindo D, Lendinez F, Hernandez A, Lopez-Robledillo JC, Dadban A, Requena L, Paller AS (2010) Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. J Am Acad Dermatol 62:489–495
Cavalcante MP, Brunelli JB, Miranda CC, Novak GV, Malle L, Aikawa NE, Jesus AA, Silva CA (2016) CANDLE syndrome: chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature-a rare case with a novel mutation. Eur J Pediatr 175:735–740
Brehm A, Liu Y, Sheikh A, Marrero B, Omoyinmi E, Zhou Q, Montealegre G, Biancotto A, Reinhardt A, Almeida de Jesus A, et al. (2015) Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J Clin Invest 125:4196–4211
Garg A, Hernandez MD, Sousa AB, Subramanyam L, Martinez de Villarreal L, dos Santos HG, Barboza O (2010) An autosomal recessive syndrome of joint contractures, muscular atrophy, microcytic anemia, and panniculitis-associated lipodystrophy. J Clin Endocrinol Metab 95:E58–E63
Kunimoto K, Kimura A, Uede K, Okuda M, Aoyagi N, Furukawa F, Kanazawa N (2013) A new infant case of Nakajo-Nishimura syndrome with a genetic mutation in the immunoproteasome subunit: an overlapping entity with JMP and CANDLE syndrome related to PSMB8 mutations. Dermatology 227:26–30
Ramot Y, Czarnowicki T, Maly A, Navon-Elkan P, Zlotogorski A (2011) Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome: a case report. Pediatr Dermatol 28:538–541
Tanaka M, Miyatani N, Yamada S, Miyashita K, Toyoshima I, Sakuma K, Tanaka K, Yuasa T, Miyatake T, Tsubaki T (1993) Hereditary lipo-muscular atrophy with joint contracture, skin eruptions and hyper-gamma-globulinemia: a new syndrome. Intern Med 32:42–45
McDermott A, Jesus AA, Liu Y, Kim P, Jacks J, Montealegre Sanchez GA, Chen Y, Kannan A, Schnebelen A, Emanuel PD, et al. (2013) A case of proteasome-associated auto-inflammatory syndrome with compound heterozygous mutations. J Am Acad Dermatol 69:e29–e32
Buchbinder D, Montealegre Sanchez GA, Goldbach-Mansky R, Hsieh L, Mahajeran A, Nugent D, Puthenveetil G, Soni A, Stites J, Wacha L, Shulman A (2015) Pulmonary hypertension in two patients with CANDLE syndrome. Clinical Immunology Society 2015 Annual Meeting Houston, Texas
Ciechanover A (2012) Intracellular protein degradation: from a vague idea through the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Neurodegener Dis 10:7–22
Brehm A, Kruger E (2015) Dysfunction in protein clearance by the proteasome: impact on autoinflammatory diseases. Semin Immunopathol 37:323–333
Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, Goudin N, Fremond ML, Nitschke P, Molina TJ, et al. (2014) Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest 124:5516–5520
Omoyinmi E, Melo Gomes S, Nanthapisal S, Woo P, Standing A, Eleftheriou D, Klein N, Brogan PA (2015) Stimulator of interferon genes-associated vasculitis of infancy. Arthritis Rheumatol 67:808
Munoz J, Rodiere M, Jeremiah N, Rieux-Laucat F, Oojageer A, Rice GI, Rozenberg F, Crow YJ, Bessis D (2015) Stimulator of interferon genes-associated vasculopathy with onset in infancy: a mimic of childhood Granulomatosis with Polyangiitis. JAMA Dermatol 151:872–877
Chia J, Eroglu FK, Ozen S, Orhan D, Montealegre-Sanchez G, de Jesus AA, Goldbach-Mansky R, Cowen EW (2016) Failure to thrive, interstitial lung disease, and progressive digital necrosis with onset in infancy. J Am Acad Dermatol 74:186–189
Burdette DL, Vance RE (2013) STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol 14:19–26
Keating SE, Baran M, Bowie AG (2011) Cytosolic DNA sensors regulating type I interferon induction. Trends Immunol 32:574–581
Montealegre Sanchez GA, Reinhardt A, Brogan P, Berkun Y, Brown D, Chira P, Gao L, Chapelle D, Plass N, Kim H, et al. (2013) Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperatures (CANDLE): clinical characterization and initial response to Janus Kinase inhibition with Baricitinib. American College of Rheumatology Arthritis and Rheumatism, San Diego, pp. S758–S759
Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G, et al. (2015) Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A 167A:296–312
Goutieres F, Aicardi J, Barth PG, Lebon P (1998) Aicardi-Goutieres syndrome: an update and results of interferon-alpha studies. Ann Neurol 44:900–907
Orcesi S, La Piana R, Fazzi E (2009) Aicardi-Goutieres syndrome. Br Med Bull 89:183–201
Dale RC, Tang SP, Heckmatt JZ, Tatnall FM (2000) Familial systemic lupus erythematosus and congenital infection-like syndrome. Neuropediatrics 31:155–158
Abdel-Salam GM, El-Kamah GY, Rice GI, El-Darouti M, Gornall H, Szynkiewicz M, Aymard F, Zaki MS, Abdel-Aleem AK, Lebon P, Crow YJ (2010) Chilblains as a diagnostic sign of aicardi-goutieres syndrome. Neuropediatrics 41:18–23
Schmid M, Kreil A, Jessner W, Homoncik M, Datz C, Gangl A, Ferenci P, Peck-Radosavljevic M (2005) Suppression of haematopoiesis during therapy of chronic hepatitis C with different interferon alpha mono and combination therapy regimens. Gut 54:1014–1020
Sanford M, Lyseng-Williamson KA (2011) Subcutaneous recombinant interferon-beta-1a (Rebif(R)): a review of its use in the treatment of relapsing multiple sclerosis. Drugs 71:1865–1891
Ramantani G, Kohlhase J, Hertzberg C, Innes AM, Engel K, Hunger S, Borozdin W, Mah JK, Ungerath K, Walkenhorst H, et al. (2010) Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutieres syndrome. Arthritis Rheum 62:1469–1477
Hochberg MC (1997) Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 40:1725
Okon LG, Werth VP (2013) Cutaneous lupus erythematosus: diagnosis and treatment. Best Pract Res Clin Rheumatol 27:391–404
Halla JT, Schrohenloher RE, Volanakis JE (1980) Immune complexes and other laboratory features of pleural effusions: a comparison of rheumatoid arthritis, systemic lupus erythematosus, and other diseases. Annals Int Med 92:748–752
Jesus AA, Goldbach-Mansky R (2014) IL-1 blockade in autoinflammatory syndromes. Ann Rev Med 65:223–244
Beachboard DC, Horner SM (2016) Innate immune evasion strategies of DNA and RNA viruses. Curr Opin Microbiol 32:113–119
Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, et al. (2006a) Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet 38:917–920
Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, et al. (2006b) Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet 38:910–916
Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, et al. (2009) Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet 41:829–832
Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, et al. (2012) Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet 44:1243–1248
Rice GI, del Toro Duany Y, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, Bader-Meunier B, Baildam EM, Battini R, Beresford MW, et al. (2014) Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet 46:503–509
Oda H, Nakagawa K, Abe J, Awaya T, Funabiki M, Hijikata A, Nishikomori R, Funatsuka M, Ohshima Y, Sugawara Y, et al. (2014) Aicardi-Goutieres syndrome is caused by IFIH1 mutations. Am J Hum Genet 95:121–125
Chowdhury D, Beresford PJ, Zhu P, Zhang D, Sung JS, Demple B, Perrino FW, Lieberman J (2006) The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol Cell 23:133–142
Ahn J, Urist M, Prives C (2004) The Chk2 protein kinase. DNA Repair (Amst) 3:1039–1047
Yang YG, Lindahl T, Barnes DE (2007) Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 131:873–886
Stetson DB, Ko JS, Heidmann T, Medzhitov R (2008) Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134:587–598
Gall A, Treuting P, Elkon KB, Loo YM, Gale M Jr, Barber GN, Stetson DB (2012) Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity 36:120–131
Wahba L, Amon JD, Koshland D, Vuica-Ross M (2011) RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol Cell 44:978–988
Nadel J, Athanasiadou R, Lemetre C, Wijetunga NA, OB P, Sato H, Zhang Z, Jeddeloh J, Montagna C, Golden A, Seoighe C, Greally JM (2015) RNA:DNA hybrids in the human genome have distinctive nucleotide characteristics, chromatin composition, and transcriptional relationships. Epigenetics Chromatin 8:46
Gunther C, Kind B, Reijns MA, Berndt N, Martinez-Bueno M, Wolf C, Tungler V, Chara O, Lee YA, Hubner N, et al. (2015) Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. J Clin Invest 125:413–424
Mackenzie KJ, Carroll P, Lettice L, Tarnauskaite Z, Reddy K, Dix F, Revuelta A, Abbondati E, Rigby RE, Rabe B, et al. (2016) Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J 35:831–844
Kretschmer S, Wolf C, Konig N, Staroske W, Guck J, Hausler M, Luksch H, Nguyen LA, Kim B, Alexopoulou D, et al. (2015) SAMHD1 prevents autoimmunity by maintaining genome stability. Ann Rheum Dis 74:e17
Pokatayev V, Hasin N, Chon H, Cerritelli SM, Sakhuja K, Ward JM, Morris HD, Yan N, Crouch RJ (2016) RNase H2 catalytic core Aicardi-Goutieres syndrome-related mutant invokes cGAS-STING innate immune-sensing pathway in mice. J Exp Med 213:329–336
Sze A, Belgnaoui SM, Olagnier D, Lin R, Hiscott J, van Grevenynghe J (2013) Host restriction factor SAMHD1 limits human T cell leukemia virus type 1 infection of monocytes via STING-mediated apoptosis. Cell Host Microbe 14:422–434
Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR (2015) RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349:1115–1120
Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ, et al. (2014) The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9:1482–1494
Akwa Y, Hassett DE, Eloranta ML, Sandberg K, Masliah E, Powell H, Whitton JL, Bloom FE, Campbell IL (1998) Transgenic expression of IFN-alpha in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J Immunol 161:5016–5026
Lee-Kirsch MA, Gong M, Schulz H, Ruschendorf F, Stein A, Pfeiffer C, Ballarini A, Gahr M, Hubner N, Linne M (2006) Familial chilblain lupus, a monogenic form of cutaneous lupus erythematosus, maps to chromosome 3p. Am J Hum Genet 79:731–737
Lee-Kirsch MA, Chowdhury D, Harvey S, Gong M, Senenko L, Engel K, Pfeiffer C, Hollis T, Gahr M, Perrino FW, Lieberman J, Hubner N (2007) A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J Mol Med (Berl) 85:531–537
Ravenscroft JC, Suri M, Rice GI, Szynkiewicz M, Crow YJ (2011) Autosomal dominant inheritance of a heterozygous mutation in SAMHD1 causing familial chilblain lupus. Am J Med Genet A 155A:235–237
König N, Fiehn C, Wolf C, Schuster M, Costa EC, Tüngler V, Alvarez HA, Chara O, Engel K, Goldbach-Mansky R, Günther C, Lee-Kirsch M (2016) Familial chilblain lupus due to a gain-of-function mutation in STING. Ann Rheum Dis. doi:10.1136/annrheumdis-2016-209841
Gunther C, Berndt N, Wolf C, Lee-Kirsch MA (2015) Familial chilblain lupus due to a novel mutation in the exonuclease III domain of 3' repair exonuclease 1 (TREX1). JAMA Dermatol 151: 426-431. doi:10.1001/jamadermatol.2014.3438
Rice GI, Rodero MP, Crow YJ (2015) Human disease phenotypes associated with mutations in TREX1. J Clin Immunol 35:235–243
Peschke K, Friebe F, Zimmermann N, Wahlicht T, Schumann T, Achleitner M, Berndt N, Luksch H, Behrendt R, Lee-Kirsch MA, Roers A, Gunther C (2014) Deregulated type I IFN response in TREX1-associated familial chilblain lupus. J Invest Dermatol 134:1456–1459
Sugiura K, Takeichi T, Kono M, Ito Y, Ogawa Y, Muro Y, Akiyama M (2012) Severe chilblain lupus is associated with heterozygous missense mutations of catalytic amino acids or their adjacent mutations in the exonuclease domains of 3'-repair exonuclease 1. J Invest Dermatol 132:2855–2857
Richards A, van den Maagdenberg AM, Jen JC, Kavanagh D, Bertram P, Spitzer D, Liszewski MK, Barilla-Labarca ML, Terwindt GM, Kasai Y, et al. (2007) C-terminal truncations in human 3'-5' DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet 39:1068–1070
Schuh E, Ertl-Wagner B, Lohse P, Wolf W, Mann JF, Lee-Kirsch MA, Hohlfeld R, Kumpfel T (2015) Multiple sclerosis-like lesions and type I interferon signature in a patient with RVCL. Neurol Neuroimmunol Neuroinflamm 2:e55
Jang MA, Kim EK, Now H, Nguyen NT, Kim WJ, Yoo JY, Lee J, Jeong YM, Kim CH, Kim OH, et al. (2015) Mutations in DDX58, which encodes RIG-I, cause atypical singleton-Merten syndrome. Am J Hum Genet 96:266–274
Rutsch F, MacDougall M, Lu C, Buers I, Mamaeva O, Nitschke Y, Rice GI, Erlandsen H, Kehl HG, Thiele H, et al. (2015) A specific IFIH1 gain-of-function mutation causes singleton-Merten syndrome. Am J Hum Genet 96:275–282
Bursztejn AC, Briggs TA, del Toro Duany Y, Anderson BH, O'Sullivan J, Williams SG, Bodemer C, Fraitag S, Gebhard F, Leheup B, et al. (2015) Unusual cutaneous features associated with a heterozygous gain-of-function mutation in IFIH1: overlap between Aicardi-Goutieres and singleton-Merten syndromes. Br J Dermatol 173:1505–1513
Lintner KE, YL W, Yang Y, Spencer CH, Hauptmann G, Hebert LA, Atkinson JP, Yu CY (2016) Early components of the complement classical activation pathway in human systemic autoimmune diseases. Front Immunol 7:36
Vergani D, Wells L, Larcher VF, Nasaruddin BA, Davies ET, Mieli-Vergani G, Mowat AP (1985) Genetically determined low C4: a predisposing factor to autoimmune chronic active hepatitis. Lancet 2:294–298
Samano ES, Ribeiro Lde M, Gorescu RG, Rocha KC, Grumach AS (2004) Involvement of C4 allotypes in the pathogenesis of human diseases. Rev Hosp Clin Fac Med Sao Paulo 59:138–144
Mampaso F, Ecija J, Fogue L, Moneo I, Gallego N, Leyva-Cobian F (1981) Familial C1q deficiency in 3 siblings with glomerulonephritis and Rothmund-Thomson syndrome. Nephron 28:179–185
Topaloglu R, Bakkaloglu A, Slingsby JH, Mihatsch MJ, Pascual M, Norsworthy P, Morley BJ, Saatci U, Schifferli JA, Walport MJ (1996) Molecular basis of hereditary C1q deficiency associated with SLE and IgA nephropathy in a Turkish family. Kidney Int 50:635–642
Santer DM, Hall BE, George TC, Tangsombatvisit S, Liu CL, Arkwright PD, Elkon KB (2010) C1q deficiency leads to the defective suppression of IFN-alpha in response to nucleoprotein containing immune complexes. Journal of immunology 185: 4738-4749. doi:10.4049/jimmunol.1001731
Slingsby JH, Norsworthy P, Pearce G, Vaishnaw AK, Issler H, Morley BJ, Walport MJ (1996) Homozygous hereditary C1q deficiency and systemic lupus erythematosus. A new family and the molecular basis of C1q deficiency in three families. Arthritis Rheum 39:663–670
Vassallo G, Newton RW, Chieng SE, Haeney MR, Shabani A, Arkwright PD (2007) Clinical variability and characteristic autoantibody profile in primary C1q complement deficiency. Rheumatology 46:1612–1614
American College of Rheumatology (2016) Image Library
Malar rash [digital image] (2012) Retrieved from http://www.mollysfund.org/2012/11/the-lupus-butterfly-rash-or-malar-rash-information-you-need-to-know/
Shiau CJ, Abi Daoud MS, Wong SM, Crawford RI (2015) Lymphocytic panniculitis: an algorithmic approach to lymphocytes in subcutaneous tissue. J Clin Pathol 68:954–962
Keane MP, Lynch JP III (2000) Pleuropulmonary manifestations of systemic lupus erythematosus. Thorax 55:159–166
Rosen A, Casciola-Rosen L (2016) Autoantigens as partners in initiation and propagation of autoimmune rheumatic diseases. Annu Rev Immunol 34:395–420
Mahajan A, Herrmann M, Munoz LE (2016) Clearance deficiency and cell death pathways: a model for the pathogenesis of SLE. Front Immunol 7:35
Botto M (1998) C1q knock-out mice for the study of complement deficiency in autoimmune disease. Exp Clin Immunogenet 15:231–234
Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T (2000) Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet 25:177–181
Fismen S, Mortensen ES, Rekvig OP (2011) Nuclease deficiencies promote end-stage lupus nephritis but not nephritogenic autoimmunity in (NZB x NZW) F1 mice. Immunol Cell Biol 89:90–99
Sisirak V, Sally B, D'Agati V, Martinez-Ortiz W, Ozcakar ZB, David J, Rashidfarrokhi A, Yeste A, Panea C, Chida AS, et al. (2016) Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell 166:88–101
Gaipl US, Beyer TD, Heyder P, Kuenkele S, Bottcher A, Voll RE, Kalden JR, Herrmann M (2004) Cooperation between C1q and DNase I in the clearance of necrotic cell-derived chromatin. Arthritis Rheum 50:640–649
Casciola-Rosen LA, Anhalt G, Rosen A (1994) Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med 179:1317–1330
Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, Kaplan MJ (2016) Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 22:146–153
Biermann MH, Veissi S, Maueroder C, Chaurio R, Berens C, Herrmann M, Munoz LE (2014) The role of dead cell clearance in the etiology and pathogenesis of systemic lupus erythematosus: dendritic cells as potential targets. Expert Rev Clin Immunol 10:1151–1164
Fransen JH, Hilbrands LB, Ruben J, Stoffels M, Adema GJ, van der Vlag J, Berden JH (2009a) Mouse dendritic cells matured by ingestion of apoptotic blebs induce T cells to produce interleukin-17. Arthritis Rheum 60:2304–2313
Scaffidi P, Misteli T, Bianchi ME (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418:191–195
Fransen JH, Hilbrands LB, Jacobs CW, Adema GJ, Berden JH, Van der Vlag J (2009b) Both early and late apoptotic blebs are taken up by DC and induce IL-6 production. Autoimmunity 42:325–327
Munoz LE, Janko C, Grossmayer GE, Frey B, Voll RE, Kern P, et al. Remnants of secondarily necrotic cells fuel inflammation in systemic lupus erythematosus. Arthritis and rheumatism. 2009;60(6):1733–42
Kono DH, Haraldsson MK, Lawson BR, Pollard KM, Koh YT, Du X, Arnold CN, Baccala R, Silverman GJ, Beutler BA, Theofilopoulos AN (2009) Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc Natl Acan Sci USA 106:12061–12066
Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A (2002) Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 416:603–607. doi:10.1038/416603a
Nangaku M, Couser WG (2005) Mechanisms of immune-deposit formation and the mediation of immune renal injury. Clin Exp Nephrol 9:183–191
Bouts YM, Wolthuis DF, Dirkx MF, Pieterse E, Simons EM, van Boekel AM, Dieker JW, van der Vlag J (2012) Apoptosis and NET formation in the pathogenesis of SLE. Autoimmunity 45:597–601
Ricklin D, Hajishengallis G, Yang K, Lambris JD (2010) Complement: a key system for immune surveillance and homeostasis. Nat Immunol 11:785–797
Farrera C, Fadeel B (2013) Macrophage clearance of neutrophil extracellular traps is a silent process. J Immunol 191:2647–2656
Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE, Zychlinsky A (2010) Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acan Sci USA 107:9813–9818
Kiefer K, Oropallo MA, Cancro MP, Marshak-Rothstein A (2012) Role of type I interferons in the activation of autoreactive B cells. Immunol Cell Biol 90:498–504
Martinelli S, Urosevic M, Daryadel A, Oberholzer PA, Baumann C, Fey MF, Dummer R, Simon HU, Yousefi S (2004) Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J Biol Chem 279:44123–44132
Knight JS, Carmona-Rivera C, Kaplan MJ (2012) Proteins derived from neutrophil extracellular traps may serve as self-antigens and mediate organ damage in autoimmune diseases. Front Immunol 3:380
Trinchieri G (2010) Type I interferon: friend or foe? J Exp Med 207:2053–2063
Briggs TA, Rice GI, Daly S, Urquhart J, Gornall H, Bader-Meunier B, Baskar K, Baskar S, Baudouin V, Beresford MW, et al. (2011) Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat Genet 43:127–131
Lausch E, Janecke A, Bros M, Trojandt S, Alanay Y, De Laet C, Hubner CA, Meinecke P, Nishimura G, Matsuo M, et al. (2011) Genetic deficiency of tartrate-resistant acid phosphatase associated with skeletal dysplasia, cerebral calcifications and autoimmunity. Nat Genet 43:132–137
Eloranta ML, Franck-Larsson K, Lovgren T, Kalamajski S, Ronnblom A, Rubin K, Alm GV, Ronnblom L (2010) Type I interferon system activation and association with disease manifestations in systemic sclerosis. Ann Rheum Dis 69:1396–1402
Higgs BW, Liu Z, White B, Zhu W, White WI, Morehouse C, Brohawn P, Kiener PA, Richman L, Fiorentino D, Greenberg SA, Jallal B, Yao Y (2011) Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann Rheum Dis 70:2029–2036
Cappelletti C, Baggi F, Zolezzi F, Biancolini D, Beretta O, Severa M, Coccia EM, Confalonieri P, Morandi L, Mora M, Mantegazza R, Bernasconi P (2011) Type I interferon and toll-like receptor expression characterizes inflammatory myopathies. Neurology 76:2079–2088
Urbonaviciute V, Furnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F, Bianchi ME, Kirschning C, Wagner H, Manfredi AA, et al. (2008) Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med 205:3007–3018
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, NIAID, and NIAMS. The authors would like to thank Dr. Ann Marschuk-Rothstein and Dr. Adriana Almeida de Jesus for helpful discussions.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Disclosures
HK does not have any disclosures. GMS has received study support from SOBI, Regeneron, and Lilly. RGM has received study support from SOBI, Novartis, Regeneron and Lilly.
Rights and permissions
About this article

Cite this article
Kim, H., Sanchez, G.A.M. & Goldbach-Mansky, R. Insights from Mendelian Interferonopathies: Comparison of CANDLE, SAVI with AGS, Monogenic Lupus. J Mol Med 94, 1111–1127 (2016). https://doi.org/10.1007/s00109-016-1465-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-016-1465-5