
Abstract
Chemoresistance remains a major problem in the treatment of gastric cancer patients. Hence, novel pharmacological agents that can overcome drug resistance are urgently required. Whether simvastatin can sensitize the gastric cancer to the antitumor effects of capecitabine in vitro and in vivo was investigated. The effect of simvastatin on the proliferation of gastric cancer cells was examined by mitochondrial dye-uptake 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide method, apoptosis by esterase staining, NF-κB activation by DNA binding assay, and protein expression by western blot analysis. The effect of simvastatin on the tumor growth in xenograft mouse model of human gastric cancer was also examined. Simvastatin suppressed the proliferation of gastric cancer cells, enhanced the apoptotic effects of capecitabine, suppressed the constitutive activation of NF-κB, and abrogated the expression of cyclooxygenase-2 (COX-2), cyclin D1, Bcl-2, survivin, CXC motif receptor 4, and MMP-9 proteins. In a xenograft mouse model, we observed that the administration of simvastatin alone (5 mg/kg body weight, intraperitoneal thrice/week) significantly suppressed the growth of the tumor and this effect was further potentiated by capecitabine treatment. As compared to the vehicle control, simvastatin also suppressed the expression of NF-κB-regulated gene products such as cyclin D1, COX-2, ICAM-1, MMP-9, survivin, Bcl-xL, and XIAP in tumor tissues. Overall, our results demonstrate that simvastatin can enhance the effects of capecitabine through suppression of NF-κB-regulated markers of proliferation, invasion, angiogenesis, and metastasis.
Key message
-
Simvastatin suppressed the proliferation and augmented apoptotic effects of capecitabine.
-
Simvastatin suppressed constitutive activation of NF-κB and its regulated genes.
-
Simvastatin enhanced the tumor growth inhibitory effects of capecitabine.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gastric cancer in spite of its decreasing incidence remains one of the most common causes of cancer-related deaths worldwide [1]. In addition to the surgery, various chemotherapeutic agents have been used in the gastric cancer patients, but still the median progression for survival remains 7–10 months [2]. Development of resistance to the chemotherapeutic drugs like capecitabine is frequently encountered in gastric cancer patients and contributes to the poor prognosis and overall median survival rate [3, 4]. Hence, novel treatment modalities to enhance the effects of chemotherapeutic drugs and reduce their resistance are required.
The pro-inflammatory transcription factor, NF-κB, and its regulated gene products play a critical role in the proliferation, metastasis, and chemoresistance of gastric cancer [4]. In the cells, NF-κB is composed of homodimers and heterodimers derived from five distinct subunits: RelA (p65), c-Rel, RelB, p50 (NF-κB1), and p52 (NF-κB2). All family members share a highly conserved Rel homology domain (∼300aa) responsible for DNA binding, dimerization domain, and interaction with IκBs, the intracellular inhibitor of NF-κB [5]. In the resting stage, majority of NF-κB complexes are localized predominantly in an inactive form in the cytoplasm by binding to a family of inhibitory proteins, the IκBs. Phosphorylation of these conserved serine residues in response to stimulator leads to the immediate polyubiquitination of IκB proteins by the SCF-β-TrCP complex. This modification subsequently targets IκB proteins for rapid degradation by the 26S proteasome. Activation of the NF-κB signaling cascade results in complete degradation of IκB, allowing translocation of NF-κB to the nucleus, where it induces gene transcription [6]. A number of NF-κB-regulated gene products, including cyclin D1, CXC motif receptor 4 (CXCR4), and cyclooxygenase-2 (COX-2), have been closely linked with the initiation, progression, and also the development of chemoresistance in gastric cancer [4–6]. Interestingly, Long and coworkers have also reported that NF-κB is activated in gastric carcinoma tissues and can be considered a prognostic marker of chemotherapy for human stage IV gastric carcinoma [7]. Also, the gastric mucosa of the individuals infected with Helicobacter pylori, a major risk factor for gastric cancer show upregulated NF-κB pathway and Th1-type cytokine responses, which can alter the overall integrity of the gut epithelial barrier [8]. Thus, the identification of novel pharmacological blockers of NF-κB activation cascade pathway can be a useful therapeutic strategy to circumvent chemoresistance in the gastric cancer patients.
In the present study, we determined whether simvastatin, a cholesterol-lowering drug [9], can sensitize the human gastric cancer to capecitabine in vitro and in a xenograft mouse model. The potential anti-carcinogenic effects of simvastatin alone or in combination with various anticancer therapies have been documented in a number of tumor cell lines and mouse models, including colorectal [10], hepatocellular [11], prostate [12], breast [13], and hematological malignancies [14]. How simvastatin exerts its anticancer effects is not fully understood, but it has been found to modulate various signaling cascades including mitogen-activated protein kinases [15, 16], PI3-K/Akt [17], NF-κB [18–20], STAT3 [14], cell cycle-dependent kinases [21], Rho-dependent kinase [22, 23], and Ras-related C3 botulinum toxin substrate 1 [24].
However, no prior reports exist in literature elaborating the potential anticancer effects of simvastatin in the gastric cancer alone or in combination with the chemotherapeutic agents. Hence, we investigated whether simvastatin can potentiate the anticancer effects of capecitabine in gastric cancer and through what molecular mechanism(s). Our findings suggest for the first time that simvastatin can suppress the proliferation of gastric cancer cells, potentiate capecitabine-induced apoptosis, and also the anticancer efficacy of capecitabine in the human xenograft gastric cancer model through the suppression of NF-κB-regulated gene products.
Materials and methods
Reagents
Simvastatin with purity >98 % was purchased from Alexis Biochemicals (San Diego, CA, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Tris base, glycine, NaCl, SDS, BSA, β-actin antibody, and corn oil were purchased from Sigma-Aldrich (St. Louis, MO). Simvastatin was dissolved in dimethylsulfoxide as a 50-mM stock solution and stored at 4 °C for in vitro and in corn oil for in vivo experiments, respectively. Further dilution was done in cell culture medium. RPMI-1640 media, fetal bovine serum (FBS), 0.4 % trypan blue vital stain, and antibiotic–antimycotic mixture were obtained from Invitrogen (Carlsbad, CA). Antibodies against p65, MMP-9, Bcl-2, Bcl-xL, XIAP, COX-2, cyclin D1, survivin, and VEGF were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). CXCR4 antibody was obtained from Abcam (Cambridge, MA, USA). CD31 antibody was purchased from and Cell Signaling Technology (Danvers, MA). Ki-67 antibody was purchased from BD PharMingen, Inc. (San Diego, CA). Goat anti-rabbit-horse radish peroxidase (HRP) conjugate and goat anti-mouse HRP were purchased from Invitrogen (Carlsbad, CA). Capecitabine was obtained from Duheng International Trading Company Ltd. Shanghai, China, and dissolved in sterile phosphate buffered saline (PBS) on the day of use.
Cell lines
The gastric cancer cell lines (SNU-5, SNU-16, MKN45 and AGS) were kindly provided by Prof. Patrick Tan from DUKE-NUS, Singapore. The gastric cancer cell lines were cultured in RPMI-1640 media supplemented with 10 % FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin.
Western blot analysis
For detection of various proteins in gastric cancer cells, 2 × 106/ml cells were treated with simvastatin for different time intervals and western blot analysis was performed as described previously [25].
Cell proliferation assay
The effect of simvastatin on cell proliferation was determined by the MTT uptake method as described previously [25].
Live and dead assay
To investigate whether simvastatin could enhance the apoptotic effects of capecitabine in gastric cancer cells, we used a LIVE/DEAD cell viability assay kit (Invitrogen), which is used to determine intracellular esterase activity and plasma membrane integrity [4]. Briefly, gastric cancer cells (5,000/well) were incubated in chamber slides, pretreated with simvastatin for 4 h, and treated with capecitabine for 24 h. Cells were then stained with the assay reagents for 30 min at room temperature. Cell viability was determined under a fluorescence microscope by counting live (green) and dead (red) cells.
Flow cytometric analysis
To determine the effect on the cell cycle, cells were exposed to combination of simvastatin for 4 h and treated with capecitabine for 24 h. Thereafter, cells were washed, fixed with 70 % ethanol, and incubated for 30 min at 37 °C with 0.1 % R Nase A in PBS. Cells were then washed again, resuspended, and stained in PBS containing 25 μg/ml propidium iodide for 30 min at room temperature. Cell distribution across the cell cycle was analyzed with a CyAn ADP flow cytometer (Dako Cytomation).
RNA isolation and reverse transcription
Total cellular RNA was extracted from untreated and simvastatin-treated cells using TRIZOL reagent (Invitrogen, Carlsbad, CA, USA) as described previously [25].
Real-time polymerase chain reaction
Real-time polymerase chain reaction (PCR) for Bcl-2, Bcl-xl, cyclin D1, VEGF, and Mcl-1 genes was performed as described previously [25].
Xenograft gastric tumor mouse model
All procedures involving animals were reviewed and approved by NUS Institutional Animal Care and Use Committee. Six-week-old athymic nu/nu female mice (Animal Resource Centre, Australia) were implanted subcutaneously in the right flank with SNU-5 cells (3 × 106 cells/100 μl saline). When tumors have reached 0.25 cm in diameter, the mice were randomized into the following treatment groups (n = 5/group): (a) untreated control (corn oil, 100 μL daily); (b) simvastatin (5 mg/kg bodyweight, suspended in corn oil, intraperitoneal [i.p.] injection) thrice/week; (c) capecitabine alone (60 mg/kg bodyweight, suspended in corn oil, twice weekly by gavage; and (d) combination (simvastatin, 5 mg/kg bodyweight, suspended in corn oil, i.p. injection thrice/week and capecitabine, 60 mg/kg bodyweight, suspended in corn oil, twice weekly by gavage). Therapy was continued for 4 weeks, and the animals were euthanized 1 week later. Primary tumors were excised and the final tumor volume was measured during the course of experiment and calculated using the formula V = 4 / 3πr 3: r is the radius of the tumor. Half of the tumor tissue was fixed in formalin and embedded in paraffin for immunohistochemistry analysis. The other half was snap frozen in liquid nitrogen and stored at −80 °C.
Immunohistochemical analysis of tumor samples
Solid tumors from control and treatment groups were fixed with 10 % phosphate-buffered formalin, processed, and embedded in paraffin. Sections were cut and deparaffinized in xylene, and dehydrated in graded alcohol, and finally hydrated in water. Antigen retrieval was performed by boiling the slide in 10 mM sodium citrate (pH 6.0) for 30 min. Immunohistochemistry was performed following manufacturer instructions (DAKO LSAB kit). Briefly, endogenous peroxidases were quenched with 3 % hydrogen peroxide. Non-specific binding was blocked by incubation in the blocking reagent in the LSAB kit (Dako, Carpinteria, CA) according to the manufacturer’s instructions. Sections were incubated overnight with primary antibodies as follows: anti-p65, anti-COX-2, anti-VEGF, anti-MMP-9, anti-Ki-67, and anti-CD31 (each at 1:100 dilutions). Slides were subsequently washed several times in Tris-buffered saline with 0.1 % Tween 20 and were incubated with biotinylated linker for 30 min, followed by incubation with streptavidin conjugate. Immunoreactive species were detected using 3,3-diaminobenzidine tetrahydrochloride as a substrate. Sections were counterstained with Gill’s hematoxylin and mounted under glass cover slips. Images were taken using an Olympus BX51 microscope (magnification, 40×).
Measurement of NF-κB activation in gastric cancer cells and tumor samples
To determine NF-κB activation, we performed DNA binding assay using TransAM NF-κB p65 transcription factor assay kit (Active Motif, Carlsbad, CA, USA) as described previously[4].
Statistical analysis
Statistical analysis was performed by Student’s t test and one-way analysis of variance. A p value of less than 0.05 was considered statistically significant.
Results
Simvastatin suppresses the proliferation and enhances the apoptotic effect of capecitabine in gastric cancer cells
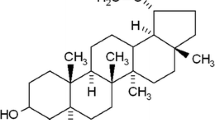
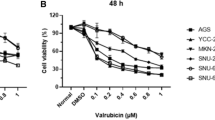
The aim of this study was to determine whether simvastatin, a cholesterol-lowering drug (with chemical structure shown in Fig. 1a), has a potential role in the treatment of gastric cancer either alone or in combination with capecitabine and if so, through what mechanism(s). We first analyzed the anticancer effect of simvastatin on the proliferation of four different gastric cancer cell lines. Simvastatin inhibited the growth of all three human gastric cancer cells (SNU-16, MKN-45, AGS, and SNU-5) in a dose- and time-dependent manner (Fig. 1b). Whether simvastatin can also enhance the apoptotic effect of capecitabine against gastric cancer cells was also determined. We use flow cytometric analysis and an esterase staining method (live/dead assay) to establish whether simvastatin can potentiate the apoptosis induced by capecitabine. As shown in Fig. 1c, d, the suboptimal doses of simvastatin (10 μM) or capecitabine (10 μM) that had minimum effect on apoptosis alone produced significant increase in apoptosis when used in combination against gastric cancer cells.

Simvastatin inhibits the proliferation and enhances the apoptotic effects of capecitabine in gastric cancer cells in vitro. a The chemical structure of simvastatin. b MTT assay results showed dose-dependent suppression of cell proliferation in four different gastric cancer cell lines treated with simvastatin. Points, mean of triplicate; bars, SE. Data are a representative of two independent experiments. c, d Flow cytometric analysis and Live/Dead assay results indicate that simvastatin (ST, 10 μM) treatment in combination with capecitabine (CAP, 10 μM) for 24 h can enhance apoptosis in gastric cancer cells. Data indicated as percentage proportions of apoptotic gastric cancer cells for Live/Dead assay. Values are mean ± SE of triplicate. Data are a representative of two independent experiments
Simvastatin inhibits the constitutive and inducible NF-κB activation in gastric cancer cells
We next examined how simvastatin enhances the apoptotic effects of capecitabine in gastric cancer cells. NF-κB has been found to be constitutively activated in gastric cancer and mediates resistance to apoptosis [4, 26–28]. Whether simvastatin induces downregulation of constitutive NF-κB activation in SNU-5 cells was investigated by using an ELISA-based DNA binding assay. The assay results indicate that the treatment with simvastatin inhibited constitutive NF-κB activation in SNU-5 cells in a dose- and time-dependent manner (Fig. 2a, b). We have previously reported that the chemotherapeutic agent capecitabine can cause NF-κB activation in a dose-dependent manner in MKN-45 cells, with significant activation observed at 25 μM [4]. Hence, we next determined whether simvastatin treatment can also modulate capecitabine-induced NF-κB activation in MKN-45 cells. Moreover, we found that simvastatin was also able to inhibit capecitabine-induced NF-κB activation in a dose-dependent manner in MKN-45 cells (Fig. 2c), thereby indicating that it is a potent suppressor of both constitutive and inducible NF-κB activation in gastric cancer cells. The master transcription factor NF-κB regulates the expression of a wide plethora of genes involved in the proliferation, survival, metastasis, angiogenesis, and chemoresistance in gastric cancer [4]. Whether simvastatin exposure can also modulate the expression of various NF-κB-regulated gene products was also examined. We found that the simvastatin suppressed the constitutive expression of antiproliferative (cyclin D1, COX-2), anti-apoptotic (Bcl-2, survivin), and invasive/metastatic (MMP-9, CXCR4) protein expression in a time-dependent manner in SNU-5 cells (Fig. 2d). Simvastatin also induced the cleavage of PARP in SNU-5 cells (Fig. 2d). Interestingly, we also noticed that there was a time-dependent decrease in the expression of various genes namely Bcl-2, Bcl-xL, survivin, VEGF, and Mcl-1 upon simvastatin treatment with maximal inhibitory effect observed at 48 h (Fig. 2e). Based on these intriguing findings in vitro, we proceeded to study the effect of simvastatin and capecitabine alone and in combination in a xenograft mouse model.

DNA binding assay results showing that simvastatin suppresses the constitutive activation of NF-κB in SNU-5 cells in a dose- and time-dependent manner. a SNU-5 (1 × 106) cells were treated with indicated concentrations of simvastatin for 8 h and nuclear extracts were prepared and assayed for NF-κB activation by ELISA-linked DNA binding assay. b SNU-5 (1 × 106) cells were treated with 50 μM simvastatin for 0, 2, 4, 6, and 8 h, and nuclear extracts were prepared and assayed for NF-κB activation by ELISA-linked DNA binding assay. c MKN-45 (1 × 106) cells were pretreated with simvastatin (10, 20, and 25 μM) for 12 h, stimulated with capecitabine 25 μM for 4 h, and then the nuclear extracts were prepared and assayed for NF-κB activation by ELISA-linked DNA binding assay. *p < 0.05. d Simvastatin suppressed the constitutive expression of gene products involved in proliferation, metastasis, and anti-apoptosis in gastric cancer cells. SNU-5 (1 × 106) cells were treated with 50 μM simvastatin for the indicated time points and western blot was performed as described under “Materials and methods” section. *p < 0.05. e Simvastatin suppressed the expression of various genes involved in tumor progression in gastric cancer cells. SNU-5 (1 × 106) cells were treated with 50 μM simvastatin for the indicated time points and real-time PCR analyses was performed as described under “Materials and methods” section. *p < 0.05
Simvastatin potentiates the antitumor effects of capecitabine in a xenograft gastric cancer mouse model
We examined the therapeutic efficacy of simvastatin and capecitabine either alone or in combination on the growth of subcutaneously implanted human gastric cancer cells in nude mice. The experimental protocol is depicted in Fig. 3a. SNU-5 cells were implanted subcutaneously in the right flank of nude mice. When tumors have reached 0.25 cm in diameter after a week, the mice were randomized into four groups and started the treatment as per the experimental protocol. The treatment was continued for 4 weeks and animals were sacrificed after 5 weeks. We found that simvastatin alone when given at 5 mg/kg body weight significantly inhibited the growth of the tumor (p < 0.001 when compared to control) (Fig. 3b, c). Capecitabine alone was also found to be quite effective (p < 0.001 when compared to control; p > 0.05 when compared to simvastatin alone group), and the combination of the two drugs was significantly more potent in reducing the tumor burden. The tumor volume in the combination of simvastatin and capecitabine group was significantly lower than simvastatin alone group (p < 0.001) or capecitabine alone group (p < 0.001) on day 35 (Fig. 3c, d).

Simvastatin potentiates the anticancer effect of capecitabine to inhibit growth of gastric cancer in xenograft mouse model. a Schematic representation of experimental protocol described in “Materials and methods.” Group I was given corn oil (100 μl, p.o., daily), group II was given simvastatin (5 mg/kg body weight, i.p. thrice/week), group III was given capecitabine (60 mg/kg body weight, twice weekly by gavage), and group IV was given simvastatin (5 mg/kg body weight, i.p. thrice/week) and capecitabine (60 mg/kg body weight, twice weekly by gavage). b Necropsy photographs of mice bearing subcutaneously implanted SNU-5 cells; c Tumor volumes in mice measured during the course of experiment and calculated using the formula V = 4 / 3πr 3. d Tumor volumes in mice measured on the last day of the experiment at autopsy using Vernier calipers and calculated using the formula V = 4 / 3πr 3 (n = 5). Columns, mean; bars, SE. ***p < 0.001
Simvastatin inhibits the expression of markers of proliferation and angiogenesis in gastric tumor tissues
While Ki-67-positive index is used as a marker for cell proliferation, the CD31 level is considered as marker for microvessel density. Whether simvastatin and capecitabine can affect these markers in gastric tumor tissues was also determined. Figure 4a shows that both simvastatin and capecitabine alone downregulated the expression of Ki-67 in tumor tissues and the combination of the two was most effective (p < 0.001). Also, when examined for CD31 expression levels, we found that both agents individually substantially reduced the CD31 expression and the combination of two was most effective (p < 0.001) (Fig. 4b).

Simvastatin enhances the effect of capecitabine against tumor cell proliferation and angiogenesis in gastric cancer. a Left panel, immunohistochemical analysis of proliferation marker Ki-67 indicates the inhibition of gastric cancer cell proliferation following simvastatin exposure either alone or in combination with capecitabine-treated groups of animals. A right panel, quantification of Ki-67+ cells as described in “Materials and methods.” Columns, mean of triplicate; bars, SE. ***p < 0.001. b Left panel, immunohistochemical analysis of CD31 for microvessel density in gastric cancer tumors indicates the inhibition of angiogenesis by either simvastatin alone and in combination with capecitabine; b right panel, quantification of CD31+ microvessel density as described in “Materials and methods.” Columns, mean of triplicate; bars, SE. ***p < 0.001
Simvastatin suppressed the expression of NF-κB-regulated gene products in gastric tumor tissues
We also investigated the effect of simvastatin and capecitabine on NF-κB levels in gastric tumor tissue. Figure 5a shows that simvastatin either alone or in combination was quite effective in suppressing the constitutive activation of NF-κB in gastric cancer tissues, whereas capecitabine treatment alone had no significant effect on constitutive NF-κB activation in gastric tissue as evident by DNA binding analysis (Fig. 5a).

Simvastatin enhances the effect of capecitabine against the activation of NF-κB and expression of NF-κB-regulated gene products in gastric cancer tissue samples. a Detection of NF-κB by DNA binding assay in tumor tissue samples showed the significant inhibition of NF-κB by combination, *p < 0.05; ***p < 0.001. b Western blot showing that combination of simvastatin and capecitabine inhibit the expression of NF-κB-dependent gene products involved in the proliferation (cyclin D1, COX-2, ICAM-1), invasion (MMP-9), and metastasis (CXCR4) in gastric tumor tissues. c Western blot showing that combination of simvastatin and capecitabine inhibits the expression of NF-κB-dependent gene products involved in the survival (Bcl-2, Bcl-xL, survivin, XIAP) and angiogenesis (VEGF) in gastric tumor tissues. Samples from three mice in each group were analyzed and representative data are shown
NF-κB is known to regulate the expression of number of proteins, including those involved in proliferation (cyclin D1, COX-2), invasion/metastasis (ICAM-1, MMP-9, CXCR4) and survival (Bcl-2, Bcl-xL, survivin, XIAP) [6]. Whether simvastatin can affect the expression of these diverse NF-κB-regulated gene products in tumor tissues was examined by western blot analysis. We found that treatment with combination of simvastatin and capecitabine was quite effective in substantially downregulating the expression of various gene products involved in the gastric cancer growth, survival, invasion, and metastasis (Fig. 5b, c).
The observed suppression of NF-κB, COX2, VEGF, and MMP-9 by western blot analysis was further confirmed by immunohistochemical methods. As shown in Fig. 6, these gene products were significantly downregulated in gastric tumor samples treated with simvastatin in combination with capecitabine. Overall, the data indicates that simvastatin can suppress the expression of various NF-κB-regulated gene products involved in the proliferation, survival, invasion, and angiogenesis, thereby causing inhibition of tumor growth in gastric cancer xenograft mouse model.

Immunohistochemical analysis of nuclear p65, COX-2, MMP-9, and VEGF showed the inhibition of these diverse biomarkers by either simvastatin alone or in combination with capecitabine in gastric tumor tissues. Percentage, positive staining for the given biomarker
Discussion
Despite significant improvements in the available treatment regimens following the introduction of targeted therapies, gastric cancer remains the second most lethal cancers, with only less than 20 % of patients surviving up to 5 years due to the existing problems of chemoresistance and tumor recurrence. Thus, novel pharmacological agents that are efficacious and can significantly enhance the effects of existing drugs are urgently needed. The goal of the present study was to investigate whether simvastatin, a cholesterol-lowering drug, can enhance the antitumor efficacy of capecitabine against human gastric cancer. We observed that simvastatin inhibited the proliferation of gastric cancer cells, enhanced capecitabine-induced apoptosis, modulated both constitutive as well the inducible NF-κB activation, and attenuated the expression of various NF-κB-regulated gene products. We also found that in a xenograft mouse model, simvastatin effectively suppressed the growth of gastric cancer alone and in combination with capecitabine.
We first noticed that simvastatin treatment can inhibit the proliferation of various gastric cancer cell lines in a dose- and time-dependent manner. Interestingly, the antiproliferative effects of simvastatin have not been evaluated previously in gastric cancer cells, although it has been found to suppress the proliferation of other gastrointestinal cancers. For example, in a recent study, Kodach and coworkers reported that simvastatin can reduce the proliferation and augment the chemosensitivity of colorectal cancer cells via the bone morphogenetic protein pathway [29]. Also, simvastatin has been found to attenuate the growth and malignant potential of human esophageal adenocarcinoma [30] and hepatic cancer cells [31], but most of these studies were restricted only to cell lines with no in vivo evidence. We observed that simvastatin caused the downregulation of cell proliferative gene products such as cyclin D1, which may explain its potent antiproliferative effects in gastric cancer.
We further observed that simvastatin when used in combination with capecitabine can cause increased apoptosis in three different gastric cancer cells. This is very intriguing because simvastatin has never been previously reported to induce apoptosis in gastric cancer cells either alone or in combination with the chemotherapeutic agents. We also noticed that this effect may be mediated due to the downregulation of cell survival proteins such as Bcl-2 and survivin in gastric cancer cells. Moreover, we also found that both the constitutive and capecitabine-induced NF-κB activation was suppressed by simvastatin in gastric cancer cells. We had previously reported that both constitutive and chemotherapy-induced NF-κB activation in chronic myeloid leukemia cells can be abrogated by simvastatin [18, 19], but its potential effect on NF-κB signaling cascade in gastric cancer cells has never been evaluated before. Also, simvastatin was observed to downregulate the expression of various invasive, metastatic, and angiogenic gene products (ICAM-1, MMP-9, CXCR4, and VEGF) which further support its application for gastric cancer treatment.
We also noticed for the first time that the intraperitoneal administration of simvastatin alone inhibited the growth of human gastric tumors when examined in vivo in a xenograft mouse model. Tumor growth was inhibited by more than 50 % on treatment with simvastatin and capecitabine alone, respectively. Also, when the two drugs were used in combination, they were found to be much more effective in modulating tumor growth. When investigated for the molecular mechanism(s) by which simvastatin exerts its effects in xenograft model, we noticed that the expression of proliferation marker Ki67 as well as microvessel density indicator CD31 was attenuated upon simvastatin treatment. Further analyses revealed that simvastatin caused the downregulation of NF-κB and various proteins (Cyclin D1, COX-2, survivin, Bcl-xL, XIAP, ICAM-1, MMP-9, and VEGF) regulated by NF-κB which play a pivotal role in the proliferation, survival, angiogenesis, and metastasis of gastric cancer. All of these observed effects were further enhanced upon capecitabine treatment. These findings are also consistent with a previous study from our group in which vitamin E-gamma tocotrienol was found to potentiate the effect of chemotherapy in gastric cancer through the modulation of NF-κB signaling cascade [4].
Simvastatin has been used in combination therapy with several chemotherapeutic agents/targeted therapies such as farnesyl transferase inhibitors, EGFR inhibitors geftinib and cetuximab, doxorubicin, non-steroidal anti-inflammatory drugs, and vitamin-E gamma tocotrienol in diverse tumor cell lines and in vivo cancer models [10, 13, 16, 32–34] but so far its effects either alone or in combination with anticancer therapies in gastric cancer mouse models has never been studied before. Recently conducted couple of population-based case–control studies clearly indicate that statins uptake can reduce the risk of gastric cancer [35], and our findings provide further scientific evidence that simvastatin has significant potential for the treatment of gastric cancer and its effects can be further enhanced by capecitabine. Finally, a phase III clinical study of capecitabine/CDDP chemotherapy plus a low dose of simvastatin is already in progress in Korea (http://www.clinicaltrials.gov) and may further help to substantiate our preclinical findings as reported here.
References
Jemal A, Siegel R, Xu J, Ward E (2010) Cancer statistics, 2010. CA Cancer J Clin 60:277–300
Zhao AG, Li T, You SF, Zhao HL, Gu Y, Tang LD, Yang JK (2008) Effects of Wei Chang An on expression of multiple genes in human gastric cancer grafted onto nude mice. World J Gastroenterol 14:693–700
Kim HK, Choi IJ, Kim CG, Kim HS, Oshima A, Michalowski A, Green JE (2011) A gene expression signature of acquired chemoresistance to cisplatin and fluorouracil combination chemotherapy in gastric cancer patients. PLoS One 6:e16694
Manu KA, Shanmugam MK, Ramachandran L, Li F, Fong CW, Kumar AP, Tan P, Sethi G (2012) First evidence that gamma-tocotrienol inhibits the growth of human gastric cancer and chemosensitizes it to capecitabine in a xenograft mouse model through the modulation of NF-kappaB pathway. Clin Cancer Res 18:2220–2229
Li F, Sethi G (2010) Targeting transcription factor NF-kappaB to overcome chemoresistance and radioresistance in cancer therapy. Biochim Biophys Acta 1805:167–180
Sethi G, Tergaonkar V (2009) Potential pharmacological control of the NF-κB pathway. Trends Pharmacol Sci 30:313–321
Long YM, Ye S, Rong J, Xie WR (2008) Nuclear factor kappa B: a marker of chemotherapy for human stage IV gastric carcinoma. World J Gastroenterol 14:4739–4744
Yang YJ, Chuang CC, Yang HB, Lu CC, Sheu BS (2012) Lactobacillus acidophilus ameliorates H. pylori-induced gastric inflammation by inactivating the Smad7 and NFkappaB pathways. BMC Microbiol 12:38
Thompson JS, Sood A, Arora R (2010) Statins and cancer: a potential link? Am J Ther 17:e100–e104
Lee J, Lee I, Han B, Park JO, Jang J, Park C, Kang WK (2011) Effect of simvastatin on cetuximab resistance in human colorectal cancer with KRAS mutations. J Natl Cancer Inst 103:674–688
Kah J, Wustenberg A, Keller AD, Sirma H, Montalbano R, Ocker M, Volz T, Dandri M, Tiegs G, Sass G (2012) Selective induction of apoptosis by HMG-CoA reductase inhibitors in hepatoma cells and dependence on p53 expression. Oncol Rep 28:1077–1083
Park YH, Seo SY, Lee E, Ku JH, Kim HH, Kwak C (2012) Simvastatin induces apoptosis in castrate resistant prostate cancer cells by deregulating NF-kB pathway. J Urol. 189: 1547–1552
Gopalan A, Yu W, Sanders BG, Kline K (2013) Eliminating drug resistant breast cancer stem-like cells with combination of simvastatin and gamma-tocotrienol. Cancer Lett 328:285–296
Oh B, Kim TY, Min HJ, Kim M, Kang MS, Huh JY, Kim Y, Lee DS (2013) Synergistic killing effect of imatinib and simvastatin on imatinib-resistant chronic myelogenous leukemia cells. Anticancer Drugs 24:20–31
Gopalan A, Yu W, Sanders BG, Kline K (2012) Simvastatin inhibition of mevalonate pathway induces apoptosis in human breast cancer cells via activation of JNK/CHOP/DR5 signaling pathway. Cancer Lett. 329: 9–16
Pelaia G, Gallelli L, Renda T, Fratto D, Falcone D, Caraglia M, Busceti MT, Terracciano R, Vatrella A, Maselli R et al (2012) Effects of statins and farnesyl transferase inhibitors on ERK phosphorylation, apoptosis and cell viability in non-small lung cancer cells. Cell Prolif 45:557–565
Kochuparambil ST, Al-Husein B, Goc A, Soliman S, Somanath PR (2011) Anticancer efficacy of simvastatin on prostate cancer cells and tumor xenografts is associated with inhibition of Akt and reduced prostate-specific antigen expression. J Pharmacol Exp Ther 336:496–505
Ahn KS, Sethi G, Aggarwal BB (2007) Simvastatin potentiates TNF-alpha-induced apoptosis through the down-regulation of NF-kappaB-dependent antiapoptotic gene products: role of IkappaBalpha kinase and TGF-beta-activated kinase-1. J Immunol 178:2507–2516
Ahn KS, Sethi G, Aggarwal BB (2008) Reversal of chemoresistance and enhancement of apoptosis by statins through down-regulation of the NF-kappaB pathway. Biochem Pharmacol 75:907–913
Ahn KS, Sethi G, Chaturvedi MM, Aggarwal BB (2008) Simvastatin, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, suppresses osteoclastogenesis induced by receptor activator of nuclear factor-kappaB ligand through modulation of NF-kappaB pathway. Int J Cancer 123:1733–1740
Tu YS, Kang XL, Zhou JG, Lv XF, Tang YB, Guan YY (2011) Involvement of Chk1-Cdc25A-cyclin A/CDK2 pathway in simvastatin induced S-phase cell cycle arrest and apoptosis in multiple myeloma cells. Eur J Pharmacol 670:356–364
Kidera Y, Tsubaki M, Yamazoe Y, Shoji K, Nakamura H, Ogaki M, Satou T, Itoh T, Isozaki M, Kaneko J et al (2010) Reduction of lung metastasis, cell invasion, and adhesion in mouse melanoma by statin-induced blockade of the Rho/Rho-associated coiled-coil-containing protein kinase pathway. J Exp Clin Cancer Res 29:127
Relja B, Meder F, Wang M, Blaheta R, Henrich D, Marzi I, Lehnert M (2011) Simvastatin modulates the adhesion and growth of hepatocellular carcinoma cells via decrease of integrin expression and ROCK. Int J Oncol 38:879–885
Miller T, Yang F, Wise CE, Meng F, Priester S, Munshi MK, Guerrier M, Dostal DE, Glaser SS (2011) Simvastatin stimulates apoptosis in cholangiocarcinoma by inhibition of Rac1 activity. Dig Liver Dis 43:395–403
Ramachandran L, Manu KA, Shanmugam MK, Li F, Siveen KS, Vali S, Kapoor S, Abbasi T, Surana R, Smoot DT et al (2012) Isorhamnetin inhibits proliferation and invasion and induces apoptosis through the modulation of peroxisome proliferator-activated receptor gamma activation pathway in gastric cancer. J Biol Chem 287:38028–38040
Camp ER, Li J, Minnich DJ, Brank A, Moldawer LL, MacKay SL, Hochwald SN (2004) Inducible nuclear factor-kappaB activation contributes to chemotherapy resistance in gastric cancer. J Am Coll Surg 199:249–258
Yu LL, Dai N, Yu HG, Sun LM, Si JM (2010) Akt associates with nuclear factor kappaB and plays an important role in chemoresistance of gastric cancer cells. Oncol Rep 24:113–119
Nam SY, Ko YS, Jung J, Yoon J, Kim YH, Choi YJ, Park JW, Chang MS, Kim WH, Lee BL (2011) A hypoxia-dependent upregulation of hypoxia-inducible factor-1 by nuclear factor-kappaB promotes gastric tumour growth and angiogenesis. Br J Cancer 104:166–174
Kodach LL, Jacobs RJ, Voorneveld PW, Wildenberg ME, Verspaget HW, van Wezel T, Morreau H, Hommes DW, Peppelenbosch MP, van den Brink GR et al (2011) Statins augment the chemosensitivity of colorectal cancer cells inducing epigenetic reprogramming and reducing colorectal cancer cell ‘stemness’ via the bone morphogenetic protein pathway. Gut 60:1544–1553
Sadaria MR, Reppert AE, Yu JA, Meng X, Fullerton DA, Reece TB, Weyant MJ (2011) Statin therapy attenuates growth and malignant potential of human esophageal adenocarcinoma cells. J Thorac Cardiovasc Surg 142:1152–1160
Relja B, Meder F, Wilhelm K, Henrich D, Marzi I, Lehnert M (2010) Simvastatin inhibits cell growth and induces apoptosis and G0/G1 cell cycle arrest in hepatic cancer cells. Int J Mol Med 26:735–741
Han JY, Lee SH, Yoo NJ, Hyung LS, Moon YJ, Yun T, Kim HT, Lee JS (2011) A randomized phase II study of gefitinib plus simvastatin versus gefitinib alone in previously treated patients with advanced non-small cell lung cancer. Clin Cancer Res 17:1553–1560
Sadeghi-Aliabadi H, Minaiyan M, Dabestan A (2010) Cytotoxic evaluation of doxorubicin in combination with simvastatin against human cancer cells. Res Pharm Sci 5:127–133
Xiao H, Yang CS (2008) Combination regimen with statins and NSAIDs: a promising strategy for cancer chemoprevention. Int J Cancer 123:983–990
Leung HW, Chan AL, Lo D, Leung JH, Chen HL (2012) Common cancer risk and statins: a population-based case–control study in a Chinese population. Expert Opin Drug Saf. 12:19−27
Acknowledgments
This work was supported by grant from National Medical Research Council of Singapore [R-184-000-211-213] to GS. APK was supported by grants from Singapore Ministry of Education Tier 2 [MOE2012-T2-2-139], Academic Research Fund Tier 1 [R-184-000-228-112], and Cancer Science Institute of Singapore, Experimental Therapeutics I Program [Grant R-713-001-011-271].
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Both KAM and MKS contributed equally to this work.
Rights and permissions
About this article
Cite this article
Manu, K.A., Shanmugam, M.K., Li, F. et al. Simvastatin sensitizes human gastric cancer xenograft in nude mice to capecitabine by suppressing nuclear factor-kappa B-regulated gene products. J Mol Med 92, 267–276 (2014). https://doi.org/10.1007/s00109-013-1095-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-013-1095-0