
Abstract
During innate immune responses, proteostasis is greatly impacted by synthesis of pathogen proteins as well as by inflammatory tissue damage through radicals or other damaging molecules released by phagocytes. An adequate adaptation of cellular clearance pathways to the increased burden of damaged proteins is thus of fundamental importance for cells and tissues to prevent protein aggregation, inclusion body formation, and ultimately cell death. We here review the current understanding of the pivotal role of the ubiquitin proteasome system (UPS) in this proteostasis network. The proteolytic capacity of the UPS can be adjusted by differential gene expression, the incorporation and maturation kinetics of alternative active sites, and the attachment of different regulators. Dysregulation of this fine-tuning is likely to induce cell death but seen more often to promote inflammation as well. The link between proteostasis impairment and inflammation may play a crucial role in autoinflammation as well as in age-related diseases and currently uncharacterized diseases. Recent studies on proteasome-associated autoinflammatory syndromes (PRAAS) discovered that IFN signaling drives the inflammation caused by reduction of degradation capacity. Elucidation of these syndromes will reveal further insights in the understanding of inadequate immune responses. Knowledge related to the diversity of this degradation system will raise the awareness of potential pitfalls in the molecular diagnostics of autoinflammatory syndromes and may help to identify novel drug targets.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Autoinflammation is primarily the result of alterations in the innate immune system and shows a broad range of clinical symptoms. Great advances in genotyping efficiency, sequencing technology, and bioinformatical evaluation of patients with autoinflammatory syndromes have revealed single-gene mutations for example in critical components relating to danger sensing and cytokine regulation of the innate immune response, for example, the interleukin (IL)-1, IL-10, NF-κB, and interferon (IFN) signaling cascades [1, 2]. Interestingly, mutations or variants in components of the protein clearance pathways such as the cytokine-induced immunoproteasome subunit PSMB8 (LMP7/β5i) or components of the autophagy pathway were also identified, clearly linking proteinopathies with innate immune responses [3–5].
Cellular proteostasis is a delicate balance between protein synthesis and protein quality control with protein degradation. Perturbations in the cell physiology through environmental stimuli require a fast and coordinated adjustment of translational systems, chaperone supply, and various proteolytic systems. It is increasingly realized that during innate immune responses proteostasis is severely challenged by the production of radicals and/or the massive synthesis of pathogen proteins. The adequate adaptation of their cellular clearance pathways to the increased requirement for protein degradation is thus of fundamental importance for cells and tissues, which are otherwise faced with the threat of an accumulation of faulty, damaged or misfolded proteins, followed by protein aggregation, inclusion body formation, and ultimately cell death. The cells choke on their protein trash as shown for neurodegenerative disorders as classical example for proteinopathies. An impairment of proteostasis systems reflected by intra- and extracellular protein deposits accompanied by oxidative stress, neuroinflammation, and progressive degeneration of specific brain regions all represent hallmarks of these diseases [6–8]. A further example of disturbance in proteostasis in disease is the aging-related sporadic inclusion body myositis, where abnormal intracellular protein accumulation and aggregation is symptomatic combined with proteasome dysregulation and endoplasmic reticulum (ER) stress. Patients suffer from inflammatory myopathy characterized by progressive weakness and wasting of muscle [4]. Restoration of proteostasis network function therefore leads to aggregate detoxification and clearance via chaperone systems and proteolysis by the UPS, autophagy, and/or phagocytosis as an important extracellular pathway. Lysosomal degradation via autophagy has been shown to be crucial for clearance of prion aggregates in skeletal muscle [9] in particular because prions tend to inhibit the ubiquitin proteasome system (UPS) via stabilization of the closed barrel formation [10].
In this review, we will focus on the UPS protein clearance pathway, the inhibition of which, either by aging, mutation, or protein interaction, has a dramatic impact on the proteostasis in cells and leads to proteotoxic stress followed by diverse stress responses such as the unfolded protein response (UPR) with chaperone induction, induction of autophagy, apoptosis, and inflammation via the innate immune response. In particular, the proteasome associated autoinflammatory syndromes (PRAAS), in which the UPS is impaired in all cells of the body, clearly reflects how dynamic adaption of the protein degradation capacity is essential for cell surveillance and a regulated immune response.
The ubiquitin proteasome system
Regulated proteolysis of ubiquitylated and/or oxidative damaged proteins via the UPS plays a key role governing the vitality of eukaryotic cells and tissues, whereby the availability of short-lived proteins is determined and the accumulation of misfolded and damaged proteins is prevented to ensure protein homeostasis. The UPS fulfills the essential regulation of numerous important cellular processes, such as gene transcription, DNA repair, apoptosis, cell cycle, cell differentiation, and signaling, as well as the generation of antigenic peptides presented by major histocompatibility (MHC) class I molecules [11, 12].
Most of the proteins to be degraded are modified with lysine 48 (K48)-linked poly-ubiquitin chains that serve as a signal for degradation. This modification is conjugated to proteasomal substrates by a three-step thioester cascade involving the E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzymes, and E3-ubiquitin ligases [13]. In line with its multitude of functions, the proteasome appears in mammals in a variety of compositions to allow for adaptation to changing proteolytic requirements (Fig. 1). The modular architecture of the proteasome consists of a 20S catalytic core complex associated with one or two regulator complexes. The barrel-shaped 20S proteasome is formed by four stacked rings of seven subunits each with the arrangement of α1–α7 (β1–β7)2 α1–α7. With the β1, β2, and β5 subunits, the two inner β-rings harbor six catalytically active sites with caspase-like, tryptic-like, and chymotryptic-like activities, respectively [14]. Several proteasome isoforms consisting of alternative compositions can exist in parallel to the above-described standard proteasome (SP). Constitutively in hematopoetic cells and in other cells in response to infections or cytokines like type I or type II interferons (IFNs), the immunosubunits β1i/LMP2/PSMB9, β2i/Mecl1/PSMB10, and β5i/LMP7/PSMB8 are expressed and rapidly incorporated into nascent proteasome complexes to form immunoproteasomes (IP). There is, however, also the possibility that just one or two of the immunosubunits are incorporated and mixed-type proteasomes (MP) are formed. In addition, a further alternative catalytic subunit namely β5t/PSMB11 is solely expressed in thymic cortical epithelial cells forming the thymoproteasome (TP), and finally, the spermatoproteasome containing an alternative α-subunit is expressed in the testis during spermatogenesis. Alongside these different 20S isoforms, the regulatory complexes 19S, PA28αβ, PA28γ, and PA200 bind to one or both sides of the core particle, open the catalytic pore, and form several complexes such as the 26S (20S + 19S), 30S (20S + 2 × 19S), 20S-PA28, 20S-PA200, and hybrid proteasomes (HP) (20S + 19S + PA28 and 20S + 19S + PA200) [15–18]. It was reported that PA28αβ preferentially binds to IP [19]. While the catalytic activity is located in the 20S proteasome core complex, the 19S regulatory complex governs the recognition, unfolding, and access of ubiquitin-conjugated substrates into the catalytic cavity. Thus, only 19S containing proteasome isoforms (SP, IP, PA28-HP) are involved in the degradation of ubiquitin-conjugated substrates, and their impact in the development of diseases will be further discussed in more detail.

Proteasome diversity upon cellular requirements. In mammalian cells, a variety of proteasomes is formed according to cell type and challenging status to ensure an appropriate adaption to protein degradation requirements. 20S proteasomes are barrel-shaped and consist of four seven-membered rings in the order of α1–α7 (β1–β7)2 α1–α7. The stepwise assembly of the 20S proteasome is assisted by the proteasome assembly chaperones (PAC) 1–4 and the proteasome maturation protein (POMP). After 16S precursor dimerization and completed maturation of the β-subunits β1, 2, 5, 6, and 7, the latent inactive 20S proteasome is activated through the binding of regulatory particles such as 19S, PA28αβ, PA28γ, and PA200 on one or both sides of the proteasome, which open the catalytic pore. According to regulator combinations, the 26S/30S (20S + 19S/20S + 2x19S), the 20S-PA28, 20S-PA200, and the hybrid proteasomes with two differing regulators (20S + 19S + PA28 or 20S + 19S + PA200) can be formed. Besides the standard proteasome with the catalytic subunits β1, 2, and 5, alternative catalytic subunits such as β1i, β2i, β5i, and β5t can be incorporated, forming the immuno-proteasome (β1i, 2i, 5i), the thymo-proteasome (β1i, 2i, 5t), and mixed-type proteasome (β1, 2, 5i or β1, 2i, 5i). During spermatogenesis, the alternative α-subunit α4s is expressed, leading to spermato-proteasome formation [16–18, 58]. The asterisk refers to the main isoforms expressed in most cell types and are described in more detail in the text
The proteolytic capacity of the UPS can be tailored to particular proteolytic requirements by differential gene expression of specific subsets of UPS factors and the assembly of different proteasome isoforms. Depending on the cell type or the tissue context, different proteasome modules can be expressed and coexist in cells. Postmitotic cells like neurons or muscle cells mainly contain SP, whereas immune cells like myeloid or lymphoid cells mainly contain IP or HP. The UPS can be adapted to proteotoxic stress by the Nrf1/Nrf2/ARE-activated pathway, which induces the SP and certain UPS-related genes [20–22].
IPs and HPs in turn are permanently expressed in immune cells and can be induced not only by cytokine or developmental signaling, in particular by IFNs, but also by heat stress or during the course of neurodegenerative diseases in almost all other cell types. The UPS is responsible for the generation of the vast majority of intracellular pathogen-derived and self-peptides presented by MHC class I molecules at the cell surface to cytotoxic T lymphocytes. This function of the adaptive immune system is generally aided by cytokine-mediated induction of IP, which enhances MHC class I antigen presentation by improved antigen processing [11]. An additional IP function has been uncovered in maintaining protein homeostasis under cytokine-induced oxidative stress [21]. IFNs induce the intracellular production of radicals thus increasing the content of oxidant-damaged proteins and increased sensitivity to cytokine-induced cell death. Such oxidant-damaged proteins are immediately ubiquitinated and require the high proteolytic capacity of IP for their efficient removal. As nascent polypeptides are particularly sensitive to oxidation, IFN-signaling enhances the rate of defective nascent proteins and concomitantly MHC class I antigen presentation, placing IP function at the interface of innate and adaptive immune response. IP-expressing cells such as phagocytes including dendritic cells or microglia are thus better equipped to handle cytokine-induced oxidative protein aggregation. The particular function of HP is still under investigation. However, different studies indicate the involvement of PA28 and HP in the removal of damaged proteins under oxidative stress [20, 23].
Interestingly, LMP7 (PSMB8) knockout mice do not obviously show any lipodystrophy or autoinflammatory defect and are physically undistinguishable from wild-type mice [24], but they are more susceptible to some infections, and pathogen clearance is prolonged or inadequate. In particular, an example for the CNS is that IP deficiency was found to result in significantly increased clinical scores by studying autoimmune encephalomyelitis (EAE) in a mouse model [21]. IP deficiency was further associated with severe heart muscle injury with large inflammatory lesions and severe myocardial tissue damage in a mouse model for Coxsackie virus B3-induced myocarditis [25]. This protective role is also evidenced by studies showing that IP-deficient mice severely suffer from impaired stress responses, survival, and/or clearance rates upon infection or inflammation [26–29] or develop severe LPS-induced hepatitis [21]. Moreover, IP dysfunction is connected with (auto)inflammation and autoimmunity, often accompanied by altered cytokine patterns, such as diabetes or Sjögren syndrome [30–33].
Taken together, these data evidence that UPS-controlled proteostasis is important for immune responses to ensure both a balanced response and protection for non-infected cells.
Proteasome-associated autoinflammatory syndromes
More recently, several previously described autoinflammatory syndromes, such as Nakajo-Nishimura syndrome (NNS) [34], joint contractures, muscle atrophy, microcytic anemia and panniculitis-induced lipodystrophy (JMP) syndrome [35], Japanese autoinflammatory syndrome with lipodystrophy (JASL) [36], and chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) [5], were found to have a mutated PSMB8/β5i in common and are now classified as a spectrum of diseases named proteasome-associated autoinflammatory syndrome (PRAAS).
The clinical presentation of PRAAS patients is diverse in severity and shows a broad range of symptoms, including a pernio-like rash and repetitive spiking fever with progressive lipodystrophy starting in early infancy. Other reported symptoms include nodular erythema with infiltration and induration, erythema on the eyelids, long clubbed fingers and toes with joint contractures, progressive partial lipomuscular atrophy and emaciation mainly of the upper body, hepatosplenomegaly and basal ganglia calcification, chronic anemia, and delayed physical development [5, 34–39]. A detailed summary of the clinical presentation was reviewed by McDermott et al. [40].
To date, an autosomal recessive inheritance of four missense mutations and one non-sense mutation in PSMB8 (p.T75M; p.G201V; p.A92T; p.M117V; p.C135X) have been implicated to cause PRAAS [5, 34–37, 41]. The reported mutations have different impacts on the proteasome by modulation of gene expression, subunit folding and maturation, assembly of the core complex, and/or structural alterations of the proteolytic pocket [5, 34, 36], but all result in reduced proteasome activity [35, 36] which is probably insufficient to cope with a higher load of damaged proteins. In collaboration with the group of R. Goldbach-Mansky [5], we started to investigate an increasing number of patients with autoinflammatory syndromes similar to CANDLE but without the described mutations in PSMB8. From the knowledge about the diversity of the proteasome system, we hypothesize the presence of additional mutation sites in further proteasome components, which may affect the assembly, the activity, and even the regulation of proteasome complexes or other UPS-related components. There is indeed evidence from our collaboration that there are mutations in proteasome components other than PSMB8.
These findings strongly suggest that the disturbance of the overall proteasome activity and capacity in response to environmental stress is crucial, not just a disturbance of the inducible catalytically subunit β5i. We therefore strongly advise for diagnostic sequencing to broaden the spectrum of candidate genes up to all 20S subunits, assembly helpers, and regulatory particle subunits (Table 1).
Laboratory findings and pathogenesis
A strongly reduced chymotryptic-like activity was observed in Epstein-Barr virus (EBV)-transformed B cells from PRAAS patients compared to healthy controls [35, 36], and an accumulation of poly-ubiquitylated proteins can be detected in skin sections, EBV-transformed B cells, and fibroblasts from patients [34, 36]. In the skeletal muscle cells from a deceased suspected CANDLE patient, intramitochondrial paracrystalline inclusions and cytoplasmatic and myeloid bodies were observed [42], indicating accumulation of damaged/aggregated proteins. Aggregates and inclusions increase cellular sensitivity to apoptosis [21], and this could be a cause of muscle loss later in life.
Interestingly, adipocyte differentiation seemed to be reduced after PSMB8 siRNA treatment of preadipocytes, suggesting that a high proteasomal activity is needed for adipocyte differentiation [36].
In skin biopsy, Kitamura et al. observed a much higher interleukin-6 (IL-6) expression (tenfold) compared to a healthy control patient. They further showed an IL-6 hyperactivation of the patient’s EBV-B cells (PSMB8 G201V +/+) after ionomycin/PMA stimulation, which could be partially rescued by p38 inhibition or PSMB8 WT retroviral overexpression. In another study using mouse macrophages, inhibition of the proteasome with lactacystin results in an increased p38 and JNK phosphorylation [43], supporting the results from Kitamura et al. Arima et al. detected higher p38 phosphorylation in patients’ fibroblasts as well and found no deregulation of NF-κB, JNK1/2/3, and ERK1/2 [34]. In contrast, Liu et al. showed that cytokine analysis of blood serum revealed only moderately elevated levels of monocyte chemotactic protein 1 (MCP1), IL-6, and IL-1 receptor antagonist. However, a characteristic persistent IFN signature evidenced by high levels of IP-10 (CXCL10) was observed and verified by microarray analysis of whole blood, revealing 48 IFN-regulated genes. Furthermore, the downstream mediator of IFN signaling, STAT-1, showed a much stronger phosphorylation after IFNγ stimulation of patient monocytes compared to a healthy control patient [5]. In line with Liu et al., reported abnormally high levels of IL-6 [34, 35, 37], IL-8, IP-10, and IFN-γ in serum of affected patients [35, 37]. This together with the accumulation of ubiquitylated proteins strongly suggests that reduced proteasome activity leads to proteotoxic stress, which is linked by an as yet unidentified pathway to the innate immune response via IFN signaling and p38.
Model of pathomechanism
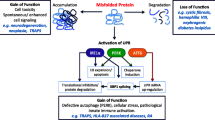
We here suggest a model of the pathomechanism involved in PRAAS (Fig. 2), an overview of observed findings which will need to be further elaborated in a cell type-specific manner. Because PRAAS flares coincide with infections and other stressful events [5], we suggest environmental stress as the trigger, initiating overwhelming inflammation. The skin and cells from the hematopoietic system, such as dendritic cells and macrophages, establish the first line of defense. A bacterial or viral infection leads to activation of pattern recognition receptors (PRR) such as Toll-like receptors (TLRs) via identification of pathogen-associated molecular patterns (PAMP), such as lipopolysaccharides or nucleic acids. Multiple signaling cascades can be induced after pathogen recognition, which result in expression of genes involved in the specific immune response such as cytokines, particularly IFN type I, enzymes for production of reactive oxygen species (ROS), reactive nitric species (RNS), and antimicrobiotics [44, 45]. This initial “normal” inflammation leads to a higher level of defective ribosomal products (DRiPs) and oxidant-damaged proteins due to temporal attenuation of global protein synthesis as result of eIF2a phosphorylation and release of ROS such as superoxide anions, hydrogen peroxide, and hydroxylradicals [46, 47]. Due to the cell’s inability to adapt the proteasomal capacity to levels required to degrade the raising amount of impaired proteins, aggregates of damaged proteins are formed [21, 47, 48]. Furthermore, ER stress is induced through the activity of ROS, and the unfolded protein response (UPR) is activated. Four different pathways are induced by ER stress; these are the pancreatic ER kinase (PERK) pathway, the inositol-requiring transmembrane kinase/endonuclease 1 (IRE1) pathway, the activating transcription factor 6 (ATF6) pathway, and the inflammasome pathway [49–52]. We can, however, exclude the inflammasome pathway as a main mechanism in PRAAS because patients are nonresponders for IL-1β antagonistic drugs [5]. The detailed mechanism which leads from accumulation of damaged proteins in the cytosol and ER to the induction of IFN type I expression needs to be further elucidated, but with this induction, a vicious circle of overwhelming inflammation starts.

Overwhelming inflammation caused by proteasome impairment. Sensing of stressors such as virus, bacteria, and low temperatures through pattern recognition receptors (PPRs) leads to the induction of the innate immune response with secretion of cytokines and activation and expression of NOX (NADPH-oxidase) enzymes. Reactive oxygen species (ROS) are produced, which lead to an increased load of oxidative damaged and misfolded foreign- and self-proteins in the cytosol and endoplasmic reticulum (ER). An impairment of the upregulation of the proteasomal degradation capacity results in the formation of damaged protein aggregates. This effect is enhanced by further autocrine and paracrine IFNα/β and IFNγ responses. The accumulation of damaged proteins induces ER stress with the following unrelated protein response (UPR) acting via the pancreatic ER kinase (PERK) pathway, the inositol-requiring transmembrane kinase/endonuclease 1 (IRE1) pathway, and the activating transcription factor 6 (ATF6) pathway PERK. Furthermore, through a still unknown mechanism, proteasome impairment triggers IFN type I expression and leads to a vicious circle of IFN signaling and response
Diagnostics for proteasome-associated autoinflammatory syndromes
Many patients pass through a long period of misdiagnosis associated with variable but not often beneficial treatments because the full features of PRAAS develop over time. First described for NNS but also suitable for CANDLE, JMP and JALS, a tentative set of eight features is used as criteria for clinical diagnosis, whereby at least five of these features need to be fulfilled. The criteria are the following: 1. autosomal recessive inheritance (parental consanguinity and/or familial occurrence), 2. pernio-like purplish rash on hands and feet (appearing in winter since infancy), 3. haunting nodular erythema with infiltration and induration (sometimes circumscribed), 4. repetitive spiking fever (periodic, not necessarily), 5. long clubbed fingers and toes with joint contractures, 6. progressive partial lipomuscular atrophy and emaciation (marked in the upper part of the body), 7. Hepatosplenomegaly, and 8. basal ganglia calcification [53]. It is clear that there is an urgent need for improved diagnosis in the early infancy before full symptoms are apparent, helping to avoid misdiagnosis like dermatosis, lupus profundus, systemic lupus erythematous, Weber-Christian disease, cryopyrin-associated periodic syndrome, and inclusion body myositis [53].
For differential diagnosis, a genomic confirmation is indicated, but this is normally time consuming. There are two features observed in patient blood samples that could serve for differential diagnostics, a strong IFN signature [5] and impaired proteolytic activity [35, 36]. A strong IFN signature is characteristic for PRAAS patients [5], but this is not a unique feature, as STING-associated vasculopathy with onset in infancy, Aicardi-Goutières-syndrome, severe lupus, ISG15 deficiency, and some undifferentiated interferonopathies show similar induction of IFN-responsive genes [54–57]. The establishment of an additional diagnostic-approved proteasome-specific assay is therefore essential to verify a significantly reduced proteolytic activity in at least one of the three protease activities in PBMCs. From our own data with preclinical mouse models and cell culture, we assume that syndromes with increased interferon signature but without proteasome defects may display induced proteolytic activity due to the induction of IP [21]. If both features, induced IFN signature and significantly reduced proteasome activity, are present, a final confirmation of PRAAS could be achieved by identification of a disease-related mutation in one or two proteasome components such as those listed in Table 1. To date, only mutations in the 20S proteasome component PSMB8/β5i have been identified, but it is worth expanding sequencing to include regulatory particle components, as the full catalytic capacity of the 20S core particle is only possible by interaction with regulators. Mutations can affect not only coding sequence regions but also regulatory sequences, splice sites, or the translational control of proteasome subunits.
Treatment
Some of the symptoms such as skin lesions, fever, and acute-phase reactants can be alleviated by application of high doses of systemic steroids. But these treatments fail to halt the progression of lipodystrophy and even worsens the central obesity and can cause severe side effects including growth retardation and glaucoma. Administration of various antirheumatic and immunosuppressive drugs, like IL-1 receptor antagonists, IL-6 receptor antagonists, and TNF-α inhibitors, either have no effect or showed only a temporal clinical improvement. Many other immunosuppressant drugs have been used such as methotrexate, hydroxychloroquine, azathioprine, cyclosporine, tacrolimus, colchicine, and dapsone, but they proved ineffective in most patients [5, 40, 53].
Because laboratory investigations identified IFN signaling as the most deregulated pathway in PRAAS patients, inhibition of this pathway seems to be the most promising target for therapeutic treatment. Preliminary investigations with the biological drug group of JAK inhibitors are promising. The JAK inhibitor tofacitinib (mainly inhibiting JAK3) has been shown to decrease phosphorylation of STAT-1, a downstream signaling molecule of IFNα/β and IFNγ receptors which is constitutively upregulated in CANDLE patients [5]. In an ongoing compassionate run by the National Institute of Health (NIH) administration of the JAK1/2 inhibitor baricitinib, which is in phase three clinical development for rheumatoid arthritis and phase two development for psoriasis and diabetic nephropathy, produced clinical improvement for CANDLE patients (NCT01724580) [40] (oral communication from R. Goldbach-Mansky).
Conclusions
The accumulation of protein aggregates (misfolded or damaged proteins) is common to proteinopathies and proteasome dysfunction disorders (PRAAS), and it seems likely that in both cases, this misbalance in proteostasis triggers an inflammatory outcome and cell death. Recent publications showed that some proteinopathies [3–5] are accompanied with the dysregulation of the proteasome or autophagy, and this may also be true for other proteinopathies. More investigations are needed to clarify the exact signaling pathway; however, recent studies on autoinflammation caused by mutations of proteasomal components indicate the involvement of IFN type I signaling. A detailed knowledge about the molecular pathway linking proteotoxic stress to type I IFN production will likely disclose novel targets for therapeutics in several inflammatory diseases driven by intracellular stress coupled with IFN production. Besides the development of new drugs, the development of diagnostic tools to differentiate inflammation types and to monitor activity of different clearance pathways would be invaluable.
References
Liu Y, Jesus AA, Marrero B et al (2014) Activated STING in a vascular and pulmonary syndrome. N Engl J Med 371:507–18. doi:10.1056/NEJMoa1312625
Sanchez GAM, de Jesus AA, Goldbach-Mansky R (2013) Monogenic autoinflammatory diseases: disorders of amplified danger sensing and cytokine dysregulation. Rheum Dis Clin North Am 39:701–34. doi:10.1016/j.rdc.2013.08.001
Yamanaka K, Sasagawa Y, Ogura T (2012) Recent advances in p97/VCP/Cdc48 cellular functions. Biochim Biophys Acta 1823:130–7. doi:10.1016/j.bbamcr.2011.07.001
Askanas V, Engel WK (2006) Inclusion-body myositis: a myodegenerative conformational disorder associated with Abeta, protein misfolding, and proteasome inhibition. Neurology 66:S39–48. doi:10.1212/01.wnl.0000192128.13875.1e
Liu Y, Ramot Y, Torrelo A et al (2012) Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum 64:895–907. doi:10.1002/art.33368
LaFerla FM (2010) Pathways linking Abeta and tau pathologies. Biochem Soc Trans 38:993–5. doi:10.1042/BST0380993
Sulistio YA, Heese K (2015) The ubiquitin-proteasome system and molecular chaperone deregulation in Alzheimer’s disease. Mol Neurobiol. doi:10.1007/s12035-014-9063-4
LaFerla FM, Green KN, Oddo S (2007) Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci 8:499–509. doi:10.1038/nrn2168
Joshi-Barr S, Bett C, Chiang W-C et al (2014) De novo prion aggregates trigger autophagy in skeletal muscle. J Virol 88:2071–82. doi:10.1128/JVI. 02279-13
Deriziotis P, André R, Smith DM et al (2011) Misfolded PrP impairs the UPS by interaction with the 20S proteasome and inhibition of substrate entry. EMBO J 30:3065–77. doi:10.1038/emboj.2011.224
Ebstein F, Kloetzel P-M, Krüger E, Seifert U (2012) Emerging roles of immunoproteasomes beyond MHC class I antigen processing. Cell Mol Life Sci 69:2543–58. doi:10.1007/s00018-012-0938-0
Krüger E, Kloetzel P-M (2012) Immunoproteasomes at the interface of innate and adaptive immune responses: two faces of one enzyme. Curr Opin Immunol 24:77–83. doi:10.1016/j.coi.2012.01.005
Ciechanover A (2013) Intracellular protein degradation: from a vague idea through the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Bioorg Med Chem 21:3400–10. doi:10.1016/j.bmc.2013.01.056
Groll M, Ditzel L, Löwe J et al (1997) Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 386:463–71. doi:10.1038/386463a0
Vigneron N, Van den Eynde BJ (2014) Proteasome subtypes and regulators in the processing of antigenic peptides presented by class I molecules of the major histocompatibility complex. Biomolecules 4:994–1025. doi:10.3390/biom4040994
Gu ZC, Enenkel C (2014) Proteasome assembly. Cell Mol Life Sci 71:4729–45. doi:10.1007/s00018-014-1699-8
Sahara K, Kogleck L, Yashiroda H, Murata S (2014) The mechanism for molecular assembly of the proteasome. Adv Biol Regul 54:51–8. doi:10.1016/j.jbior.2013.09.010
Kniepert A, Groettrup M (2014) The unique functions of tissue-specific proteasomes. Trends Biochem Sci 39:17–24. doi:10.1016/j.tibs.2013.10.004
Fabre B, Lambour T, Garrigues L et al (2015) Deciphering preferential interactions within supramolecular protein complexes: the proteasome case. Mol Syst Biol 11:771, doi: 10.15252/msb.20145497
Kriegenburg F, Poulsen EG, Koch A et al (2011) Redox control of the ubiquitin-proteasome system: from molecular mechanisms to functional significance. Antioxid Redox Signal 15:2265–99. doi:10.1089/ars.2010.3590
Seifert U, Bialy LP, Ebstein F et al (2010) Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell 142:613–24. doi:10.1016/j.cell.2010.07.036
Ebstein F, Voigt A, Lange N et al (2013) Immunoproteasomes are important for proteostasis in immune responses. Cell 152:935–7. doi:10.1016/j.cell.2013.02.018
Pickering AM, Koop AL, Teoh CY et al (2010) The immunoproteasome, the 20S proteasome and the PA28αβ proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem J 432:585–94. doi:10.1042/BJ20100878
Fehling H, Swat W, Laplace C et al (1994) MHC class I expression in mice lacking the proteasome subunit LMP-7. Science 265(80):1234–1237. doi:10.1126/science.8066463
Opitz E, Koch A, Klingel K et al (2011) Impairment of immunoproteasome function by β5i/LMP7 subunit deficiency results in severe enterovirus myocarditis. PLoS Pathog 7:e1002233. doi:10.1371/journal.ppat.1002233
Strehl B, Joeris T, Rieger M et al (2006) Immunoproteasomes are essential for clearance of Listeria monocytogenes in nonlymphoid tissues but not for induction of bacteria-specific CD8+ T cells. J Immunol 177:6238–6244. doi:10.4049/jimmunol.177.9.6238
Ishii K, Hisaeda H, Duan X et al (2006) The involvement of immunoproteasomes in induction of MHC class I-restricted immunity targeting Toxoplasma SAG1. Microbes Infect 8:1045–53. doi:10.1016/j.micinf.2005.10.023
Hussong SA, Kapphahn RJ, Phillips SL et al (2010) Immunoproteasome deficiency alters retinal proteasome’s response to stress. J Neurochem 113:1481–90. doi:10.1111/j.1471-4159.2010.06688.x
Chou B, Hisaeda H, Shen J et al (2008) Critical contribution of immunoproteasomes in the induction of protective immunity against Trypanosoma cruzi in mice vaccinated with a plasmid encoding a CTL epitope fused to green fluorescence protein. Microbes Infect 10:241–50. doi:10.1016/j.micinf.2007.11.010
Zaiss DMW, Bekker CPJ, Gröne A et al (2011) Proteasome immunosubunits protect against the development of CD8 T cell-mediated autoimmune diseases. J Immunol 187:2302–9. doi:10.4049/jimmunol.1101003
Eleftheriadis T, Pissas G, Antoniadi G et al (2013) CD8+ T-cell auto-reactivity is dependent on the expression of the immunoproteasome subunit LMP7 in exposed to lipopolysaccharide antigen presenting cells and epithelial target cells. Autoimmunity 46:439–45. doi:10.3109/08916934.2013.801460
Krause S, Kuckelkorn U, Dörner T et al (2006) Immunoproteasome subunit LMP2 expression is deregulated in Sjogren’s syndrome but not in other autoimmune disorders. Ann Rheum Dis 65:1021–7. doi:10.1136/ard.2005.045930
Hayashi T, Faustman D (2000) Defective function of the proteasome in autoimmunity: involvement of impaired NF-kappaB activation. Diabetes Technol Ther 2:415–28
Arima K, Kinoshita A, Mishima H et al (2011) Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc Natl Acad Sci U S A 108:14914–9. doi:10.1073/pnas.1106015108
Agarwal AK, Xing C, DeMartino GN et al (2010) PSMB8 encoding the β5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet 87:866–72. doi:10.1016/j.ajhg.2010.10.031
Kitamura A, Maekawa Y, Uehara H et al (2011) A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J Clin Invest 121:4150–60. doi:10.1172/JCI58414DS1
McDermott A, Jesus AA, Liu Y et al (2013) A case of proteasome-associated auto-inflammatory syndrome with compound heterozygous mutations. J Am Acad Dermatol 69:e29–e32
Mégarbané A, Sanders A, Chouery E et al (2002) An unknown autoinflammatory syndrome associated with short stature and dysmorphic features in a young boy. J Rheumatol 29:1084–7
Torrelo A, Patel S, Colmenero I et al (2010) Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. J Am Acad Dermatol 62:489–95. doi:10.1016/j.jaad.2009.04.046
McDermott A, Jacks J, Kessler M et al (2014) Proteasome-associated autoinflammatory syndromes: advances in pathogeneses, clinical presentations, diagnosis, and management. Int J Dermatol. doi:10.1111/ijd.12695
Kluk J, Rustin M, Brogan PA et al (2013) CANDLE syndrome: a report of a novel mutation and review of the literature. Br J Dermatol. doi:10.1111/bjd.12600
Oyanagi K, Sasaki K, Ohama E et al (1987) An autopsy case of a syndrome with muscular atrophy, decreased subcutaneous fat, skin eruption and hyper gamma-globulinemia: peculiar vascular changes and muscle fiber degeneration. Acta Neuropathol 73:313–9
Qureshi N, Perera P-Y, Shen J et al (2003) The proteasome as a lipopolysaccharide-binding protein in macrophages: differential effects of proteasome inhibition on lipopolysaccharide-induced signaling events. J Immunol 171:1515–1525. doi:10.4049/jimmunol.171.3.1515
Kawai T, Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol 11:373–84. doi:10.1038/ni.1863
Katsuyama M (2010) NOX/NADPH oxidase, the superoxide-generating enzyme: its transcriptional regulation and physiological roles. J Pharmacol Sci 114:134–46
Yewdell JW (2011) DRiPs solidify: progress in understanding endogenous MHC class I antigen processing. Trends Immunol 32:548–58. doi:10.1016/j.it.2011.08.001
Warnatsch A, Bergann T, Krüger E (2013) Oxidation matters: the ubiquitin proteasome system connects innate immune mechanisms with MHC class I antigen presentation. Mol Immunol 55:106–9. doi:10.1016/j.molimm.2012.10.007
Szeto J, Kaniuk NA, Canadien V et al (2014) ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy 2:189–199. doi:10.4161/auto.2731
Senft D, Ronai ZA (2015) UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. doi:10.1016/j.tibs.2015.01.002
Urano F, Wang X, Bertolotti A et al (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287:664–6
Menu P, Mayor A, Zhou R et al (2012) ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis 3:e261. doi:10.1038/cddis.2011.132
Lerner AG, Upton J-P, Praveen PVK et al (2012) IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab 16:250–64. doi:10.1016/j.cmet.2012.07.007
Kanazawa N (2012) Nakajo-Nishimura syndrome: an autoinflammatory disorder showing pernio-like rashes and progressive partial lipodystrophy. Allergol Int 61:197–206. doi:10.2332/allergolint.11-RAI-0416
Zhang X, Bogunovic D, Payelle-Brogard B et al (2014) Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature 517:89–93. doi:10.1038/nature13801
Baechler EC, Batliwalla FM, Karypis G et al (2003) Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A 100:2610–5. doi:10.1073/pnas.0337679100
Crow YJ (2013) Aicardi-Goutières syndrome. Handb Clin Neurol 113:1629–35. doi:10.1016/B978-0-444-59565-2.00031-9
Briggs TA, Rice GI, Daly S et al (2011) Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat Genet 43:127–31. doi:10.1038/ng.748
Qian M-X, Pang Y, Liu CH et al (2013) Acetylation-mediated proteasomal degradation of core histones during DNA repair and spermatogenesis. Cell 153:1012–24. doi:10.1016/j.cell.2013.04.032
Stadtmueller BM, Kish-Trier E, Ferrell K et al (2012) Structure of a proteasome Pba1-Pba2 complex: implications for proteasome assembly, activation, and biological function. J Biol Chem 287:37371–82. doi:10.1074/jbc.M112.367003
Fricke B, Heink S, Steffen J et al (2007) The proteasome maturation protein POMP facilitates major steps of 20S proteasome formation at the endoplasmic reticulum. EMBO Rep 8:1170–5. doi:10.1038/sj.embor.7401091
Hirano Y, Hendil KB, Yashiroda H et al (2005) A heterodimeric complex that promotes the assembly of mammalian 20S proteasomes. Nature 437:1381–5. doi:10.1038/nature04106
Paraskevopoulos K, Kriegenburg F, Tatham MH et al (2014) Dss1 is a 26S proteasome ubiquitin receptor. Mol Cell 56:453–61. doi:10.1016/j.molcel.2014.09.008
Kim Y-C, DeMartino GN (2011) C termini of proteasomal ATPases play nonequivalent roles in cellular assembly of mammalian 26S proteasome. J Biol Chem 286:26652–66. doi:10.1074/jbc.M111.246793
Cascio P (2014) PA28αβ: the enigmatic magic ring of the proteasome? Biomolecules 4:566–84. doi:10.3390/biom4020566
De Graaf N, van Helden MJG, Textoris-Taube K et al (2011) PA28 and the proteasome immunosubunits play a central and independent role in the production of MHC class I-binding peptides in vivo. Eur J Immunol 41:926–35. doi:10.1002/eji.201041040
Mao I, Liu J, Li X, Luo H (2008) REGgamma, a proteasome activator and beyond? Cell Mol Life Sci 65:3971–80. doi:10.1007/s00018-008-8291-z
Lehmann A, Niewienda A, Jechow K et al (2010) Ecm29 fulfils quality control functions in proteasome assembly. Mol Cell 38:879–88. doi:10.1016/j.molcel.2010.06.016
Gorbea C, Goellner GM, Teter K et al (2004) Characterization of mammalian Ecm29, a 26S proteasome-associated protein that localizes to the nucleus and membrane vesicles. J Biol Chem 279:54849–61. doi:10.1074/jbc.M410444200
Tomko RJ Jr, Hochstrasser M (2013) Molecular architecture and assembly of the eukaryotic proteasome. Annu Rev Biochem. doi:10.1146/annurev-biochem-060410-150257
Acknowledgments
The authors thank R. Goldbach-Mansky for great collaboratory work and for reviewing the manuscript and K. E. Gilling for critical reading of the manuscript. This research was supported by the Deutsche Forschungsgemeinschaft SFB740 and SFB TR 43 to EK.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the Special Issue on The Inflammasome and Autoinflammatory Diseases - Guest Editors: Seth L. Masters, Tilmann Kallinich and Seza Ozen
Rights and permissions
About this article

Cite this article
Brehm, A., Krüger, E. Dysfunction in protein clearance by the proteasome: impact on autoinflammatory diseases. Semin Immunopathol 37, 323–333 (2015). https://doi.org/10.1007/s00281-015-0486-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-015-0486-4