
Abstract
Recent studies have identified a novel bone−kidney endocrine axis that maintains phosphate homeostasis. When phosphate is in excess, fibroblast growth factor-23 (FGF23) is secreted from bone and acts on the kidney to promote phosphate excretion into urine and suppress vitamin D synthesis, thereby inducing negative phosphate balance. One critical feature of FGF23 is that it requires Klotho, a single-pass transmembrane protein expressed in renal tubules, as an obligate coreceptor to bind and activate FGF receptors. Several hereditary disorders that exhibit inappropriately high serum FGF23 levels are associated with phosphate wasting and impaired bone mineralization. In contrast, defects in either FGF23 or Klotho are associated with phosphate retention and a premature-aging syndrome. The aging-like phenotypes in Klotho-deficient or FGF23-deficient mice can be rescued by resolving hyperphosphatemia with dietary or genetic manipulation, suggesting a novel concept that phosphate retention accelerates aging. Phosphate retention is universally observed in patients with chronic kidney disease (CKD) and identified as a potent risk of death in epidemiological studies. Thus, the bone−kidney endocrine axis mediated by FGF23 and Klotho has emerged as a novel target of therapeutic interventions in CKD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The blood phosphate level is determined by counterbalance between absorption of dietary phosphate from the intestine, mobilization from bone (the major reservoir of calcium and phosphate in the body), and excretion from the kidney into urine [1]. These processes are coordinately regulated by several endocrine factors. Vitamin D and parathyroid hormone (PTH), which have been extensively studied as hormones that regulate calcium metabolism [2], are also involved in phosphate metabolism. The active form of vitamin D (1,25-dihydroxyvitamin D3) is synthesized in the kidney and acts on the intestine to increase absorption of dietary calcium and phosphate. It also acts on bone to stimulate osteoclastogenesis and promote mobilization of calcium and phosphate from the reservoir, thereby increasing blood levels of both calcium and phosphate. PTH acts on the kidney to promote both vitamin D synthesis and phosphaturia (phosphate excretion into urine). As a result, unlike vitamin D, PTH can selectively increase blood calcium levels without concomitant increase in blood phosphate levels [3].
Recent studies have identified fibroblast growth factor-23 (FGF23) as a novel hormone that lowers blood phosphate levels [4–6]. When phosphate is in excess, FGF23 is secreted from bone and acts on the kidney to induce phosphaturia and suppress vitamin D synthesis, thereby inducing a negative phosphate balance to maintain phosphate homeostasis [6–9]. FGF23 requires Klotho protein as a coreceptor for high affinity binding to cognate FGF receptors (FGFRs). The purpose of this review was to overview recent progress in our understanding of endocrine regulation of phosphate metabolism by FGF23 and Klotho and to discuss its potential role in the pathophysiology of chronic kidney disease (CKD).
FGF23
FGF23 was originally identified as a gene mutated in patients with autosomal dominant hypophosphatemic rickets (ADHR) [5]. Patients with ADHR carry missense mutations in the FGF23 gene that confer resistance to proteolytic degradation of the FGF23 protein [10]. As a result, ADHR patients exhibit high serum FGF23 levels. Because FGF23 has an activity that induces negative phosphate balance, ADHR patients exhibit phosphate-wasting phenotypes such as hypophosphatemia and rickets. Although the protease(s) that inactivates FGF23 remains to be identified, it cleaves FGF23 at the 176RXXR179 motif and generates two inactive fragments [11]. Missense mutations in this critical motif (R176Q, R179Q/W) make the protein resistant to the protease and increase the half-life of FGF23 in the blood [12]. Several other phosphate-wasting syndromes associated with FGF23 excess have been identified, including tumor-induced osteomalacia (TIO), X-linked hypophosphatemia (XLH), and autosomal recessive hypophosphatemic rickets (ARHR). TIO is caused by FGF23-producing tumors [13]. XLH and ARHR are caused by mutations in the PHEX (a phosphate-regulating gene with homologies to endopeptidases on the X-chromosome) gene [14] and the DMP-1 (dentin matrix protein-1) gene [15, 16], respectively. These two genes are expressed in osteocytes in the bone where FGF23 is primarily produced and secreted. Recent animal studies have demonstrated that PHEX and DMP-1 may be involved in the regulation of FGF23 gene expression. Hyp mice, which have deletions in the Phex gene, show high FGF23 expression in the bone, high serum levels of FGF23, hypophosphatemia, and impaired bone mineralization, as observed in XLH patients [17, 18]. DMP-1 knockout mice also exhibit high FGF23 expression and phosphate-wasting phenotypes, as observed in ARHR patients [15]. The precise mechanism by which PHEX and DMP-1 proteins suppress expression of the FGF23 gene remains to be determined.
In contrast to patients with ADHR, ARHR, XLH, and TIO that exhibit phosphate-wasting phenotypes, patients with familial tumoral calcinosis (FTC) exhibit phosphate-retention phenotypes including hyperphosphatemia and ectopic calcification associated with low serum FGF23. FTC is caused by mutations in the GALNT3 gene that encodes a glycosyl transferase called UDP-N-acetyl-α-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase-3 (ppGaNTase-T3) [19]. This enzyme is required for O-glycosylation of FGF23 at Thr178, which resides within the cleavage motif of FGF23 [20]. It is likely that loss of O-glycosylation at Thr178 increases susceptibility of FGF23 to proteolytic degeneration, resulting in low serum levels of FGF23. Mice that completely lack FGF23 (Fgf23 -/- mice) develop severe phosphate-retention phenotypes characterized by extensive soft tissue calcification and hyperphosphatemia [21]. These studies on phosphate-wasting and phosphate-retention syndromes in mice and humans have provided unequivocal evidence indicating that FGF23 is an essential hormone for maintaining phosphate homeostasis.
Although FGF23 belongs to the FGF ligand superfamily [22], phylogenetic and sequence analyses have segregated FGF23 and two additional FGFs (FGF19 and FGF21) from the other FGF family members [23]. These three atypical FGFs—namely, FGF19, FGF21, and FGF23—are collectively called endocrine FGFs because they function as endocrine factors, unlike the other classic FGFs that basically function as paracrine and/or autocrine factors [24]. The molecular basis behind the endocrine mode of action may lie in the fact that these endocrine FGFs have low affinity to heparan sulfate (HS). In general, FGF ligands have a conserved core region with 12 antiparallel β strands, where the HS-binding domain resides [25–27]. However, the HS-binding domain of endocrine FGFs deviates from that of the other paracrine-acting FGFs and prohibits formation of hydrogen bonding between HS and amino acid residues in the HS-binding domain, which is the basis of affinity to HS [28, 29]. This unique structural feature reduces affinity of endocrine FGFs to HS and allows them to escape from HS-rich extracellular matrices and enter into systemic circulation. Although the low affinity of endocrine FGFs to HS may be advantageous for the endocrine mode of action, it may be disadvantageous for signal transduction through FGFRs because HS is required for high-affinity binding of FGFs to FGFRs. FGFRs are single-pass transmembrane receptor tyrosin kinases that dimerize upon binding FGFs. HS participates in the FGF-FGFR interaction and promotes formation of a 2:2:2 FGF-FGFR-HS signaling complex, which is essential for efficient activation of FGFR tyrosine kinase [30]. Thus, endocrine FGFs may require a cofactor(s) other than and/or in addition to HS to secure efficient dimerization and activation of FGFR. In fact, FGF23 cannot activate FGF signaling in most cultured cells, even when they express FGFRs endogenously, whereas classic FGFs such as FGF2 can activate FGF signaling in these cells [31]. Identity of the putative cofactor(s) required for FGF23 to activate FGFRs was not clear until it was realized that the phenotypes of FGF23-deficient mice are identical with those of Klotho-deficient mice.
Klotho
The klotho gene, named after a Greek goddess who spins the thread of life, was originally identified as a gene mutated in a mouse strain that inherits a premature-aging syndrome in an autosomal recessive manner [32]. Mice defective in klotho gene expression develop multiple aging-like phenotypes around 3–4 weeks after birth, including growth retardation, hypogonadotropic hypogonadism, rapid thymus atrophy [33], skin atrophy, sarcopenia, vascular calcification, osteopenia [34], pulmonary emphysema [35–37], cognition impairment [38], hearing disturbance [39], and motor neuron degeneration [40], and die around 2 months of age. In contrast, transgenic mice that overexpress Klotho live longer than wild-type mice [41]. Thus, the Klotho gene may be an aging suppressor gene that extends life span when overexpressed and accelerates aging when disrupted [42]. Furthermore, polymorphisms in the human KLOTHO gene are associated with life span [43] as well as bone mineral density [44–46], high-density lipoprotein (HDL) cholesterol level, blood pressure, stroke [47], coronary artery disease [48], and cognitive function [49], suggesting that Klotho may be involved in the regulation of aging processes in humans. The Klotho gene encodes a single-pass transmembrane protein that belongs to a family 1 glycosidase [50] and is expressed primarily in renal tubules in the kidney and choroid plexus in the brain [32]. Although recombinant Klotho protein was reported to have weak β-glucuronidase activity in vitro [51], physiological relevance of the β-glucuronidase activity in vivo was not clear.
The clue to understanding Klotho protein function was the fact that FGF23-deficient mice and Klotho-deficient mice develop identical phenotypes. FGF23-deficient mice not only exhibit phosphate retention but also develop multiple aging-like phenotypes [21], which is reminiscent of Klotho-deficient mice. Conversely, Klotho-deficient mice not only develop a premature-aging syndrome but also exhibit hyperphosphatemia [52, 53], which is reminiscent of FGF23-deficient mice. These observations suggested that Klotho and FGF23 might function in a common signal transduction pathway. In fact, Klotho protein forms a constitutive binary complex with several FGF receptor isoforms (FGFR1c, 3c, 4) and significantly increases the affinity of these FGFRs specifically to FGF23 [31]. Thus, Klotho protein functions as an obligate coreceptor for FGF23. This finding was later confirmed in an independent study [54]. The fact that FGF23 requires Klotho protein as a coreceptor explains why Klotho-deficient mice, FGF23-deficient mice, and mice lacking both Klotho and FGF23 [55] develop identical phenotypes. It also explain why extremely high serum FGF23 levels of Klotho-deficient mice [54] do not cause any adverse effects in Klotho-deficient mice [55]. In addition, kidney-specific expression of Klotho explains why FGF23 can identify the kidney as its target organ among many other tissues that express multiple FGFR isoforms. Klotho protein function is to compensate for the low affinity of FGF23 to heparan sulfate and specifically support FGFR activation with FGF23, which represents a novel mechanism for confining target organs in redundant ligand-receptor interactions.
Endocrine regulation of phosphate metabolism
The bone−kidney endocrine axis mediated by FGF23 and Klotho has emerged as the major regulator of phosphate homeostasis [4, 9, 24, 56–58]. FGF23 has an activity that reduces the number of sodium-phosphate cotransporter type-2a (NaPi-2a) on the brush border membrane of proximal tubules, thereby promoting renal phosphate excretion [59–62]. Thus, FGF23 functions as a phosphaturic hormone. In addition, FGF23 suppresses synthesis and promotes degradation of 1,25-dihydroxyvitamin D3 in proximal tubules [63]. FGF23 down-regulates expression of the Cyp27b1 gene, which encodes 1α-hydroxylase, the enzyme that synthesizes the active form of vitamin D (1,25-dihydroxyvitamin D3) from its inactive precursor (25-hydroxyvitamin D3). Furthermore, FGF23 up-regulates expression of the Cyp24 gene that encodes 24-hydroxylase, the enzyme that hydrolyzes and inactivates 1,25-dihydroxyvitamin D3. Thus, FGF23 functions as a counterregulatory hormone for vitamin D [8]. The ability of FGF23 to reduce serum 1,25-dihydroxyvitamin D3 levels also contributes to induction of negative phosphate balance through reducing phosphate absorption from the intestine. Importantly, 1,25-dihydroxyvitamin D3 up-regulates expression of the FGF23 gene [63] and closes a negative feedback loop (Fig. 1). Disruption of this negative feedback loop results in high serum 1,25-dihydroxyvitamin D3 levels, as observed in Klotho-deficient mice, FGF23-deficient mice, and FTC patients. Recently, a patient carrying a homozygous missense mutation in the KLOTHO gene (H193R) was reported [64]. The patient exhibited phosphate-retention phenotypes similar to FTC patients, indicating that the H193R mutation is a loss-of-function mutation. This is the first case in humans exhibiting phosphate retention due to a defect in Klotho protein.

The bone−kidney−parathyroid endocrine axes mediated by fibroblast growth factor-23 (FGF23) and Klotho. Active form of vitamin D (1,25-dihydroxyvitamin D3) binds to vitamin D receptor (VDR) in the bone (osteocytes). The ligand-bound VDR forms a heterodimer with a nuclear receptor RXR and transactivates expression of the FGF23 gene. FGF23 secreted from bone acts on the Klotho-FGF receptor (FGFR) complex expressed in the kidney (the bone−kidney axis) and parathyroid gland (the bone−parathyroid axis). In the kidney, FGF23 suppresses synthesis of active vitamin D by down-regulating expression of the Cyp27b1 gene and promotes its inactivation by up-regulating expression of the Cyp24 gene, thereby closing a negative feedback loop for vitamin D homeostasis. In the parathyroid gland, FGF23 suppresses production and secretion of parathyroid hormone (PTH). PTH binds to the PTH receptor (PTHR) expressed on renal tubular cells, leading to up-regulation of the Cyp27b1 gene expression. Thus, suppression of PTH by FGF23 reduces expression of the Cyp27b1 gene and serum levels of 1,25-dihydroxyvitamin D3. This closes another long negative feedback loop for vitamin D homeostasis
It should be noted that Klotho protein is expressed much more abundantly in distal convoluted tubules than in proximal tubules, whereas both phosphate reabsorption and vitamin D synthesis take place in proximal tubules. This discrepancy has raised two possibilities that are not mutually exclusive. One possibility is that, although Klotho expression levels in proximal tubules are not as high as distal convoluted tubules, FGF23 may signal through the FGFR-Klotho complex on proximal tubules and directly regulate NaPi-2a expression and vitamin D synthesis. In this case, the function of Klotho protein abundantly expressed in distal convoluted tubules must be addressed. The other possibility is that FGF23 may act first on distal convoluted tubules and then generate a secondary signal that instructs proximal tubules to reduce phosphate reabsorption and vitamin D synthesis. Recent animal studies may support the latter possibility. Despite the fact that proximal tubules primarily express FGFR3, knockout of the Fgfr3 gene in Hyp mice, which have elevated serum FGF23 levels, failed to rescue their phosphate-wasting phenotypes [65]. Furthermore, it was reported that activation of the FGF signaling pathway was detectable only in distal convoluted tubules after injection of FGF23 into mice [66]. These findings suggest that activity of FGF23 may be independent of FGF signaling activation in the proximal tubule.
PTH plays an important role not only in calcium metabolism but also in phosphate homeostasis. Like FGF23, PTH has an activity that induces phosphaturia [3]. However, in contrast to FGF23, PTH up-regulates expression of the Cyp27b1 gene and increases serum 1,25-dihydroxyvitamin D3 levels [2]. Recent studies showed that the parathyroid gland is one of the few organs that express a decent amount of Klotho protein endogenously, indicating that the parathyroid may be a target organ of FGF23. In fact, FGF23 down-regulates PTH expression and suppresses PTH secretion in vivo and in vitro [67, 68]. The ability of FGF23 to reduce serum PTH levels may further enhance the activity of FGF23 as a counterregulatory hormone for vitamin D and contribute to a long negative feedback loop involving bone, kidney, and parathyroid gland (Fig. 1). However, it should be noted that patients with CKD typically exhibit secondary hyperparathyroidism associated with high serum FGF23 levels, which seemingly contradicts the ability of FG23 to suppress PTH secretion and production. Considering that Klotho expression is positively regulated by vitamin D [53], one possible explanation is that low serum vitamin D levels in CKD patients may reduce Klotho expression not only in the kidney (discussed below) but also in the parathyroid glands and make these organs resistant to FGF23.
Phosphate toxicity
Defects in either Klotho or FGF23 disrupt the negative feedback loops that maintain phosphate and vitamin D homeostasis, resulting in high serum phosphate and vitamin D levels. High serum vitamin D promotes intestinal absorption of calcium and induces hypercalcemia as well. Importantly, this metabolic state characterized by high serum phosphate, calcium, and vitamin D levels is associated with a premature aging syndrome, as observed in Klotho-deficient mice and FGF23-deficient mice. These observations imply that phosphate, calcium, and/or vitamin D may be toxic when retained and thus accelerate aging. Several animal studies have supported this notion. First, vitamin-D-deficient diet not only restored serum phosphate and calcium levels but also rescued several aging-like phenotypes in Klotho-deficient mice and FGF23-deficient mice [53, 69]. Second, ablation of vitamin D activity in Klotho-deficient mice and FGF23-deficient mice by disrupting the Cyp27b1 gene [70, 71] or vitamin D receptor gene [72] also rescued hyperphosphatemia, hypercalcemia, and the premature aging syndrome. Lastly, low phosphate diet rescued shortened life span and vascular calcification in FGF23-deficient mice and Klotho-deficient mice [69, 73]. These studies provide evidence that the premature aging syndrome caused by defects in the bone−kidney endocrine axis is due to retention of phosphate, calcium, and/or vitamin D. It should be noted that low phosphate diet rescued FGF23-deficient mice despite the fact that it further increased already high serum calcium and vitamin D levels [69], suggesting that phosphate, but not calcium or vitamin D, is primarily responsible for the aging-like phenotypes. It is likely that low vitamin D diet and ablation of vitamin D activity rescued accelerated aging through reducing serum phosphate levels, although it remains to be determined whether high serum vitamin D and/or calcium levels are a prerequisite for phosphate to accelerate aging.
Chronic kidney disease
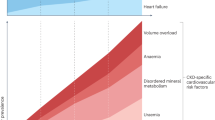
Phosphate retention is universally observed in patients with CKD. Hyperphosphatemia has been identified as a potent, independent risk of death [74]. Why does hyperphosphatemia increase mortality? One likely explanation is that high blood phosphate levels trigger vascular calcification and accelerate life-threatening complications such as cardiovascular events [75]. Vascular calcification is a very common complication in CKD and has been shown to contribute to the high morbidity and mortality in terms of cardiovascular events. The National Kidney Foundation task force indicated that the cardiovascular mortality of a 35-year-old patient on dialysis is equivalent to that of an 80-year-old healthy individual, rendering CKD to be one of the most potent accelerators of aging [76]. In addition, the American Heart Association announced that CKD should be included in the highest-risk group for cardiovascular disease and that patients with CKD should receive aggressive therapeutic measures to reduce morbidity and mortality [77]. Thus, lowering blood phosphate levels is expected to reduce vascular calcification and cardiovascular events, thereby improving prognosis of CKD patients. In fact, CKD patients with hyperphosphatemia (≥6.5 mg/dl) were reported to have higher risk for death resulting from cardiovascular disease than those with the lower serum phosphate levels (< 6.5 mg/dl) [78]. Based on these observations, control of blood phosphate levels <6.5 mg/dl has been proposed as one of the most important therapeutic goals for improving life expectancy of CKD patients.
It is likely that dysregulation of the FGF23-Klotho endocrine axis may be involved in the mechanism by which CKD patients fail to maintain phosphate homeostasis. In fact, serum FGF23 levels are increased with advancing stages of CKD [79], whereas Klotho expression in the kidney is significantly decreased in CKD patients [80] and in various animal models of chronic and acute renal damage [81, 82]. Thus, CKD may be viewed as a state of FGF23 resistance caused by Klotho deficiency. This viewpoint explains several observations on phosphate metabolism in CKD that lack mechanistic insights. For example, epidemiological studies have indicated that serum FGF23 levels increase long before serum phosphate levels increase during the progression of CKD [79]. In other words, patients with early stages of CKD require higher serum FGF23 levels than normal people to maintain normal serum phosphate levels. This may represent compensation for end-organ resistance to FGF23 due to decreased Klotho expression in the kidney. It has been also known that serum vitamin D levels decrease long before serum phosphate levels increase during CKD progression [79]. This may be a result of the secondary hyper-FGF23-emia caused by decreased renal Klotho expression, because FGF23 has an activity that lowers serum vitamin D levels. In addition, epidemiological studies have indicated that high serum FGF23 levels are associated with poor prognosis in patients undergoing dialysis [83]. This may be explained by assuming that high serum FGF23 indicates low renal Klotho expression associated with severe renal damage. It remains to be determined whether decrease in Klotho expression is one of the earliest changes in the progression of CKD.
Of note, recent animal studies have shown that Klotho functions as a renoprotective factor. Although the mechanism is yet to be determined, overexpression of Klotho ameliorated progressive renal injury in mouse models of glomerulonephritis [81] and acute kidney injury [82]. Thus, it may be of therapeutic value for CKD to preserve Klotho expression in the kidney. Klotho expression is down-regulated by angiotensin II [84, 85] and up-regulated by peroxisome proliferator-activated receptor-gamma (PPARγ) agonists such as thiazolidinediones [86]. These observations suggest that renoprotective effects of angiotensin-converting enzyme inhibitors and thiazolidinediones may be partly attributed to their potential for increasing or preserving Klotho expression in the kidney. Klotho expression is also up-regulated by 1,25-dihydroxyvitamin D3 [53]. Thus, low serum vitamin D caused by secondary hyper-FGF23-emia further reduces Klotho expression, potentially leading to deterioration spiral of Klotho expression. The benefit of vitamin D replacement therapy may be partly attributed to interruption of this vicious cycle.
In addition to functioning as an obligate coreceptor for FGF23, Klotho protein functions as a humoral factor that regulates activity of several ion channels and growth-factor receptors [41], which represents a novel function of Klotho protein. The entire extracellular domain of Klotho protein is clipped by a membrane-anchored protease ADAM10/17 on the cell surface and released into the extracellular space [87]. In fact, Klotho ectodomain (secreted Klotho protein) is detectable in the blood, urine, and cerebrospinal fluid [41, 88]. The secreted Klotho protein in turn functions as a putative sialidase that removes terminal sialic acids in the glycans of several ion channels, including a calcium channel, transient receptor potential vanilloid type isoform 5 (TRPV5) [89, 90], and a potassium channel, renal outer medullary potassium channel-1 (ROMK1) [91]. Removal of sialic acids by secreted Klotho protein on the cell surface prevents internalization of these ion channels, resulting in increase in transepithelial calcium (Ca2+) absorption and potassium (K+) secretion in distal nephrons, respectively. Thus, Klotho protein not only regulates phosphate metabolism by functioning as a coreceptor for FGF23 but also regulates calcium and potassium metabolism by functioning as a humoral factor that modifies trafficking of TRPV5 and ROMK1. Significance of the secreted Klotho protein in the regulation of calcium and potassium homeostasis and in pathophysiology in CKD remains to be determined.
It has become increasingly clear that phosphate metabolism plays a critical role in the pathophysiology in CKD and that hyperphosphatemia should be aggressively treated to improve life expectancy of CKD patients. In this context, the Klotho and FGF23 axis is expected to be a novel target of therapeutic interventions in CKD.
References
Berndt T, Kumar R (2009) Novel mechanisms in the regulation of phosphorus homeostasis. Physiology (Bethesda) 24:17–25
Dusso AS, Brown AJ, Slatopolsky E (2005) Vitamin D. Am J Physiol Renal Physiol 289:F8–F28
Berndt T, Kumar R (2007) Phosphatonins and the regulation of phosphate homeostasis. Annu Rev Physiol 69:341–359
Kuro-o M (2006) Klotho as a regulator of fibroblast growth factor signaling and phosphate/calcium metabolism. Curr Opin Nephrol Hypertens 15:437–441
White KE, Evans WE, O'Riordan JLH, Speer MC, Econs MJ, Lorenz-Depiereux B, Grabowski M, Meitinger T, Storm TM (2000) Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 26:345–348
Quarles LD (2008) Endocrine functions of bone in mineral metabolism regulation. J Clin Invest 118:3820–3828
Quarles LD (2003) FGF23, PHEX, and MEPE regulation of phosphate homeostasis and skeletal mineralization. Am J Physiol Endocrinol Metab 285:E1–E9
Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, Quarles LD (2006) Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol 17:1305–1315
Liu S, Gupta A, Quarles LD (2007) Emerging role of fibroblast growth factor 23 in a bone-kidney axis regulating systemic phosphate homeostasis and extracellular matrix mineralization. Curr Opin Nephrol Hypertens 16:329–335
Kuro-o M (2009) Klotho in chronic kidney disease-What’s new? Nephrol Dial Transplant 24:1705–1708
White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ (2001) Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int 60:2079–2086
Shimada T, Muto T, Urakawa I, Yoneya T, Yamazaki Y, Okawa K, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T (2002) Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology 143:3179–3182
Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T (2001) Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A 98:6500–6505
The HYP Consortium (1995) A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nat Genet 11:130–136
Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE (2006) Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet 38:1310–1315
Lorenz-Depiereux B, Bastepe M, Benet-Pages A, Amyere M, Wagenstaller J, Muller-Barth U, Badenhoop K, Kaiser SM, Rittmaster RS, Shlossberg AH, Olivares JL, Loris C, Ramos FJ, Glorieux F, Vikkula M, Juppner H, Strom TM (2006) DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet 38:1248–1250
Beck L, Soumounou Y, Martel J, Krishnamurthy G, Gauthier C, Goodyer CG, Tenenhouse HS (1997) Pex/PEX tissue distribution and evidence for a deletion in the 3′ region of the Pex gene in X-linked hypophosphatemic mice. J Clin Invest 99:1200–1209
Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD (2006) Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab 291:E38–E49
Garringer HJ, Fisher C, Larsson TE, Davis SI, Koller DL, Cullen MJ, Draman MS, Conlon N, Jain A, Fedarko NS, Dasgupta B, White KE (2006) The role of mutant UDP-N-acetyl-alpha-D-galactosamine-polypeptide N-acetylgalactosaminyltransferase 3 in regulating serum intact fibroblast growth factor 23 and matrix extracellular phosphoglycoprotein in heritable tumoral calcinosis. J Clin Endocrinol Metab 91:4037–4042
Kato K, Jeanneau C, Tarp MA, Benet-Pages A, Lorenz-Depiereux B, Bennett EP, Mandel U, Strom TM, Clausen H (2006) Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J Biol Chem 281:18370–18377
Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T (2004) Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest 113:561–568
Yamashita T, Yoshioka M, Itoh N (2000) Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun 277:494–498
Itoh N, Ornitz DM (2008) Functional evolutionary history of the mouse Fgf gene family. Dev Dyn 237:18–27
Kuro-o M (2008) Endocrine FGFs and Klothos: emerging concepts. Trends Endocrinol Metab 19:239–245
Murzin AG, Lesk AM, Chothia C (1992) beta-Trefoil fold. Patterns of structure and sequence in the Kunitz inhibitors interleukins-1 beta and 1 alpha and fibroblast growth factors. J Mol Biol 223:531–543
Mohammadi M, Olsen SK, Ibrahimi OA (2005) Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev 16:107–137
Mohammadi M, Olsen SK, Goetz R (2005) A protein canyon in the FGF-FGF receptor dimer selects from an a la carte menu of heparan sulfate motifs. Curr Opin Struct Biol 15:506–516
Harmer NJ, Pellegrini L, Chirgadze D, Fernandez-Recio J, Blundell TL (2004) The crystal structure of fibroblast growth factor (FGF) 19 reveals novel features of the FGF family and offers a structural basis for its unusual receptor affinity. Biochemistry 43:629–640
Goetz R, Beenken A, Ibrahimi OA, Kalinina J, Olsen SK, Eliseenkova AV, Xu C, Neubert T, Zhang F, Linhardt RJ, Yu X, White KE, Inagaki T, Kliewer SA, Yamamoto M, Kurosu H, Ogawa Y, Kuro-o M, Lanske B, Razzaque MS, Mohammadi M (2007) Molecular insights into the Klotho-dependent, endocrine mode of action of FGF19 subfamily members. Mol Cell Biol 27:3417–3428
Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, Yayon A, Linhardt RJ, Mohammadi M (2000) Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell 6:743–750
Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro-o M (2006) Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 281:6120–6123
Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima Y (1997) Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390:45–51
Min D, Panoskaltsis-Mortari A, Kuro-o M, Hollander GA, Blazar BR, Weinberg KI (2007) Sustained thymopoiesis and improvement in functional immunity induced by exogenous KGF administration in murine models of aging. Blood 109:2529–2537
Kawaguchi H, Manabe N, Miyaura C, Chikuda H, Nakamura K, Kuro-o M (1999) Independent impairment of osteoblast and osteoclast differentiation in klotho mouse exhibiting low-turnover osteopenia. J Clin Invest 104:229–237
Suga T, Kurabayashi M, Sando Y, Ohyama Y, Maeno T, Maeno Y, Aizawa H, Matsumura Y, Kuwaki T, Kuro-o M, Nabeshima Y, Nagai R (2000) Disruption of the klotho gene causes pulmonary emphysema in mice. Defect in maintenance of pulmonary integrity during postnatal life. Am J Respir Cell Mol Biol 22:26–33
Sato A, Hirai T, Imura A, Kita N, Iwano A, Muro S, Nabeshima Y, Suki B, Mishima M (2007) Morphological mechanism of the development of pulmonary emphysema in klotho mice. Proc Natl Acad Sci U S A 104:2361–2365
Ishii M, Yamaguchi Y, Yamamoto H, Hanaoka Y, Ouchi Y (2008) Airspace enlargement with airway cell apoptosis in klotho mice: a model of aging lung. J Gerontol A Biol Sci Med Sci 63:1289–1298
Nagai T, Yamada K, Kim HC, Kim YS, Noda Y, Imura A, Nabeshima Y, Nabeshima T (2003) Cognition impairment in the genetic model of aging klotho gene mutant mice: a role of oxidative stress. FASEB J 17:50–52
Kamemori M, Ohyama Y, Kurabayashi M, Takahashi K, Nagai R, Furuya N (2002) Expression of Klotho protein in the inner ear. Hear Res 171:103–110
Anamizu Y, Kawaguchi H, Seichi A, Yamaguchi S, Kawakami E, Kanda N, Matsubara S, Kuro-o M, Nabeshima Y, Nakamura K, Oyanagi K (2005) Klotho insufficiency causes decrease of ribosomal RNA gene transcription activity, cytoplasmic RNA and rough ER in the spinal anterior horn cells. Acta Neuropathol (Berl) 109:457–466
Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, Shimomura I, Takayama Y, Herz J, Kahn CR, Rosenblatt KP, Kuro-o M (2005) Suppression of aging in mice by the hormone Klotho. Science 309:1829–1833
Kuro-o M (2008) Klotho as a regulator of oxidative stress and senescence. Biol Chem 389:233–241
Arking DE, Krebsova A, Macek M Sr, Macek M Jr, Arking A, Mian IS, Fried L, Hamosh A, Dey S, McIntosh I, Dietz HC (2002) Association of human aging with a functional variant of klotho. Proc Natl Acad Sci U S A 99:856–861
Kawano K, Ogata N, Chiano M, Molloy H, Kleyn P, Spector TD, Uchida M, Hosoi T, Suzuki T, Orimo H, Inoue S, Nabeshima Y, Nakamura K, Kuro-o M, Kawaguchi H (2002) Klotho gene polymorphisms associated with bone density of aged postmenopausal women. J Bone Miner Res 17:1744–1751
Ogata N, Matsumura Y, Shiraki M, Kawano K, Koshizuka Y, Hosoi T, Nakamura K, Kuro-o M, Kawaguchi H (2002) Association of klotho gene polymorphism with bone density and spondylosis of the lumbar spine in postmenopausal women. Bone 31:37–42
Zarrabeitia MT, Hernandez JL, Valero C, Zarrabeitia AL, Ortiz F, Gonzalez-Macias J, Riancho JA (2007) Klotho gene polymorphism and male bone mass. Calcif Tissue Int 80:10–14
Arking DE, Atzmon G, Arking A, Barzilai N, Dietz HC (2005) Association between a functional variant of the KLOTHO gene and high-density lipoprotein cholesterol, blood pressure, stroke, and longevity. Circ Res 96:412–418
Arking DE, Becker DM, Yanek LR, Fallin D, Judge DP, Moy TF, Becker LC, Dietz HC (2003) KLOTHO allele status and the risk of early-onset occult coronary artery disease. Am J Hum Genet 72:1154–1161
Kachiwala SJ, Harris SE, Wright AF, Hayward C, Starr JM, Whalley LJ, Deary IJ (2005) Genetic influences on oxidative stress and their association with normal cognitive ageing. Neurosci Lett 386:116–120
Mian IS (1998) Sequence, structural, functional, and phylogenetic analyses of three glycosidase families. Blood Cells Mol Dis 24:83–100
Tohyama O, Imura A, Iwano A, Freund JN, Henrissat B, Fujimori T, Nabeshima Y (2004) Klotho is a novel beta-glucuronidase capable of hydrolyzing steroid beta-glucuronides. J Biol Chem 279:9777–9784
Yoshida T, Fujimori T, Nabeshima Y (2002) Mediation of unusually high concentrations of 1, 25-dihydroxyvitamin D in homozygous klotho mutant mice by increased expression of renal 1alpha-hydroxylase gene. Endocrinology 143:683–689
Tsujikawa H, Kurotaki Y, Fujimori T, Fukuda K, Nabeshima Y (2003) Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol Endocrinol 17:2393–2403
Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T (2006) Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444:770–774
Nakatani T, Sarraj B, Ohnishi M, Densmore MJ, Taguchi T, Goetz R, Mohammadi M, Lanske B, Razzaque MS (2009) In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23)-mediated regulation of systemic phosphate homeostasis. FASEB J 23:433–441
Kurosu H, Kuro-o M (2009) The Klotho gene family as a regulator of endocrine fibroblast growth factors. Mol Cell Endocrinol 299:72–78
Kurosu H, Kuro-o M (2008) The Klotho gene family and the endocrine fibroblast growth factors. Curr Opin Nephrol Hypertens 17:368–372
Liu S, Quarles LD (2007) How fibroblast growth factor 23 works. J Am Soc Nephrol 18:1637–1647
Segawa H, Kawakami E, Kaneko I, Kuwahata M, Ito M, Kusano K, Saito H, Fukushima N, Miyamoto K (2003) Effect of hydrolysis-resistant FGF23-R179Q on dietary phosphate regulation of the renal type-II Na/Pi transporter. Pflugers Arch 446:585–592
Segawa H, Yamanaka S, Ohno Y, Onitsuka A, Shiozawa K, Aranami F, Furutani J, Tomoe Y, Ito M, Kuwahata M, Imura A, Nabeshima Y, Miyamoto K (2007) Correlation between hyperphosphatemia and type II Na-Pi cotransporter activity in klotho mice. Am J Physiol Renal Physiol 292:F769–F779
Shimada T, Urakawa I, Yamazaki Y, Hasegawa H, Hino R, Yoneya T, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T (2004) FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun 314:409–414
Miyamoto K, Ito M, Tatsumi S, Kuwahata M, Segawa H (2007) New aspect of renal phosphate reabsorption: the type IIc sodium-dependent phosphate transporter. Am J Nephrol 27:503–515
Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T (2004) FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res 19:429–435
Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, Goetz R, Mohammadi M, White KE, Econs MJ (2007) A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest 117:2692–2701
Liu S, Vierthaler L, Tang W, Zhou J, Quarles LD (2008) FGFR3 and FGFR4 do not mediate renal effects of FGF23. J Am Soc Nephrol 19:2342–2350
Farrow EG, Davis SI, Summers LJ, White KE (2009) Initial FGF23-mediated signaling occurs in the distal convoluted tubule. J Am Soc Nephrol 20:955–960
Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, Sirkis R, Naveh-Many T, Silver J (2007) The parathyroid is a target organ for FGF23 in rats. J Clin Invest 117:4003–4008
Krajisnik T, Bjorklund P, Marsell R, Ljunggren O, Akerstrom G, Jonsson KB, Westin G, Larsson TE (2007) Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol 195:125–131
Stubbs JR, Liu S, Tang W, Zhou J, Wang Y, Yao X, Quarles LD (2007) Role of hyperphosphatemia and 1,25-Dihydroxyvitamin D in vascular calcification and mortality in fibroblastic growth factor 23 null mice. J Am Soc Nephrol 18:2116–2124
Razzaque MS, Sitara D, Taguchi T, St-Arnaud R, Lanske B (2006) Premature aging-like phenotype in fibroblast growth factor 23 null mice is a vitamin D-mediated process. FASEB J 20:720–722
Ohnishi M, Nakatani T, Lanske B, Razzaque MS (2009) Reversal of mineral ion homeostasis and soft-tissue calcification of klotho knockout mice by deletion of vitamin D 1 alpha-hydroxylase. Kidney Int 75:1166–1172
Hesse M, Frohlich LF, Zeitz U, Lanske B, Erben RG (2007) Ablation of vitamin D signaling rescues bone, mineral, and glucose homeostasis in Fgf-23 deficient mice. Matrix Biol 26:75–84
Morishita K, Shirai A, Kubota M, Katakura Y, Nabeshima Y, Takeshige K, Kamiya T (2001) The progression of aging in klotho mutant mice can be modified by dietary phosphorus and zinc. J Nutr 131:3182–3188
Tonelli M, Sacks F, Pfeffer M, Gao Z, Curhan G (2005) Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation 112:2627–2633
Hruska KA, Saab G, Mathew S, Lund R (2007) Renal osteodystrophy, phosphate homeostasis, and vascular calcification. Semin Dial 20:309–315
Meyer KB, Levey AS (1998) Controlling the epidemic of cardiovascular disease in chronic renal disease: report from the National Kidney Foundation Task Force on cardiovascular disease. J Am Soc Nephrol 9:S31–S42
Sarnak MJ, Levey AS, Schoolwerth AC, Coresh J, Culleton B, Hamm LL, McCullough PA, Kasiske BL, Kelepouris E, Klag MJ, Parfrey P, Pfeffer M, Raij L, Spinosa DJ, Wilson PW (2003) Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation 108:2154–2169
Ganesh SK, Stack AG, Levin NW, Hulbert-Shearon T, Port FK (2001) Association of elevated serum PO(4), Ca x PO(4) product, and parathyroid hormone with cardiac mortality risk in chronic hemodialysis patients. J Am Soc Nephrol 12:2131–2138
Gutierrez O, Isakova T, Rhee E, Shah A, Holmes J, Collerone G, Juppner H, Wolf M (2005) Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol 16:2205–2215
Koh N, Fujimori T, Nishiguchi S, Tamori A, Shiomi S, Nakatani T, Sugimura K, Kishimoto T, Kinoshita S, Kuroki T, Nabeshima Y (2001) Severely reduced production of klotho in human chronic renal failure kidney. Biochem Biophys Res Commun 280:1015–1020
Haruna Y, Kashihara N, Satoh M, Tomita N, Namikoshi T, Sasaki T, Fujimori T, Xie P, Kanwar YS (2007) Amelioration of progressive renal injury by genetic manipulation of Klotho gene. Proc Natl Acad Sci U S A 104:2331–2336
Sugiura H, Yoshida T, Tsuchiya K, Mitobe M, Nishimura S, Shirota S, Akiba T, Nihei H (2005) Klotho reduces apoptosis in experimental ischaemic acute renal failure. Nephrol Dial Transplant 20:2636–2645
Gutierrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, Smith K, Lee H, Thadhani R, Juppner H, Wolf M (2008) Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med 359:584–592
Mitani H, Ishizaka N, Aizawa T, Ohno M, Usui S, Suzuki T, Amaki T, Mori I, Nakamura Y, Sato M, Nangaku M, Hirata Y, Nagai R (2002) In vivo klotho gene transfer ameliorates angiotensin II-induced renal damage. Hypertension 39:838–843
Saito K, Ishizaka N, Mitani H, Ohno M, Nagai R (2003) Iron chelation and a free radical scavenger suppress angiotensin II-induced downregulation of klotho, an anti-aging gene, in rat. FEBS Lett 551:58–62
Zhang H, Li Y, Fan Y, Wu J, Zhao B, Guan Y, Chien S, Wang N (2008) Klotho is a target gene of PPAR-gamma. Kidney Int 74:732–739
Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR (2007) Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc Natl Acad Sci U S A 104:19796–19801
Imura A, Iwano A, Tohyama O, Tsuji Y, Nozaki K, Hashimoto N, Fujimori T, Nabeshima Y (2004) Secreted Klotho protein in sera and CSF: implication for post-translational cleavage in release of Klotho protein from cell membrane. FEBS Lett 565:143–147
Chang Q, Hoefs S, van der Kemp AW, Topala CN, Bindels RJ, Hoenderop JG (2005) The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science 310:490–493
Cha SK, Ortega B, Kurosu H, Rosenblatt KP, Kuro-o M, Huang CL (2008) Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc Natl Acad Sci U S A 105:9805–9810
Cha SK, Hu MC, Kurosu H, Kuro-o M, Moe O, Huang CL (2009) Regulation of ROMK1 channel and renal K + excretion by Klotho. Mol Pharmacol. doi:10.1124/mol.109.055780