
Abstract
Fibroblast growth factor (FGF23) plasma levels are elevated in cardiac and renal failure and correlate with poor clinical prognosis of those disorders. Both disorders are associated with inflammation and activation of the inflammatory transcription factor NFκB. An excessive FGF23 level is further observed in Klotho-deficient mice. The present study explored a putative sensitivity of FGF23 expression to transcription factor NFκB, which is known to upregulate Orai1, the Ca2+ channel accomplishing store-operated Ca2+ entry (SOCE). In osteoblastic cells (UMR106) and immortalized primary periosteal (IPO) cells, protein abundance was determined by Western blotting, and in UMR106 cells, transcript levels were quantified by RT-PCR, cytosolic Ca2+ activity utilizing Fura-2-fluorescence, and SOCE from Ca2+ entry following store depletion by thapsigargin. As a result, UMR106 and IPO cells expressed Ca2+ channel Orai1. SOCE was lowered by NFκB inhibitor wogonin as well as by Orai1 inhibitors 2-APB and YM58483. UMR106 cell Fgf23 transcripts were increased by stimulation of SOCE and Ca2+ ionophore ionomycin and decreased by Orai inhibitors 2-APB, YM58483 and SK&F96365, by Orai1 silencing, as well as by NFκB inhibitors wogonin, withaferin A, and CAS 545380-34-5. In conclusion, Fgf23 expression is upregulated by stimulation of NFκB-sensitive, store-operated Ca2+ entry.
Key messages
-
Osteoblast UMR106 and IPO cells express Ca2+ channel Orai1.
-
Osteoblast store-operated Ca2+ entry is accomplished by NFκB-sensitive Orai1.
-
Osteoblast Fgf23 transcription is upregulated by increase in the cytosolic Ca2+ activity.
-
Fgf23 transcription is decreased by Orai inhibitors and Orai1 silencing.
-
Fgf23 transcription is lowered by NFκB inhibitors.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Fibroblast growth factor 23 (FGF23), a hormone released mainly from bone, is a powerful regulator of calcium phosphate metabolism [1]. FGF23 downregulates renal 1α hydroxylase (Cyp27b1) and upregulates 25-hydroxyvitamin D 24-hydroxylase (Cyp24a1) thus reducing the formation and enhancing the inactivation of 1,25-dihydroxyvitamin D3 (1,25(OH)2D3) [2]. As a result, FGF23 decreases the serum level of 1,25(OH)2 D3 [3, 4]. 1,25(OH)2 D3 is in turn a powerful regulator of renal and intestinal phosphate and calcium transport [5, 6]. In addition to its effect on 1,25(OH)2D3 formation, FGF23 directly decreases renal tubular phosphate reabsorption [3] and thus stimulates renal phosphate elimination [3]. FGF23 deficiency results in elevated serum phosphate, calcium, and 1,25(OH)2D3 levels with excessive vascular calcifications, rapid aging, and a profound decrease of lifespan [2].
The effect of FGF23 on 1,25(OH)2D3 formation requires αKlotho as a co-receptor [7, 8]. Mice lacking functional αKlotho similarly suffer from extensive vascular calcifications, early onset of multiple age-related disorders, and severe shortening of life span [8]. Similar to FGF23 deficiency, αKlotho deficiency is effective mainly through excessive 1,25(OH)2D3 formation, enhanced renal tubular phosphate reabsorption, and increased serum phosphate levels [9]. As a matter of fact, hyperphosphatemia fosters vascular calcification [10] and is considered a predictor of mortality [11]. FGF23 plasma levels are extremely high in Klotho-deficient mice, a result at least in part due to excessive 1,25(OH)2D3 levels.
The pathophysiological role of FGF23 has, however, remained enigmatic [12, 13]. In patients with cardiac failure [14, 15], acute renal failure [16], chronic kidney disease [7, 15, 17], diabetic nephropathy [18], and hepatic failure [19], plasma FGF23 concentrations are high and associated with accelerated disease progression, morbidity, and/or mortality. Mechanisms upregulating FGF23 in those disorders are still ill-defined.
Known regulators of FGF23 release include 1,25(OH)2D3 [7], αKlotho [20, 21], phosphate-regulating gene with homology to endopeptidase (PHEX), dentin matrix protein, or cyclin D binding myb-like protein 1 (DMP-1), sustained phosphate load even without hyperphosphatemia, a high extracellular Ca2+ concentration, and PTH [7, 22]. FGF23 plasma levels are further modified by iron deficiency [23, 24], pregnancy [25], and FGF23-secreting tumors [26].
To the best of our knowledge, nothing is known about a role of the cytosolic Ca2+ concentration ([Ca2+] i ) in FGF23 expression. Ca2+ may enter through SOCE, i.e., store-operated Ca2+ entry, which is stimulated by intracellular Ca2+ store depletion [27]. SOCE is accomplished by the four-transmembrane-spanning pore forming calcium release-activated channel (CRAC) moiety Orai1 (CRACM1) [28] and its regulator stromal interaction molecule 1 (STIM1), which senses the Ca2+ content of the endoplasmic reticulum (ER) [29]. The expression of Orai1 has previously been shown to be regulated by nuclear transcription factor NFκB [30].
The present study explored whether regulation of FGF23 release involves NFκB-sensitive Ca2+ entry.
Materials and methods
Cell culture
UMR106 rat osteosarcoma cells were cultured in Dulbecco’s modified Eagle medium (DMEM) high-glucose medium supplemented with 10 % FCS and 1 % penicillin/streptomycin under standard culture conditions. Human immortalized primary periosteal cells (IPO) were cultured in DMEM F-12 (1:1 mixture of DMEM and Ham’s F-12, high glucose) containing GlutaMAX and 10 % FCS and 1 % penicillin/streptomycin/1 % fungicide.
Cells were pretreated with 100 nM 1,25(OH)2D3 (Sigma, Schnelldorf, Germany). After 24 h, cells were in addition treated with 50 μM Orai inhibitor 2-APB (TOCRIS, Bristol, UK), 100 nM Orai inhibitor YM58483 (TOCRIS), 10 μM Orai inhibitor SK&F96365 (TOCRIS), 500 nM NFκB inhibitor withaferin A (TOCRIS), or NFκB inhibitor CAS 545380-34-5 (EMD) for another 24 h or treated with vehicle only. Cells pretreated with 100 nM 1,25(OH)2D3 for 46 h were incubated with or without 100 nM ionomycin (Sigma) or 500 nM thapsigargin (Sigma) for 2 h. Alternatively, cells were incubated with or without 100 μM NFκB inhibitor wogonin (Sigma). After 24 h, 1,25(OH)2D3 (100 nM) was added, and cells were analyzed after another 24 h. Where indicated, experiments were performed in the presence of nifedipine (100 μM) or verapamil (10 μM) (both from Sigma).
Silencing
For silencing, 1 × 105 cells (12-well plate) and 2 × 105 cells (6-well plate) were seeded 24 h before the experiment in antibiotic-free medium. Cells were transfected with 5 μl/1000 μl ON-TARGETplus RAT Orai1 siRNA (5 μM, Thermo Fisher Scientific, Waltham, MA, USA) and ON-TARGETplus Non-targeting siRNA (5 μM, Thermo Fisher Scientific) using the cationic lipid DharmaFECT 1 transfection reagent (0.5 μl/1000 μl, Thermo Fisher Scientific) according to the manufacturer’s protocol. Twenty-four hours after transfection, cells were treated with 1,25(OH)2D3 (100 nM) for another 24 h. Cells were then harvested and analyzed. To verify silencing efficiency, the Orai1 transcript level was quantified. As a result, the Orai1 mRNA level was 0.33 ± 0.05 a.u. (n = 16) in cells transfected with a negative control siRNA and 0.13 ± 0.01 a.u. (n = 16), p < 0.001 in cells transfected with a negative control siRNA.
Quantification of mRNA expression
For the mRNA expression analysis in UMR106 cells, the final volume of the RT-PCR reaction mixture was 15 μl and contained: 1 μl cDNA, 1 μM of each primer, 7.5 μl GoTaq Master Mix Green (Promega), and sterile water up to 15 μl. PCR conditions were 95 °C for 3 min, followed by 40 cycles of 95 °C for 10 s, 58 °C for 30 s, and 72 °C for 45 s. The product size was analyzed on a 2 % agarose gel. Quantitative RT-PCR was performed on a Bio-Rad iCycler iQTM Real-Time PCR Detection System (Bio-Rad Laboratories, Munich, Germany).
For the determination of Fgf23 transcripts in mouse bone, bone was homogenized in liquid nitrogen using a mortar and pestle. Total mRNA from bone was extracted with TRIzol (Invitrogen, Switzerland) followed by purification with RNeasy Mini Kit (QIAGEN, Switzerland) according to the manufacturer’s protocol. DNAse digestion was performed using the RNase-free DNase Set (QIAGEN, Switzerland). Total RNA extractions were analyzed for quality, purity, and concentration using the NanoDrop ND-1000 spectrophotometer (Wilmington, Germany). RNA samples were diluted to a final concentration of 100 ng/μl, and cDNA was prepared using the TaqMan Reverse Transcriptase Reagent Kit (Applied Biosystems, Roche, Foster City, CA). In brief, in a reaction volume of 40 μl, 300 ng of RNA was used as template and mixed with the following final concentrations of RT buffer (1×): MgCl2 (5.5 mmol/l), random hexamers (2.5 μmol/l), dNTP mix (500 μmol/l each), RNAse inhibitor (0.4 U/μl), multiscribe reverse transcriptase (1.25 U/μl), and RNAse-free water. Reverse transcription was performed with thermocycling conditions set at 25 °C for 10 min, 48 °C for 30 min, and 95 °C for 5 min on a thermocycler (Biometra, Göttingen, Germany). Quantitative real-time PCR (RT-PCR) was performed on the ABI PRISM 7700 Sequence Detection System (Applied Biosystems). Primers were chosen to spanning intron–exon boundaries to exclude genomic DNA contamination. The specificity of all primers was tested and always resulted in a single product of the expected size (data not shown). Probes were labeled with the reporter dye FAM at the 5′-end and the quencher dye TAMRA at the 3′-end (Microsynth, Balgach, Switzerland). Real-time PCR reactions were performed using KAPA PROBE FAST qPCR Kit (Kapa Biosystems, USA).
The following primers were used:
Rat Tbp (TATA box-binding protein):
forward (5′-3′): ACTCCTGCCACACCAGCC
reverse (5′-3′): GGTCAAGTTTACAGCCAAGATTCA
Rat Fgf23
forward (5′-3′): TGGCCATGTAGACGGAACAC
reverse (5′-3′): GGCCCCTATTATCACTACGGAG
Rat Orai1
forward (5′-3′): CGTCCACAACCTCAACTCC
reverse (5′-3′): AACTGTCGGTCCGTCTTAT
Rat Orai2
forward (5′-3′): GGAAGCCGTGAGCAACAT
reverse (5′-3′): CACCAGGGAGCGGTAGAA
Rat Orai3
forward (5′-3′): TTTTGGTGGGCTGGGTCA
reverse (5′-3′): TCCTGCTTGTGGCGGTCT
Rat Stim1
forward (5′-3′): CGTCCGCAACATCCACAAG
reverse (5′-3′): CCATAGGTCCTCCACGCT
Rat Stim2
forward (5′-3′): ACTTAGAAAGCCTACAAACCG
reverse (5′-3′): GCATCAGGGACAGACCAG
Rat Sgk1
forward (5′-3′): ATGTGAAGCACCCTTTCCTG
reverse (5′-3′): TAGAACAGCTCTCCGCCATT
Mouse Fgf23
forward (5′-3′): TCGAAGGTTCCTTTGTATGGAT
reverse (5′-3′): AGTGATGCTTCTGCGACAAGT
Calculated mRNA expression levels were normalized to the expression levels of Tbp (in rat-derived cell lines) or HPRT/18S (in mice) of the same cDNA sample. Relative quantification of gene expression was performed using the ΔΔCt method.
Western blotting
Orai1 protein abundance was determined in UMR106 cells, IPO cells, and mouse bone (femur). The cells were washed in ice-cold PBS. For bone preparation, a mouse was sacrificed and the femur isolated. Muscles were carefully removed and the femur snap-frozen. After thawing, the epiphyses were removed and the bone marrow was flushed out with ice-cold PBS. Using a mortar and pestle, the bone was pulverized on dry ice. RIPA lysis buffer (Cell Signaling, Frankfurt, Germany) containing phosphatase and protease inhibitor cocktail tablet (Complete mini, Roche, Mannheim, Germany) was added to the washed cells or the bone powder. The samples were incubated on ice for 30 min and then centrifuged at 14,000 rpm and 4 °C for 20 min. The supernatant was removed and used for Western blotting. Total protein (40–60 μg) was separated by SDS-PAGE, thereafter transferred to PVDF membranes and blocked in 5 % nonfat milk/Tris-buffered saline/Tween-20 (TBST) at room temperature for 1 h. Membranes were probed overnight at 4 °C with polyclonal rabbit anti-Orai1 antibody (1:700 in 5 % BSA in TBST; Proteintech, Manchester, UK). After incubation with horseradish peroxidase-conjugated anti-rabbit secondary antibody (Cell Signaling, Frankfurt, Germany; 1:2000) for 1 h at room temperature, the bands were visualized with enhanced chemiluminescence reagents (Amersham, Freiburg, Germany). Membranes were also probed with GAPDH antibody (Cell Signaling, 1:2000) as loading control. Densitometric analysis was performed using Quantity One software (Bio-Rad, Munich, Germany).
Measurement of intracellular Ca2+
To determine the cytosolic Ca2+ concentration ([Ca2+] i ), UMR106 cells were loaded with Fura-2/AM (2 μM, Molecular Probes, Göttingen, Germany) for 15 min at 37 °C. Fluorescence measurements were carried out with an inverted phase-contrast microscope (Axiovert 100, Zeiss, Oberkochen, Germany). Cells were excited alternatively at 340 or 380 nm, and the light was deflected by a dichroic mirror into either the objective (Fluar 40×/1.30 oil, Zeiss, Oberkochen, Germany) or a camera (Proxitronic, Bensheim, Germany). Emitted fluorescence intensity was recorded at 505 nm. Data acquisition was accomplished by using specialized computer software (Metafluor, Universal Imaging Downingtown, USA). As a measure for the increase in the cytosolic Ca2+ concentration, the slope and peak of the changes in the 340/380 nm ratio were determined in each experiment.
To determine SOCE, intracellular Ca2+ was measured before and after removal of extracellular Ca2+ (and addition of 0.5 mM EDTA), followed by addition of thapsigargin (1 μM) and subsequent readdition of extracellular Ca2+ to Ringer solution, composed of (in mM): 125 NaCl, 5 KCl, 1.2 MgSO4, 32.2 HEPES, 2 Na2HPO4, 0 or 2 CaCl2 and 0.5 or 0 EGTA, respectively, and 5 glucose, pH 7.4 (NaOH).
Statistics
Data are provided as means ± SEM, n represents the number of independent experiments. All data were tested for significance using unpaired Student’s t test or ANOVA. Only results with p < 0.05 were considered statistically significant.
Results
The present study explored whether NFκB-sensitive Ca2+ signaling participates in the regulation of FGF23 release.
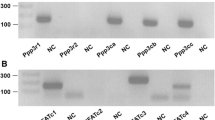
A first series of experiments explored whether Ca2+ release-activated Ca2+ channel (CRAC) moiety Orai1 and its regulator STIM1 are expressed in osteoblastic cells and could therefore participate in the signaling regulating FGF23 formation in those cells. Experiments were thus performed in UMR106 osteoblast-like cells and in immortalized primary periosteal cells (IPO). As illustrated in Fig. 1a, Orai1 was expressed in UMR106 and IPO cells as well as in bone. Moreover, Orai3 and STIM2 transcripts could readily be detected in UMR106 cells whereas the abundance of Orai2 and Stim1 was low (Fig. 1b). Orai1 transcript levels were reduced following inhibition of NFκB by wogonin in UMR106 cells (Fig. 1c).

Expression of Orai1 in bone, osteoblasts, and UMR106 cells and the effect of NFκB inhibitor wogonin. a Original Western blots showing the protein expression of Orai1 (first lane) and GAPDH (second lane), in UMR106 cells (UMR), immortalized primary periosteal cells (IPO), and bone. b Original 2 % agarose gel showing specific amplification of Orai1 (357 bp), Orai2 (292 bp), Orai3 (328 bp), Stim1 (163 bp), Stim2 (298 bp), and Tbp (91 bp) cDNA in UMR106 cells. c Arithmetic means ± SEM (n = 9) of Orai1 mRNA abundance (relative to Tbp mRNA) in UMR106 cells incubated without (white bar) or with (black bar) NFκB inhibitor wogonin (100 μM, 48 h). ***p < 0.001 indicates significant difference
Fluorescence optics was employed to explore whether Orai1 and/or NFκB impact on intracellular Ca2+ concentration ([Ca2+] i ). Store-operated Ca2+ entry (SOCE) requires depletion of intracellular Ca2+ stores which was accomplished by inhibition of the sarcoendoplasmic Ca2+ ATPase (SERCA) with thapsigargin (1 μM) in the absence of extracellular Ca2+. SOCE was estimated from the increase in [Ca2+] i following readdition of extracellular Ca2+. As illustrated in Fig. 2a, d, g, j, thapsigargin (10 μM) treatment in the absence of extracellular Ca2+ was followed by a transient elevation of [Ca2+] i reflecting depletion of intracellular stores. The subsequent addition of extracellular Ca2+ was followed by a rapid increase in [Ca2+] i reflecting store-operated Ca2+ entry (SOCE). Addition of Orai1 inhibitors 2-APB (Fig. 2a–c) or YM58483 (Fig. 2d–f) did not significantly modify the thapsigargin-induced increase in [Ca2+] i but virtually abrogated the increase in [Ca2+] i following readdition of extracellular Ca2+. Similarly, Orai1 silencing did not significantly modify the thapsigargin-induced increase in [Ca2+] i but significantly blunted the increase in [Ca2+] i following readdition of extracellular Ca2+ (Fig. 2g–i). Exposure of UMR106 cells to wogonin significantly blunted the thapsigargin-induced increase in [Ca2+] i and the increase in [Ca2+] i following readdition of extracellular Ca2+ (Fig. 2j–l).

Effect of 2-APB, YM58483, Orai1 silencing, and wogonin on SOCE in UMR106 cells. a, d, j. Representative original tracings showing intracellular Ca2+ concentrations ([Ca2+] i ) in Fura-2/AM-loaded UMR106 cells prior to and following removal of extracellular Ca2+, addition of the sarco-endoplasmic Ca2+ ATPase (SERCA) inhibitor thapsigargin (1 μM), and readdition of extracellular Ca2+, all in the absence (open circles) and presence (closed circles) of Orai inhibitor 2-APB (a, 50 μM), Orai inhibitor YM58483 (d, 100 nM), or NFκB inhibitor wogonin (j, 100 μM, 48 h). b, c, e, f, k, l. Arithmetic means ± SEM of the peak (left) and slope (right) values of [Ca2+] i increase following addition of thapsigargin reflecting Ca2+ release from intracellular stores (b, e, k) and of [Ca2+] i increase following readdition of extracellular Ca2+ reflecting store-operated Ca2+ entry (c, f, l) in UMR106 cells incubated without (white bars) or with (black bars) Orai inhibitor 2-APB (b–c, 50 μM, n = 22–60) or with the Orai inhibitor YM58483 (e–f, 100 nM, n = 40–39) or the NFκB inhibitor wogonin (k–l, 100 μM, 48 h, n = 12–31). g Representative original tracings showing intracellular Ca2+ concentrations ([Ca2+] i ) in Fura-2/AM-loaded UMR106 cells prior to and following removal of extracellular Ca2+, addition of the sarco-endoplasmic Ca2+ ATPase (SERCA) inhibitor thapsigargin (1 μM) and readdition of extracellular Ca2+, all in cells treated with unspecific siRNA (open circles) or with specific siRNA (closed circles) targeting Orai1. h, i Arithmetic means ± SEM of the peak (left) and slope (right) values of [Ca2+] i increase following addition of thapsigargin reflecting Ca2+ release from intracellular stores (h) and of [Ca2+] i increase following readdition of extracellular Ca2+ reflecting store-operated Ca2+ entry (i) in UMR106 cells treated with unspecific siRNA (white bars) or with specific siRNA targeting Orai1 (black bars).**p < 0.001, ***p < 0.001 indicate significant difference
Next, qRT-PCR was employed to determine whether Ca2+ entry and/or NFκB influence the formation of FGF23. As illustrated in Fig. 3, Fgf23 transcript levels were significantly increased by the Ca2+ ionophore ionomycin (Fig. 3a) and by thapsigargin (Fig. 3b). Conversely, blocking Ca2+ entry by the Orai blockers 2-ABP (Fig. 3c), YM58483 (Fig. 3d), or SK&F96365 (Fig. 3e) decreased the abundance of Fgf23 transcripts. In view of the limited specificity of the Orai1 blockers, additional experiments were performed with silencing of Orai1. As illustrated in Fig. 3f, silencing of Orai1 also lowered Fgf23 transcript levels in UMR106 cells. We could, however, not confirm a significant decrease of Orai1 protein abundance following Orai1 silencing (data not shown). Nevertheless, steady-state FGF23 production in UMR106 cells was dependent on [Ca2+] i which was modified by Ca2+ entry. In contrast, l-type calcium channel inhibitors did not appreciably influence Fgf23 transcript levels. The Fgf23 transcript level was 0.019 ± 0.004 a.u. in control cells (n = 9) and 0.018 ± 0.003 a.u. (n = 9) in cells treated with l-type channel inhibitor nifedipine (100 μM, 24 h). In another series, the Fgf23 transcript level was 0.011 ± 0.001 a.u. (n = 9) in control cells and 0.011 ± 0.001 a.u. (n = 9) in cells treated with l-type channel inhibitor verapamil (10 μM, 24 h).

Effect of ionomyin, thapsigargin, 2-APB, YM58483, SK&F96365, and Orai1 silencing on Fgf23 transcript levels in UMR106 cells. Arithmetic means ± SEM of Fgf23 mRNA abundance (relative to Tbp mRNA) in UMR106 cells incubated without (white bars) or with (black bars) ionomycin (a, 100 nM, 2 h, n = 12), thapsigargin (b, 500 nM, 2 h, n = 12), the Orai inhibitor 2-ABP (c, 50 μM, 24, n = 6), the Orai inhibitor YM58483 (d, 100 nM, 24 h, n = 15), and the Orai inhibitor SK&F96365 (e, 10 μM, 24 h, n = 16–19). Arithmetic means ± SEM (n = 16) of Fgf23 mRNA abundance (relative to Tbp mRNA) in UMR106 cells treated with control siRNA (siNeg, white bar) or with specific Orai1 siRNA (siOrai1, black bar) (f).*p < 0.05, **p < 0.001, ***p < 0.001 indicate significant difference (t test)

Effect of wogonin and withaferin A on Fgf23 transcript levels in UMR106 cells. Arithmetic means ± SEM of Fgf23 mRNA abundance (relative to Tbp mRNA) in UMR106 cells incubated without (white bars) or with (black bars) wogonin (a, 100 μM, 48 h, n = 8–9) and withaferin A (b, 500 nM, 24 h, n = 15–20).**p < 0.001, ***p < 0.001 indicate significant difference (t test)
Since Ca2+ entry into UMR106 cells could be attenuated by the NFκB inhibitor wogonin (Fig. 3j–l), we tested whether FGF23 formation is also NFκB-sensitive. Indeed, Fgf23 transcript levels were reduced by the NFκB inhibitors wogonin (Fig. 4a) and withaferin A (Fig. 4b). Similarly, the NFκB inhibitor CAS 545380-34-5 decreased Orai1 transcript levels, thapsigargin-induced increase in [Ca2+] i and SOCE in Fig. 5a, c as well as Fgf23 transcript levels (Fig. 5b).

Effect of NFκB inhibitor CAS 545380-34-5 on Orai1 and Fgf23 transcript levels as well as SOCE in UMR106 cells. a Arithmetic means ± SEM (n = 6) of Orai1 mRNA abundance (relative to Tbp mRNA) in UMR106 cells incubated without or with NFκB inhibitor CAS 545380-34-5 (100 nM, 24 h). b Arithmetic means ± SEM (n = 6) of Fgf23 mRNA abundance (relative to Tbp mRNA) in UMR cells incubated without or with NFκB inhibitor CAS 545380-34-5 (100 nM, 24 h). c. Representative original tracings (left panel) showing intracellular Ca2+ concentrations ([Ca2+] i ) in Fura-2/AM-loaded UMR106 cells prior to and following removal of extracellular Ca2+, addition of the sarco-endoplasmic Ca2+ ATPase (SERCA) inhibitor thapsigargin (1 μM), and readdition of extracellular Ca2+ in the absence (open circles) and presence (closed circles) of the NFκB inhibitor CAS 545380-34-5 (100 nM, 24 h). The upper right panel depicts the arithmetic means ± SEM (n = 45–72) of the peak (left) and slope (right) values of [Ca2+] i increase following addition of thapsigargin reflecting Ca2+ release from intracellular stores and the lower right panel shows the arithmetic means ± SEM (n = 45–72) of the peak (left) and slope (right) values of [Ca2+] i increase following readdition of extracellular Ca2+ reflecting store-operated Ca2+ entry in UMR106 cells incubated without (white bars) or with (black bars) NFκB inhibitor CAS 545380-34-5 (100 nM, 24 h). **p < 0.01, ***p < 0.001 indicate significant difference (t test)
Since the level of confluence has been shown to affect FGF23 production in primary osteoblasts [31], we performed experiments at different stages of confluency. In a culture of 300,000 UMR106 cells, the Fgf23 transcript levels were significantly (p < 0.01) higher in control cells (0.029 ± 0.005 a.u., n = 12) than in cells exposed 24 h to 100 μM wogonin (0.009 ± 0.001 a.u., n = 12). In a culture of 200,000 UMR106 cells, the Fgf23 transcript level was again significantly (p < 0.001) higher in control cells (0.012 ± 0.001 a.u., n = 12) than in cells exposed for 24 h to 100 μM wogonin (0.004 ± 0.000 a.u., n = 12). Finally, in a culture of 100,000 UMR106 cells, the Fgf23 transcript level was again significantly (p < 0.01) higher in control cells (0.006 ± 0.001 a.u., n = 12) than in cells exposed 24 h to 100 μM wogonin (0.002 ± 0.000 a.u., n = 12). Thus, the level of confluence influenced the basal Fgf23 transcript level but did not affect the inhibitory effect of wogonin on FGF23 formation.
Discussion
The present observations disclose a decisive role of the cytosolic Ca2+ concentration ([Ca2+] i ) for the regulation of FGF23 release. Store-operated Ca2+ entry (SOCE) apparently stimulated the transcription of Fgf23. SOCE is accomplished by the pore-forming Orai (CRAC) isoforms [28] and their regulators STIM1 and STIM2 [29]. According to the present observations, Orai1 and STIM2 are both highly expressed in osteoblastic UMR106 cells. Orai1 expression has been shown to be upregulated by the transcription factor NFκB [30]. Our study demonstrates that in UMR106 cells inhibition of NFκB reduced Orai1 transcript levels. Moreover, Fgf23 expression was also downregulated by NFκB inhibitors. The present observations do not rule out involvement of further signaling triggered by NFκB and eventually upregulating Fgf23 expression. Moreover, the present observations do not rule out that Ca2+ entry mechanisms other than Orai1 contribute to the stimulation of Fgf23 transcription.
Activation of NFκB participates in the pathophysiology of diverse inflammatory conditions [32] including chronic kidney disease [33], cardiac failure [34], and diabetic nephropathy [35]. According to the present observations, the high FGF23 plasma concentration in heart failure [14–16], chronic kidney disease [7, 15, 17], and diabetic nephropathy [18] could at least in part be secondary to upregulation of NFκB associated with those diseases. NFκB and Orai1 are upregulated by serum and glucocorticoid inducible kinase SGK1 [30], which again participates in the pathophysiology of cardiac failure and diabetic nephropathy [36]. FGF23 is further high in polycystic kidney disease [12]. It is therefore tempting to speculate that the excessive FGF23 plasma level in polycystic kidney disease results from NFκB-dependent upregulation of Orai1. Activation of NFκB may similarly contribute to enhanced FGF23 release in Klotho-deficient mice [37, 38]. It should be pointed out, however, that the FGF23 plasma level in those mice is most likely mainly due to stimulation of FGF23 formation by high 1,25(OH)2D3 [22].
The regulation of FGF23 formation by NFκB and Ca2+ entry is expected to influence mineral metabolism. FGF23 downregulates renal 1α hydroxylase expression and thus the formation of 1,25(OH)2D3 [39, 40], an effect requiring αKlotho as a co-receptor [8]. 1,25(OH)2 D3 is known to stimulate both renal and intestinal phosphate transport [5]. Moreover, FGF23 reduces renal tubular phosphate reabsorption more directly by inhibiting proximal tubular Na+-coupled phosphate transport [40]. FGF23 thus fosters phosphaturia and hypophosphatemia. In chronic kidney disease, hyperphosphatemia results in vascular calcification [41] and both Klotho and FGF23 counteract the calcification [42]. As a high serum phosphate concentration triggers vascular calcification and is associated with accelerated aging and a decreased life span [43], high FGF23 plasma levels protect against vascular calcification, aging, and early death. Along those lines, αKlotho [44] and FGF23 [45] counteract aging and lack of either, αKlotho [44] or FGF23 [40], fosters early appearance of multiple age-related disorders leading to early death. 1,25(OH)2D3 stimulates the release of FGF23 [22] and thus triggers a negative feedback loop limiting 1,25(OH)2D3 formation.
The pathophysiological impact of elevated FGF23 plasma levels is still ill-defined. While a high FGF23 plasma level is associated with high morbidity and mortality in heart and kidney failure [14–16], FGF23 neutralization increases the mortality of rats with chronic kidney disease-mineral and bone disorder [46]. In view of the present observations, it is tempting to speculate that the association between the FGF23 plasma level and morbidity and mortality reflects the stimulating effect of NFκB on inflammation on the one side and FGF23 formation on the other, thus affecting both FGF23 plasma level and disease progression.
In conclusion, the present observations disclose a novel mechanism regulating FGF23 release. FGF23 formation in UMR106 cells is stimulated by an increased cytosolic Ca2+ concentration, which is regulated by store-operated Ca2+ entry (SOCE). SOCE and FGF23 formation are upregulated by the transcription factor NFκB.
References
Hori M, Shimizu Y, Fukumoto S (2011) Minireview: fibroblast growth factor 23 in phosphate homeostasis and bone metabolism. Endocrinology 152:4–10
Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T (2004) Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest 113:561–568
Bai X, Miao D, Li J, Goltzman D, Karaplis AC (2004) Transgenic mice overexpressing human fibroblast growth factor 23 (R176Q) delineate a putative role for parathyroid hormone in renal phosphate wasting disorders. Endocrinology 145:5269–5279
Saito H, Kusano K, Kinosaki M, Ito H, Hirata M, Segawa H, Miyamoto K, Fukushima N (2003) Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate co-transport activity and 1alpha,25-dihydroxyvitamin D3 production. J Biol Chem 278:2206–2211
Brown AJ, Finch J, Slatopolsky E (2002) Differential effects of 19-nor-1,25-dihydroxyvitamin D(2) and 1,25-dihydroxyvitamin D(3) on intestinal calcium and phosphate transport. J Lab Clin Med 139:279–284
Murer H, Hernando N, Forster I, Biber J (2000) Proximal tubular phosphate reabsorption: molecular mechanisms. Physiol Rev 80:1373–1409
Hu MC, Shiizaki K, Kuro-o M, Moe OW (2013) Fibroblast growth factor 23 and Klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol 75:503–533
Kuro-o M (2010) Overview of the FGF23-Klotho axis. Pediatr Nephrol 25:583–590
Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, Razzaque MS, Rosenblatt KP, Baum MG, K-o M et al (2010) Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J 24:3438–3450
Giachelli CM (2003) Vascular calcification: in vitro evidence for the role of inorganic phosphate. J Am Soc Nephrol 14:S300–S304
Tonelli M, Sacks F, Pfeffer M, Gao Z, Curhan G (2005) Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation 112:2627–2633
Lang F, Föller M (2014) Enigmatic Cassandra: renal FGF23 formation in polycystic kidney disease. Kidney Int 85:1260–1262
Moe OW (2012) Fibroblast growth factor 23: friend or foe in uremia? J Clin Invest 122:2354–2356
Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, Gutierrez OM, Aguillon-Prada R, Lincoln J, Hare JM et al (2011) FGF23 induces left ventricular hypertrophy. J Clin Invest 121:4393–4408
Imazu M, Takahama H, Asanuma H, Funada A, Sugano Y, Ohara T, Hasegawa T, Asakura M, Kanzaki H, Anzai T et al (2014) Pathophysiological impact of serum fibroblast growth factor 23 in patients with non-ischemic cardiac disease and early chronic kidney disease. Am J Physiol Heart Circ Physiol. doi:10.1152/ajpheart.00331.2014
Christov M (2014) Fibroblast growth factor 23 in acute kidney injury. Curr Opin Nephrol Hypertens 23:340–345
Evenepoel P, Meijers B, Viaene L, Bammens B, Claes K, Kuypers D, Vanderschueren D, Vanrenterghem Y (2010) Fibroblast growth factor-23 in early chronic kidney disease: additional support in favor of a phosphate-centric paradigm for the pathogenesis of secondary hyperparathyroidism. Clin J Am Soc Nephrol 5:1268–1276
Zanchi C, Locatelli M, Benigni A, Corna D, Tomasoni S, Rottoli D, Gaspari F, Remuzzi G, Zoja C (2013) Renal expression of FGF23 in progressive renal disease of diabetes and the effect of ace inhibitor. PLoS One 8, e70775
Prie D, Forand A, Francoz C, Elie C, Cohen I, Courbebaisse M, Eladari D, Lebrec D, Durand F, Friedlander G (2013) Plasma fibroblast growth factor 23 concentration is increased and predicts mortality in patients on the liver-transplant waiting list. PLoS One 8, e66182
Juppner H, Wolf M (2012) AlphaKlotho: FGF23 coreceptor and FGF23-regulating hormone. J Clin Invest 122:4336–4339
Smith RC, O’Bryan LM, Farrow EG, Summers LJ, Clinkenbeard EL, Roberts JL, Cass TA, Saha J, Broderick C, Ma YL et al (2012) Circulating alphaKlotho influences phosphate handling by controlling FGF23 production. J Clin Invest 122:4710–4715
Masuyama R, Stockmans I, Torrekens S, Van Looveren R, Maes C, Carmeliet P, Bouillon R, Carmeliet G (2006) Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J Clin Invest 116:3150–3159
Clinkenbeard EL, Farrow EG, Summers LJ, Cass TA, Roberts JL, Bayt CA, Lahm T, Albrecht M, Allen MR, Peacock M et al (2014) Neonatal iron deficiency causes abnormal phosphate metabolism by elevating FGF23 in normal and ADHR mice. J Bone Miner Res 29:361–369
Wolf M, Koch TA, Bregman DB (2013) Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J Bone Miner Res 28:1793–1803
Kirby BJ, Ma Y, Martin HM, Buckle Favaro KL, Karaplis AC, Kovacs CS (2013) Upregulation of calcitriol during pregnancy and skeletal recovery after lactation do not require parathyroid hormone. J Bone Miner Res 28:1987–2000
Chong WH, Andreopoulou P, Chen CC, Reynolds J, Guthrie L, Kelly M, Gafni RI, Bhattacharyya N, Boyce AM, El-Maouche D et al (2013) Tumor localization and biochemical response to cure in tumor-induced osteomalacia. J Bone Miner Res 28:1386–1398
Parekh AB (2010) Store-operated CRAC channels: function in health and disease. Nat Rev Drug Discov 9:399–410
Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG (2006) Orai1 is an essential pore subunit of the CRAC channel. Nature 443:230–233
Peinelt C, Vig M, Koomoa DL, Beck A, Nadler MJ, Koblan-Huberson M, Lis A, Fleig A, Penner R, Kinet JP (2006) Amplification of CRAC current by STIM1 and CRACM1 (Orai1). Nat Cell Biol 8:771–773
Lang F, Shumilina E (2013) Regulation of ion channels by the serum- and glucocorticoid-inducible kinase SGK1. FASEB J 27:3–12
Tang WJ, Wang LF, Xu XY, Zhou Y, Jin WF, Wang HF, Gao J (2010) Autocrine/paracrine action of vitamin D on FGF23 expression in cultured rat osteoblasts. Calcif Tissue Int 86:404–410
Liu SF, Malik AB (2006) NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol 290:L622–L645
Decleves AE, Sharma K (2014) Novel targets of antifibrotic and anti-inflammatory treatment in CKD. Nat Rev Nephrol 10:257–267
Gordon JW, Shaw JA, Kirshenbaum LA (2011) Multiple facets of NF-kappaB in the heart: to be or not to NF-kappaB. Circ Res 108:1122–1132
Yi B, Hu X, Zhang H, Huang J, Liu J, Hu J, Li W, Huang L (2014) Nuclear NF-kappaB p65 in peripheral blood mononuclear cells correlates with urinary MCP-1, RANTES and the severity of type 2 diabetic nephropathy. PLoS One 9, e99633
Lang F, Bohmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V (2006) (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev 86:1151–1178
Maekawa Y, Ishikawa K, Yasuda O, Oguro R, Hanasaki H, Kida I, Takemura Y, Ohishi M, Katsuya T, Rakugi H (2009) Klotho suppresses TNF-alpha-induced expression of adhesion molecules in the endothelium and attenuates NF-kappaB activation. Endocrine 35:341–346
Zhao Y, Banerjee S, Dey N, LeJeune WS, Sarkar PS, Brobey R, Rosenblatt KP, Tilton RG, Choudhary S (2011) Klotho depletion contributes to increased inflammation in kidney of the db/db mouse model of diabetes via RelA (serine)536 phosphorylation. Diabetes 60:1907–1916
Gattineni J, Twombley K, Goetz R, Mohammadi M, Baum M (2011) Regulation of serum 1,25(OH)2Vitamin D3 levels by fibroblast growth factor 23 is mediated by FGF receptors 3 and 4. Am J Physiol Renal Physiol 301:F371–F377
Shimada T, Yamazaki Y, Takahashi M, Hasegawa H, Urakawa I, Oshima T, Ono K, Kakitani M, Tomizuka K, Fujita T et al (2005) Vitamin D receptor-independent FGF23 actions in regulating phosphate and vitamin D metabolism. Am J Physiol Renal Physiol 289:F1088–F1095
London GM, Guerin AP, Marchais SJ, Metivier F, Pannier B, Adda H (2003) Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant 18:1731–1740
Lang F, Ritz E, Voelkl J, Alesutan I (2013) Vascular calcification—is aldosterone a culprit? Nephrol Dial Transplant 28:1080–1084
Rodriguez M, Martinez-Moreno JM, Rodriguez-Ortiz ME, Munoz-Castaneda JR, Almaden Y (2011) Vitamin D and vascular calcification in chronic kidney disease. Kidney Blood Press Res 34:261–268
Kuro-o M (2010) Klotho. Pflugers Arch 459:333–343
Bernheim J, Benchetrit S (2011) The potential roles of FGF23 and Klotho in the prognosis of renal and cardiovascular diseases. Nephrol Dial Transplant 26:2433–2438
Shalhoub V, Shatzen EM, Ward SC, Davis J, Stevens J, Bi V, Renshaw L, Hawkins N, Wang W, Chen C et al (2012) FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J Clin Invest 122:2543–2553
Acknowledgments
The authors acknowledge the technical assistance of E. Faber. The study was supported by the Deutsche Forschungsgemeinschaft (La 315/15-1, Fo 695/1-1, and Fo 695/1-2) and the National Center for Competence in Research NCCR Kidney. CH was financed by the Swiss National Science Foundation.
Authors’ contribution
MF and FL made the study design. BZ, JY, ATU, HF, AF, SS, MSS, HC, DA, DS, and AD performed data collection. BZ, JY, SS, and CAW analyzed the data. MF, CAW, and FL interpreted the results. FL drafted the manuscript. FL wrote the manuscript. MF, CAW, and FL revised the manuscript content. BZ, JY, ATU, HF, AF, SS, MSS, HC, DA, DS, AD, CAW, MF, and FL read and approved the final version of the manuscript. FL takes responsibility for the integrity of the data analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Zhang, B., Yan, J., Umbach, A.T. et al. NFκB-sensitive Orai1 expression in the regulation of FGF23 release. J Mol Med 94, 557–566 (2016). https://doi.org/10.1007/s00109-015-1370-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-015-1370-3